
Multi-Omics Analysis Reveals the Transcriptional Regulatory Network of Maize Roots in Response to Nitrogen Availability

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Growth Conditions
2.2. RNA-Seq Analysis of Maize Roots under CK and LN
2.3. Ribo-Seq Analysis of Maize Roots under CK and LN
2.4. Ribo-Seq Data Analysis
2.5. Analysis of Translational Efficiency (TE)
2.6. Analysis of Upstream Open Reading Frames (uORFs)
2.7. Library Preparation and Open Chromatin Regions (OCRs) Identification of ATAC-Seq
2.8. Regulatory Network Construction Based on Transcriptome Data
2.9. Dual Luciferase Assay
2.10. Reverse Transcription Quantitative PCR (RT-qPCR)
2.11. Statistical Analysis
3. Results
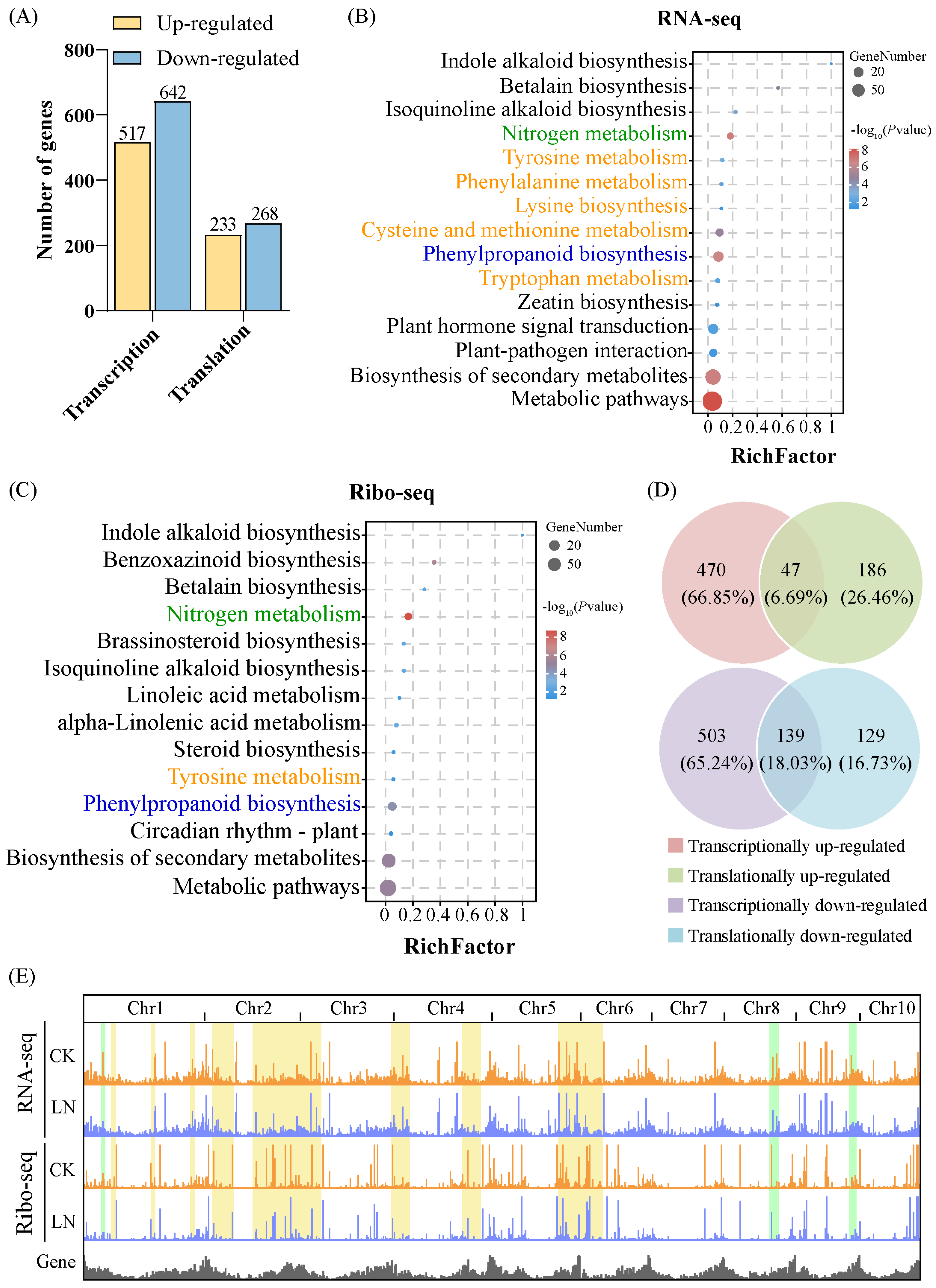
3.1. Characteristics of RNA-Seq and Ribo-Seq
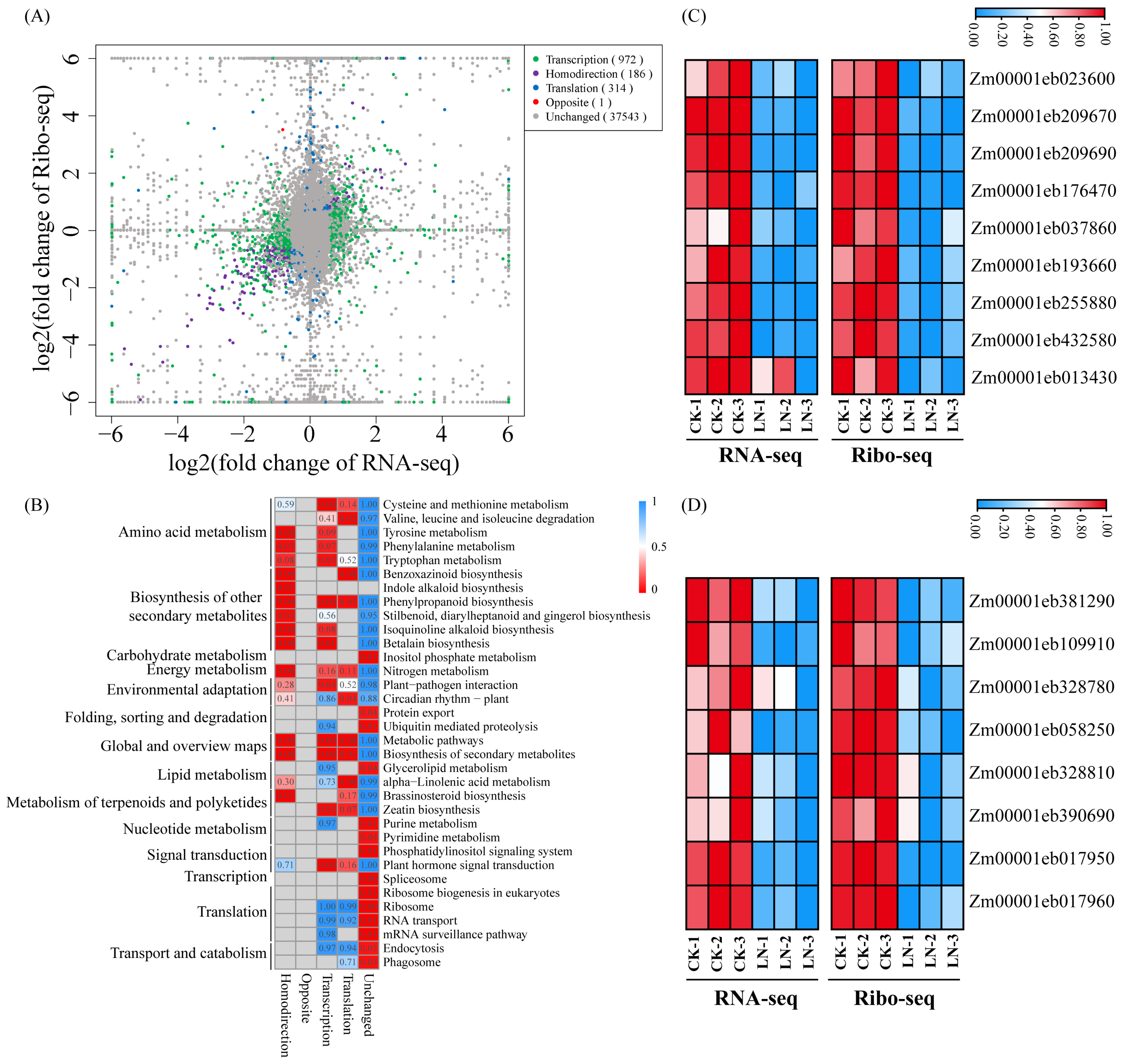
3.2. Response to LN Supplementation at the Transcription and Translation Levels
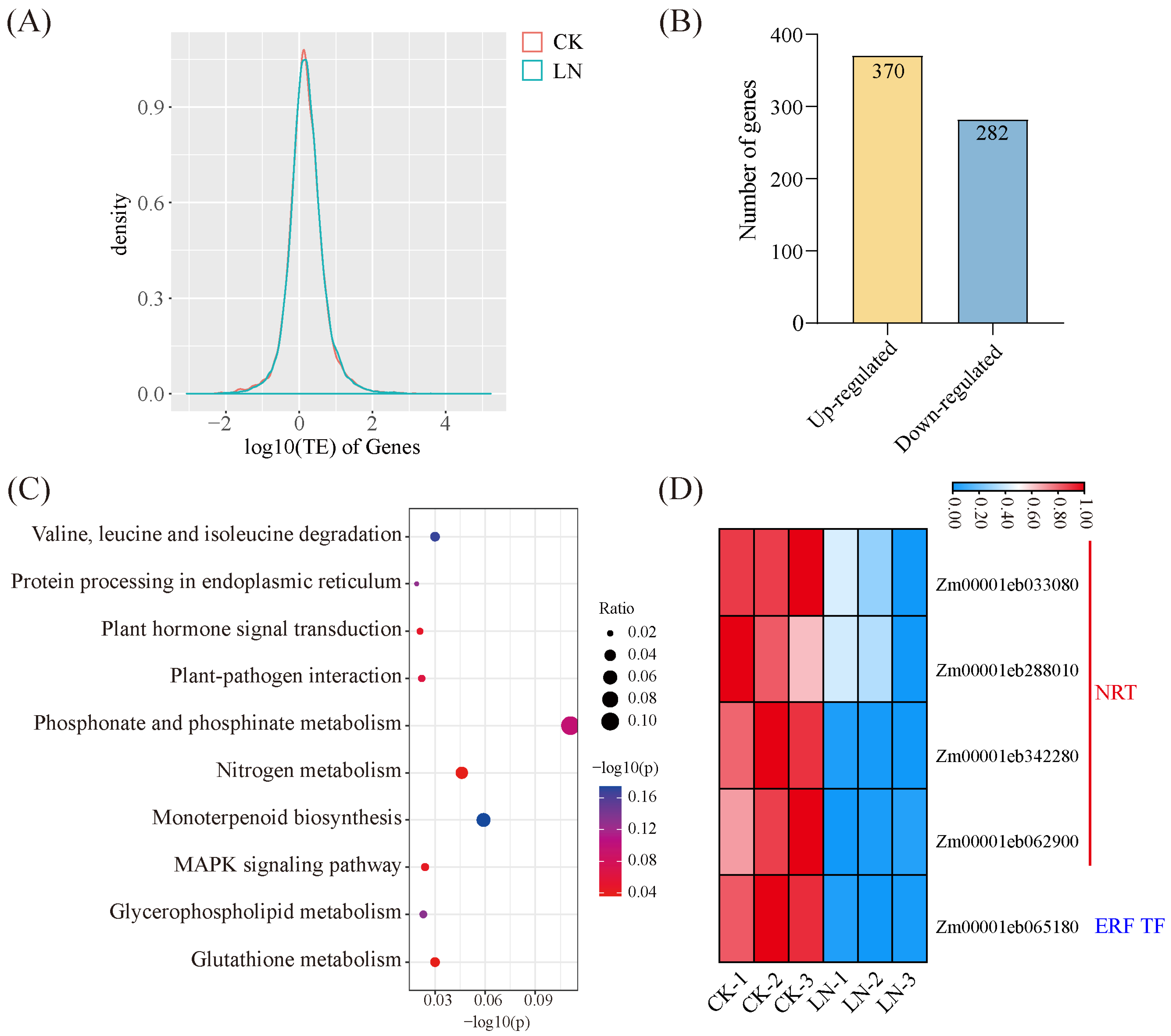
