
Genome-Wide Association Study and Candidate Gene Mining of Seed Size Traits in Soybean


 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Planting Accessions and Field Experiment Design
2.2. Phenotypic Assessment and Statistical Analysis
2.3. Genome-Wide Association Analysis
2.4. Analysis of QTN Distribution in Extreme Germplasm
2.5. Important QTNs and Haplotype Analysis
2.6. Candidate Gene Mining within the Major Loci
3. Results
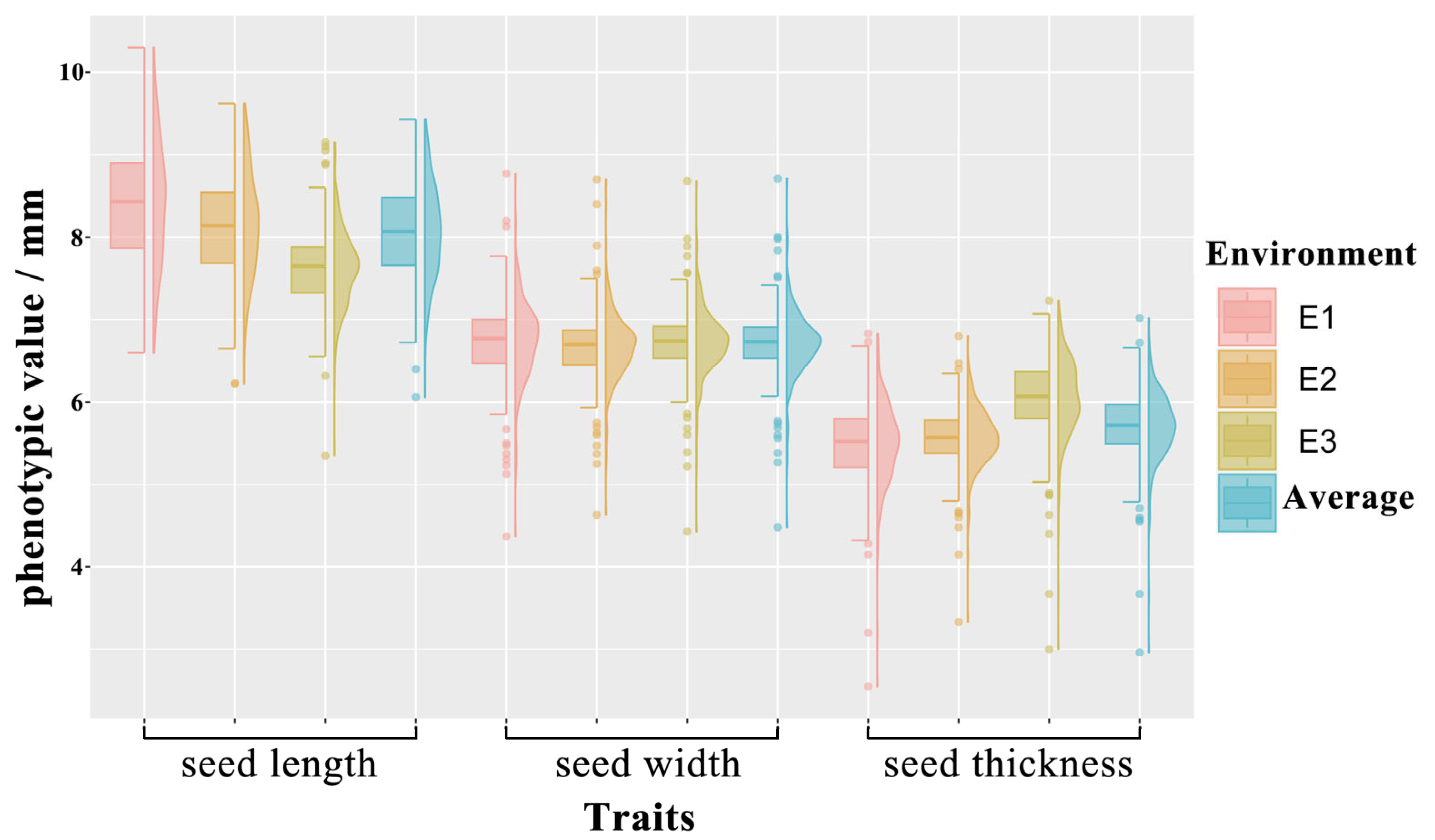
3.1. Phenotypic Evaluation of Seed Size Traits in the Mapping Panel
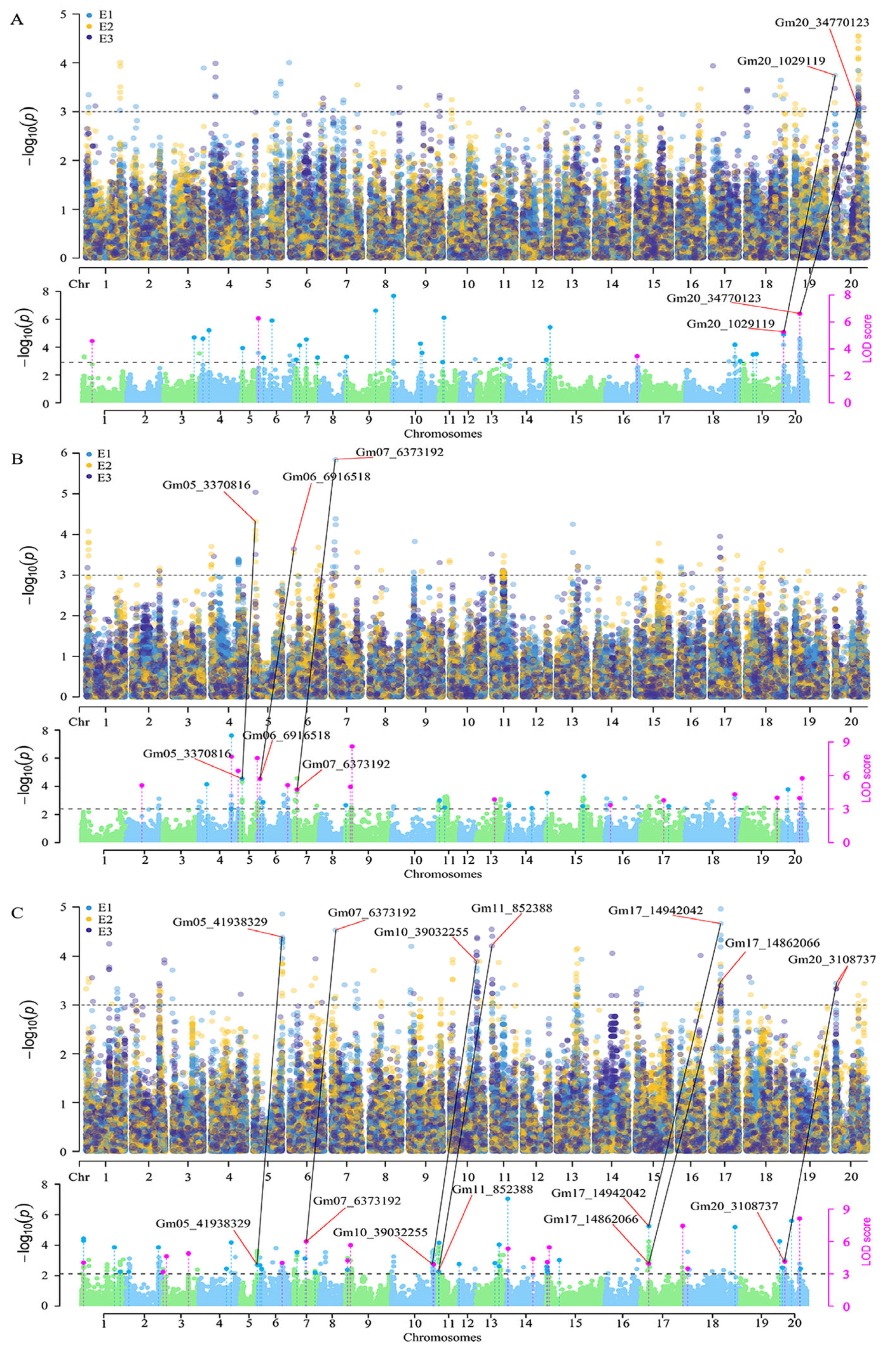
3.2. GWAS for Seed-Size Traits
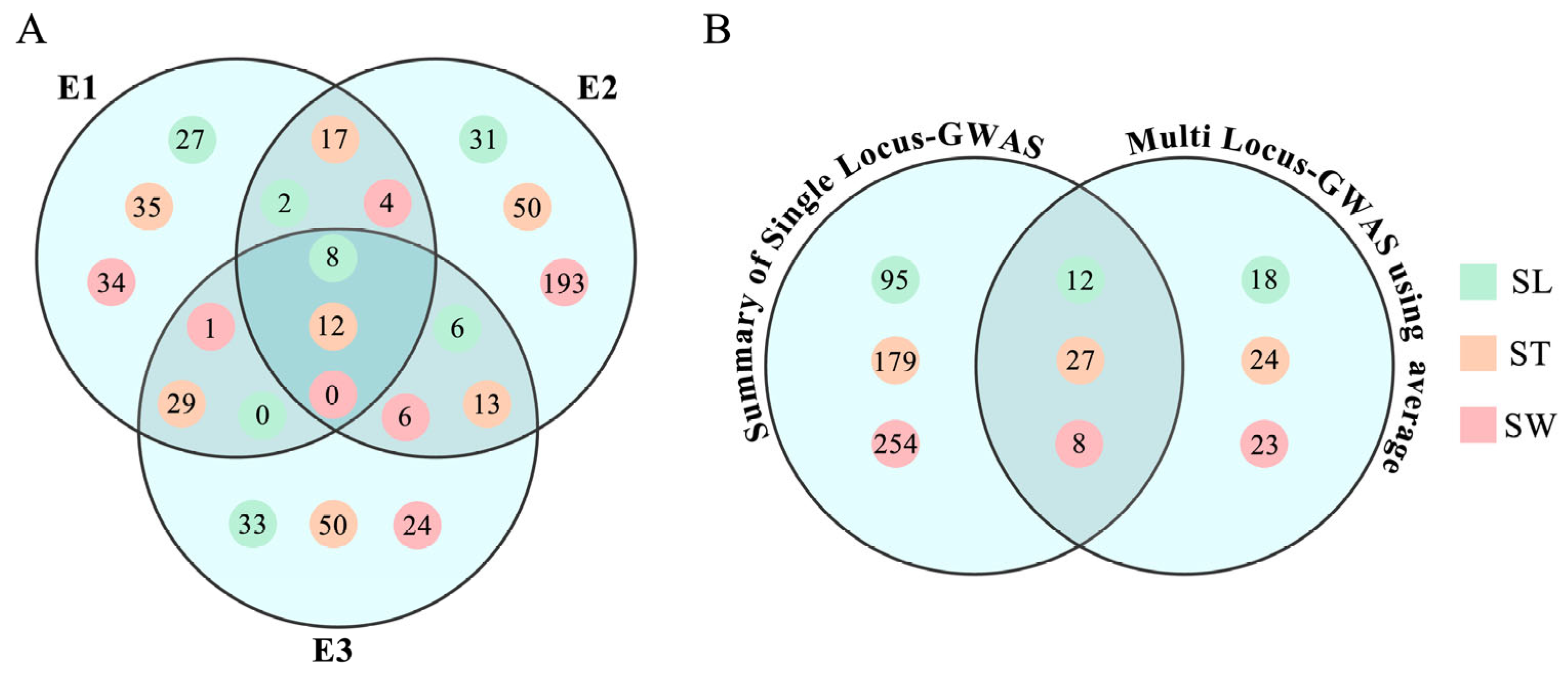
3.3. Distribution of the QTNs in Extreme Accessions
3.4. Haplotype Analysis and Excellent Allele Excavation for the Important QTNs
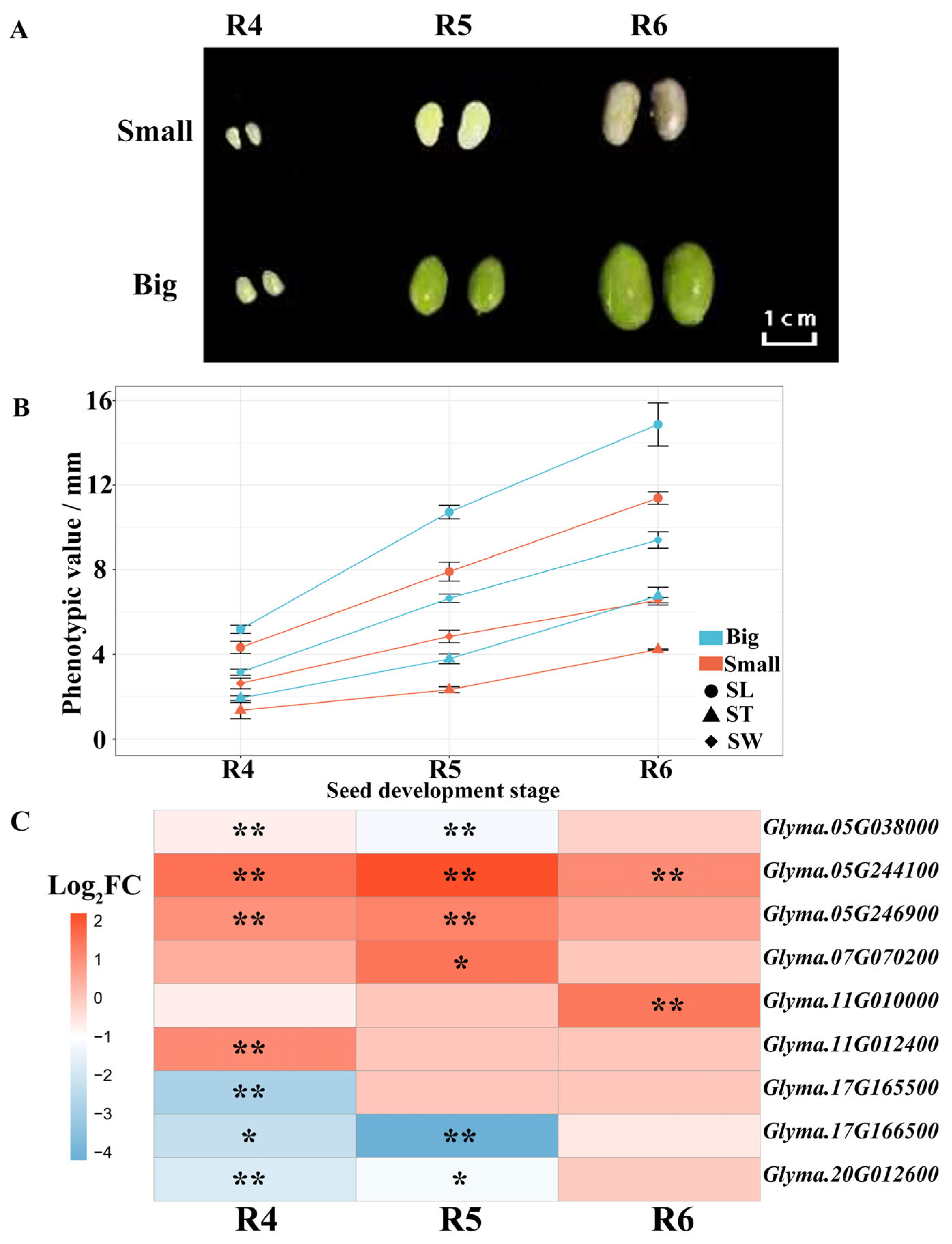
3.5. Candidate Gene Discovery for Seed Size Traits Underlying the Major Loci
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- SoyStats®2023. Available online: http://soystats.com/ (accessed on 24 April 2024).
- Adebisi, M.A.; Kehinde, T.O.; Salau, A.W.; Okesola, L.A.; Porbeni, J.B.O.; Esuruoso, A.O.; Oyekale, K.O. Influence of Different Seed Size Fractions on Seed Germination, Seedling Emergence and Seed Yield Characters in Tropical Soybean (Glycine max L. Merrill). Int. J. Agric. Res. 2013, 8, 26–33. [Google Scholar] [CrossRef]
- Morrison, M.J.; Xue, A.G. The Influence of Seed Size on Soybean Yield in Short-Season Regions. Can. J. Plant Sci. 2007, 87, 89–91. [Google Scholar] [CrossRef]
- Ray, D.D.; Sen, S.; Bhattacharyya, P.K.; Bhattacharyya, S. Study on Seed Size Variation in Soybean (Glycine max L. Merr.) and Its Correlation with Yield. Int. J. Econ. Plants 2022, 9, 204–209. [Google Scholar]
- Luo, S.; Jia, J.; Liu, R.; Wei, R.; Guo, Z.; Cai, Z.; Chen, B.; Liang, F.; Xia, Q.; Nian, H. Identification of Major QTLs for Soybean Seed Size and Seed Weight Traits Using a RIL Population in Different Environments. Front. Plant Sci. 2023, 13, 1094112. [Google Scholar] [CrossRef] [PubMed]
- Elattar, M.A.; Karikari, B.; Li, S.; Song, S.; Cao, Y.; Aslam, M.; Hina, A.; Abou-Elwafa, S.F.; Zhao, T. Identification and Validation of Major QTLs, Epistatic Interactions, and Candidate Genes for Soybean Seed Shape and Weight Using Two Related RIL Populations. Front. Genet. 2021, 12, 666440. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Ma, R.; Li, X.; Liu, J.; Jiang, A.; Tan, P.; Xiong, G.; Du, C.; Zhang, J.; Zhang, X. Construction of Genetic Map and QTL Mapping for Seed Size and Quality Traits in Soybean (Glycine max L.). Int. J. Mol. Sci. 2024, 25, 2857. [Google Scholar] [CrossRef] [PubMed]
- Jiang, A.; Liu, J.; Gao, W.; Ma, R.; Tan, P.; Liu, F.; Zhang, J. Construction of a Genetic Map and QTL Mapping of Seed Size Traits in Soybean. Front. Genet. 2023, 14, 1248315. [Google Scholar] [CrossRef]
- Yang, H.; Wang, W.; He, Q.; Xiang, S.; Tian, D.; Zhao, T.; Gai, J. Chromosome Segment Detection for Seed Size and Shape Traits Using an Improved Population of Wild Soybean Chromosome Segment Substitution Lines. Physiol. Mol. Biol. Plants 2017, 23, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Teng, W.L.; Sui, M.N.; Li, W.; Wu, D.P.; Zhao, X.; Li, H.Y.; Han, Y.P.; Li, W.B. Identification of Quantitative Trait Loci Underlying Seed Shape in Soybean across Multiple Environments. J. Agric. Sci. 2018, 156, 3–12. [Google Scholar] [CrossRef]
- Hina, A.; Cao, Y.; Song, S.; Li, S.; Sharmin, R.A.; Elattar, M.A.; Bhat, J.A.; Zhao, T. High-Resolution Mapping in Two RIL Populations Refines Major “QTL Hotspot” Regions for Seed Size and Shape in Soybean (Glycine max L.). Int. J. Mol. Sci. 2020, 21, 1040. [Google Scholar] [CrossRef]
- Li, M.; Chen, L.; Zeng, J.; Razzaq, M.K.; Xu, X.; Xu, Y.; Wang, W.; He, J.; Xing, G.; Gai, J. Identification of Additive–Epistatic QTLs Conferring Seed Traits in Soybean Using Recombinant Inbred Lines. Front. Plant Sci. 2020, 11, 566056. [Google Scholar] [CrossRef] [PubMed]
- Kumawat, G.; Xu, D. A Major and Stable Quantitative Trait Locus qSS2 for Seed Size and Shape Traits in a Soybean RIL Population. Front. Genet. 2021, 12, 646102. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Saini, M.; Taku, M.; Debbarma, P.; Mahto, R.K.; Ramlal, A.; Sharma, D.; Rajendran, A.; Pandey, R.; Gaikwad, K.; et al. Identification of Quantitative Trait Loci (QTLs) and Candidate Genes for Seed Shape and 100-Seed Weight in Soybean [Glycine max (L.) Merr.]. Front. Plant Sci. 2023, 13, 1074245. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Xiong, Q.; Cheng, T.; Li, Q.-T.; Liu, X.-L.; Bi, Y.-D.; Li, W.; Zhang, W.-K.; Ma, B.; Lai, Y.-C.; et al. A PP2C-1 Allele Underlying a Quantitative Trait Locus Enhances Soybean 100-Seed Weight. Mol. Plant 2017, 10, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Yang, C.; Yong, B.; Wang, Y.; Li, B.; Gu, Y.; Wei, S.; An, Z.; Sun, W.; Qiu, L.; et al. An Enhancing Effect Attributed to a Nonsynonymous Mutation in SOYBEAN SEED SIZE 1, a SPINDLY-like Gene, Is Exploited in Soybean Domestication and Improvement. New Phytol. 2022, 236, 1375–1392. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.X.; Paddock, K.J.; Zhang, Z.; Stacey, M.G. GmKIX8-1 Regulates Organ Size in Soybean and Is the Causative Gene for the Major Seed Weight QTL qSw17-1. New Phytol. 2021, 229, 920–934. [Google Scholar] [CrossRef] [PubMed]
- Rafalski, J.A. Association Genetics in Crop Improvement. Curr. Opin. Plant Biol. 2010, 13, 174–180. [Google Scholar] [CrossRef]
- Ibrahim, A.K.; Zhang, L.; Niyitanga, S.; Afzal, M.Z.; Xu, Y.; Zhang, L.; Zhang, L.; Qi, J. Principles and Approaches of Association Mapping in Plant Breeding. Trop. Plant Biol. 2020, 13, 212–224. [Google Scholar] [CrossRef]
- Khan, S.U.; Yangmiao, J.; Liu, S.; Zhang, K.; Khan, M.H.U.; Zhai, Y.; Olalekan, A.; Fan, C.; Zhou, Y. Genome-Wide Association Studies in the Genetic Dissection of Ovule Number, Seed Number, and Seed Weight in Brassica napus L. Ind. Crop. Prod. 2019, 142, 111877. [Google Scholar] [CrossRef]
- Tao, Y.; Zhao, X.; Wang, X.; Hathorn, A.; Hunt, C.; Cruickshank, A.W.; van Oosterom, E.J.; Godwin, I.D.; Mace, E.S.; Jordan, D.R. Large-scale GWAS in Sorghum Reveals Common Genetic Control of Grain Size among Cereals. Plant Biotechnol. J. 2020, 18, 1093–1105. [Google Scholar] [CrossRef]
- Kabange, N.R.; Dzorkpe, G.D.; Park, D.-S.; Kwon, Y.; Lee, S.-B.; Lee, S.-M.; Kang, J.-W.; Jang, S.-G.; Oh, K.-W.; Lee, J.-H. Rice (Oryza sativa L.) Grain Size, Shape, and Weight-Related QTLs Identified Using GWAS with Multiple GAPIT Models and High-Density SNP Chip DNA Markers. Plants 2023, 12, 4044. [Google Scholar] [CrossRef]
- Li, J.; Zhao, J.; Li, Y.; Gao, Y.; Hua, S.; Nadeem, M.; Sun, G.; Zhang, W.; Hou, J.; Wang, X.; et al. Identification of a Novel Seed Size Associated Locus SW9-1 in Soybean. Crop J. 2019, 7, 548–559. [Google Scholar] [CrossRef]
- Duan, Z.; Zhang, M.; Zhang, Z.; Liang, S.; Fan, L.; Yang, X.; Yuan, Y.; Pan, Y.; Zhou, G.; Liu, S.; et al. Natural Allelic Variation of GmST05 Controlling Seed Size and Quality in Soybean. Plant Biotechnol. J. 2022, 20, 1807–1818. [Google Scholar] [CrossRef]
- Shao, Z.; Shao, J.; Huo, X.; Li, W.; Kong, Y.; Du, H.; Li, X.; Zhang, C. Identification of Closely Associated SNPs and Candidate Genes with Seed Size and Shape via Deep Re-Sequencing GWAS in Soybean. Theor. Appl. Genet. 2022, 135, 2341–2351. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, Y.; Ma, R.; Huang, W.; Hou, J.; Fang, C.; Wang, L.; Yuan, Z.; Sun, Q.; Dong, X.; et al. Identification of ST1 Reveals a Selection Involving Hitchhiking of Seed Morphology and Oil Content during Soybean Domestication. Plant Biotechnol. J. 2022, 20, 1110–1121. [Google Scholar] [CrossRef]
- Duan, Z.; Li, Q.; Wang, H.; He, X.; Zhang, M. Genetic Regulatory Networks of Soybean Seed Size, Oil and Protein Contents. Front. Plant Sci. 2023, 14, 1160418. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Zhang, X.; Jia, S.; Karikari, B.; Zhang, M.; Xia, Z.; Zhao, T.; Liang, F. Genome-Wide Association among Soybean Accessions for the Genetic Basis of Salinity-Alkalinity Tolerance during Germination. Crop Pasture Sci. 2021, 72, 255–267. [Google Scholar] [CrossRef]
- Douglas Bates, M.M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using Lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar]
- Nyquist, W.E.; Baker, R.J. Estimation of Heritability and Prediction of Selection Response in Plant Populations. Crit. Rev. Plant Sci. 1991, 10, 235–322. [Google Scholar] [CrossRef]
- Cao, Y.; Jia, S.; Chen, L.; Zeng, S.; Zhao, T.; Karikari, B. Identification of Major Genomic Regions for Soybean Seed Weight by Genome-Wide Association Study. Mol. Breed. 2022, 42, 38. [Google Scholar] [CrossRef]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome Association and Prediction Integrated Tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef] [PubMed]
- Tamba, C.L.; Zhang, Y.-M. A Fast mrMLM Algorithm for Multi-Locus Genome-Wide Association Studies. bioRxiv 2018. [Google Scholar] [CrossRef]
- Wen, Y.-J.; Zhang, H.; Ni, Y.-L.; Huang, B.; Zhang, J.; Feng, J.-Y.; Wang, S.-B.; Dunwell, J.M.; Zhang, Y.-M.; Wu, R. Methodological Implementation of Mixed Linear Models in Multi-Locus Genome-Wide Association Studies. Brief. Bioinform. 2018, 19, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-B.; Feng, J.-Y.; Ren, W.-L.; Huang, B.; Zhou, L.; Wen, Y.-J.; Zhang, J.; Dunwell, J.M.; Xu, S.; Zhang, Y.-M. Improving Power and Accuracy of Genome-Wide Association Studies via a Multi-Locus Mixed Linear Model Methodology. Sci. Rep. 2016, 6, 19444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Feng, J.-Y.; Ni, Y.L.; Wen, Y.J.; Niu, Y.; Tamba, C.L.; Yue, C.; Song, Q.; Zhang, Y.M. pLARmEB: Integration of Least Angle Regression with Empirical Bayes for Multilocus Genome-Wide Association Studies. Heredity 2017, 118, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.-L.; Wen, Y.-J.; Dunwell, J.M.; Zhang, Y.-M. pKWmEB: Integration of Kruskal–Wallis Test with Empirical Bayes under Polygenic Background Control for Multi-Locus Genome-Wide Association Study. Heredity 2018, 120, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Tamba, C.L.; Ni, Y.-L.; Zhang, Y.-M. Iterative Sure Independence Screening EM-Bayesian LASSO Algorithm for Multi-Locus Genome-Wide Association Studies. PLoS Comput. Biol. 2017, 13, e1005357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-W.; Tamba, C.L.; Wen, Y.-J.; Li, P.; Ren, W.-L.; Ni, Y.-L.; Gao, J.; Zhang, Y.-M. mrMLM v4. 0.2: An R Platform for Multi-Locus Genome-Wide Association Studies. Genom. Proteom. Bioinform. 2020, 18, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Song, J.; Zhang, K.; Liu, S.; Tian, X.; Wang, Y.; Fang, Y.; Li, X.; Wang, J.; Yang, C.; et al. Identification of QTNs Controlling 100-Seed Weight in Soybean Using Multilocus Genome-Wide Association Studies. Front. Genet. 2020, 11, 689. [Google Scholar] [CrossRef]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and Visualization of LD and Haplotype Maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef]
- Tang, X.; Xue, Y.; Cao, D.; Luan, X.; Zhao, K.; Liu, Q.; Ren, Y.; Zhu, Z.; Li, Y.; Liu, X. Identification of Candidate Genes for Drought Resistance during Soybean Seed Development. Agriculture 2023, 13, 949. [Google Scholar] [CrossRef]
- Zhang, Y.; Bhat, J.A.; Zhang, Y.; Yang, S. Understanding the Molecular Regulatory Networks of Seed Size in Soybean. Int. J. Mol. Sci. 2024, 25, 1441. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, A.P. Quality of Soybean and Its Food Products. Int. Food Res. J. 2009, 16, 11–19. [Google Scholar]
- Xu, M.; Kong, K.; Miao, L.; He, J.; Liu, T.; Zhang, K.; Yue, X.; Jin, T.; Gai, J.; Li, Y. Identification of Major Quantitative Trait Loci and Candidate Genes for Seed Weight in Soybean. Theor. Appl. Genet. 2023, 136, 22. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Chen, L.; Yang, Y.; Liao, H. Genetic Analysis and Map-based Delimitation of a Major Locus qSS3 for Seed Size in Soybean. Plant Breed. 2020, 139, 1145–1157. [Google Scholar] [CrossRef]
- Dong, Y.-Y.; Ji, L.I.U.; Zhang, X.-C.; Feng, L.I.N.; Shi, F.-F.; Bo, W.; Xue, F.U.; Xue, Z.; Han, Y.-P.; Li, W.-B. Genome-Wide Association Analysis of Seed Size Traits in Soybean under Multiple Environments. Chin. J. Oil Crop Sci. 2023, 45, 111–123. [Google Scholar]
- Niu, M.; Tian, K.; Chen, Q.; Yang, C.; Zhang, M.; Sun, S.; Wang, X. A Multi-Trait GWAS-Based Genetic Association Network Controlling Soybean Architecture and Seed Traits. Front. Plant Sci. 2024, 14, 1302359. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Ma, Y.; Wu, S.; Liu, Z.; Wang, Z.; Yang, R.; Hu, G.; Zhou, Z.; Yu, H.; Zhang, M.; et al. Genome-Wide Association Studies Dissect the Genetic Networks Underlying Agronomical Traits in Soybean. Genome Biol. 2017, 18, 161. [Google Scholar] [CrossRef] [PubMed]
- Dargahi, H.; Tanya, P.; Srinives, P. Detection of Quantitative Trait Loci for Seed Size Traits in Soybean (Glycine max L.). Agric. Nat. Resour. 2015, 49, 832–843. [Google Scholar]
- Qi, X.; Li, M.-W.; Xie, M.; Liu, X.; Ni, M.; Shao, G.; Song, C.; Kay-Yuen Yim, A.; Tao, Y.; Wong, F.-L.; et al. Identification of a Novel Salt Tolerance Gene in Wild Soybean by Whole-Genome Sequencing. Nat. Commun. 2014, 5, 4340. [Google Scholar] [CrossRef]
- Salas, P.; Oyarzo-Llaipen, J.C.; Wang, D.; Chase, K.; Mansur, L. Genetic Mapping of Seed Shape in Three Populations of Recombinant Inbred Lines of Soybean (Glycine max L. Merr.). Theor. Appl. Genet. 2006, 113, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Jun, T.-H.; Freewalt, K.; Michel, A.P.; Mian, R. Identification of Novel QTL for Leaf Traits in Soybean. Plant Breed. 2014, 133, 61–66. [Google Scholar] [CrossRef]
- Li, N.; Li, Y. Signaling Pathways of Seed Size Control in Plants. Curr. Opin. Plant Biol. 2016, 33, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Xu, R.; Li, Y. Molecular Networks of Seed Size Control in Plants. Annu. Rev. Plant Biol. 2019, 70, 435–463. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wang, S.; He, C.; Zhou, B.; Ruan, Y.-L.; Shou, H. Identification of Regulatory Networks and Hub Genes Controlling Soybean Seed Set and Size Using RNA Sequencing Analysis. J. Exp. Bot. 2017, 68, 1955–1972. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Dai, A.; Wei, H.; Yang, S.; Wang, B.; Jiang, N.; Feng, X. Arabidopsis KLU Homologue GmCYP78A72 Regulates Seed Size in Soybean. Plant Mol. Biol. 2016, 90, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Yu, J.; Wang, H.; Luth, D.; Bai, G.; Wang, K.; Chen, R. Increasing Seed Size and Quality by Manipulating BIG SEEDS1 in Legume Species. Proc. Natl. Acad. Sci. USA 2016, 113, 12414–12419. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, S.; Wang, J.; Yokosho, K.; Zhou, B.; Yu, Y.-C.; Liu, Z.; Frommer, W.B.; Ma, J.F.; Chen, L.-Q.; et al. Simultaneous Changes in Seed Size, Oil Content and Protein Content Driven by Selection of SWEET Homologues during Soybean Domestication. Natl. Sci. Rev. 2020, 7, 1776–1786. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Wang, J.-W.; Li, J.; Han, B. Designing Future Crops: Challenges and Strategies for Sustainable Agriculture. Plant J. 2021, 105, 1165–1178. [Google Scholar] [CrossRef]
- Jiang, W.-B.; Huang, H.-Y.; Hu, Y.-W.; Zhu, S.-W.; Wang, Z.-Y.; Lin, W.-H. Brassinosteroid Regulates Seed Size and Shape in Arabidopsis. Plant Physiol. 2013, 162, 1965–1977. [Google Scholar] [CrossRef]
- Bartels, A. Regulation of Seed Development in Arabidopsis Thaliana. Ph.D. Thesis, Saint Louis University, Saint Louis, MO, USA, 2021. [Google Scholar]
- Dong, N.-Q.; Sun, Y.; Guo, T.; Shi, C.-L.; Zhang, Y.-M.; Kan, Y.; Xiang, Y.-H.; Zhang, H.; Yang, Y.-B.; Li, Y.-C.; et al. UDP-Glucosyltransferase Regulates Grain Size and Abiotic Stress Tolerance Associated with Metabolic Flux Redirection in Rice. Nat. Commun. 2020, 11, 2629. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Environment a | Mean (mm) | SD | Max (mm) | Min (mm) | CV (%) | h2 |
|---|---|---|---|---|---|---|---|
| Seed length | E1 | 8.40 | 0.74 | 10.30 | 6.60 | 8.76 | 0.72 |
| E2 | 8.14 | 0.63 | 9.62 | 6.22 | 7.73 | ||
| E3 | 7.63 | 0.46 | 9.15 | 5.35 | 6.09 | ||
| Average | 8.06 | 0.56 | 9.43 | 6.06 | 6.96 | ||
| Seed width | E1 | 6.74 | 0.49 | 8.77 | 4.37 | 7.23 | 0.74 |
| E2 | 6.67 | 0.41 | 8.70 | 4.63 | 6.22 | ||
| E3 | 6.73 | 0.41 | 8.68 | 4.43 | 6.04 | ||
| Average | 6.71 | 0.40 | 8.71 | 4.48 | 5.93 | ||
| Seed thickness | E1 | 5.49 | 0.49 | 6.83 | 2.55 | 8.97 | 0.75 |
| E2 | 5.57 | 0.36 | 6.80 | 3.33 | 6.55 | ||
| E3 | 6.05 | 0.49 | 7.23 | 3.00 | 8.16 | ||
| Average | 5.70 | 0.42 | 7.02 | 2.96 | 7.31 |
| Trait | Locus Name | Chromosome | Stable QTN | Genomic Interval (Mb) a | Reported QTL/QTN b |
|---|---|---|---|---|---|
| SL | SL-1 | 20 | Gm20_1029119 | 1.03–1.13 | |
| SL-2 | 20 | Gm20_34770123 | 34.77–34.87 | Seed length to width ratio 3–1 | |
| SW | SW-1 | 5 | Gm05_3370816 | 3.37–3.39 | Seed width 4–3 |
| SW-2 | 6 | Gm06_6916518 | 6.92–7.01 | Seed width 4–4 | |
| SW-3/ STW-1 | 7 | Gm07_6373192 | 6.35–6.39 | ||
| ST | ST-1 | 5 | Gm05_41938329 | 41.78–42.22 | GmST05 |
| ST-2/ STW-1 | 7 | Gm07_6373192 | 6.35–6.39 | ||
| ST-3 | 10 | Gm10_39032255 | 39.03–39.17 | ||
| ST-4 | 11 | Gm11_852388 | 0.71–0.96 | Seed height 1–2 | |
| ST-5 | 17 | Gm17_14862066 Gm17_14942042 | 14.86–15.22 | ||
| ST-6 | 20 | Gm20_3108737 | 3.09–3.41 |
| Loci | Candidate Gene | Arabidopsis Gene | Annotation Description | Biological Processes a |
|---|---|---|---|---|
| SW-1 | Glyma.05G038000 | AT5G16750 | Transducin family protein/wd-40 repeat family protein | Cell division, lipid storage |
| ST-1 | Glyma.05G244100 | AT1G18100 | Phosphatidylethanolamine-binding protein | |
| Glyma.05G246900 | AT3G17930 | Protein of unknown function | Starch biosynthetic process | |
| ST-2, SW-3 | Glyma.07G070200 | AT1G20930 | Cell division protein kinase | Cell proliferation |
| ST-4 | Glyma.11G010000 | AT2G38050,DET2 | 3-oxo-5-alpha-steroid 4-dehydrogenase family protein | Brassinosteroid biosynthetic process; brassinosteroid homeostasis |
| Glyma.11G012400 | AT4G33270 | Cell division cycle 20 (cdc20) (fizzy)-related | Cell division | |
| ST-5 | Glyma.17G165500 | AT4G37390,YDK1 | Gh3 auxin-responsive promoter | Auxin homeostasis, response to auxin stimulus |
| Glyma.17G166500 | AT2G23260 | Glucosyl/glucuronosyl transferases | Regulation of auxin metabolic process,Regulation of hormone levels | |
| SL-1 | Glyma.20G012600 | AT1G14740 | Vernalization-insensitive protein 3 | Embryo development |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, P.; Yang, Z.; Jia, S.; Chen, G.; Li, N.; Karikari, B.; Cao, Y. Genome-Wide Association Study and Candidate Gene Mining of Seed Size Traits in Soybean. Agronomy 2024, 14, 1183. https://doi.org/10.3390/agronomy14061183
Zhang P, Yang Z, Jia S, Chen G, Li N, Karikari B, Cao Y. Genome-Wide Association Study and Candidate Gene Mining of Seed Size Traits in Soybean. Agronomy. 2024; 14(6):1183. https://doi.org/10.3390/agronomy14061183
Chicago/Turabian StyleZhang, Pu, Zhiya Yang, Shihao Jia, Guoliang Chen, Nannan Li, Benjamin Karikari, and Yongce Cao. 2024. "Genome-Wide Association Study and Candidate Gene Mining of Seed Size Traits in Soybean" Agronomy 14, no. 6: 1183. https://doi.org/10.3390/agronomy14061183
APA StyleZhang, P., Yang, Z., Jia, S., Chen, G., Li, N., Karikari, B., & Cao, Y. (2024). Genome-Wide Association Study and Candidate Gene Mining of Seed Size Traits in Soybean. Agronomy, 14(6), 1183. https://doi.org/10.3390/agronomy14061183