3.3. LN Triggers Altered TE
3.4. Global Identification of uORFs and Their Influence on the Translation of mORFs
3.5. ATAC-Seq Reveals Changes in Chromatin Accessibility under LN Condition
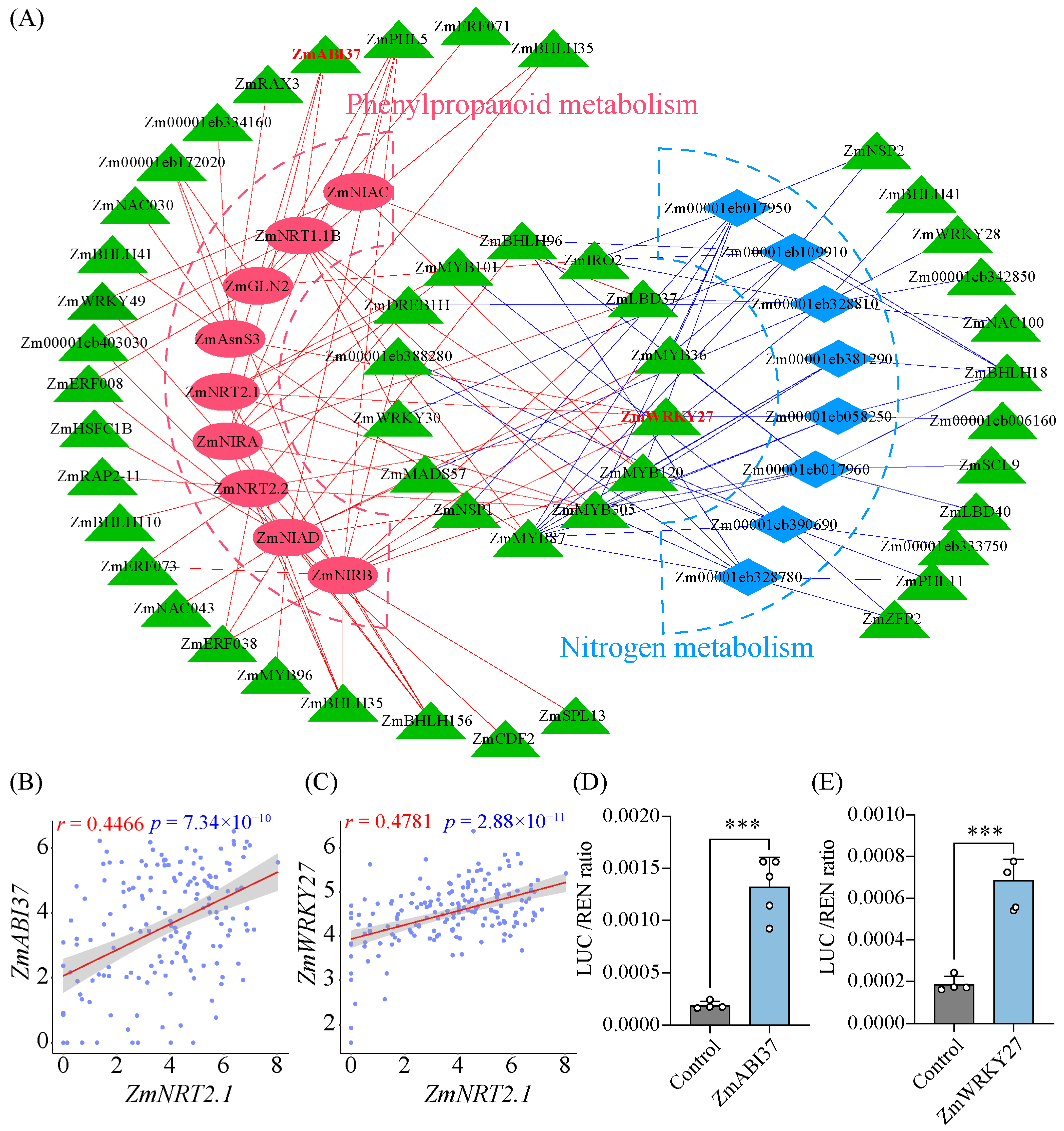
3.6. Construction of Transcriptional Regulatory Network Underlying LN Response
4. Discussion
4.1. Transcriptional and Translational Synergistically Respond to LN
4.2. Identification of Key Genes in Response to LN
4.3. Regulatory Networks Controlling Responses to LN
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Crawford, N.M.; Forde, B.G. Molecular and developmental biology of inorganic nitrogen nutrition. Arab. Book 2002, 1, e0011. [Google Scholar] [CrossRef] [PubMed]
- Kusano, M.; Fukushima, A.; Redestig, H.; Saito, K. Metabolomic approaches toward understanding nitrogen metabolism in plants. J. Exp. Bot. 2011, 62, 1439–1453. [Google Scholar] [CrossRef] [PubMed]
- Ladha, J.K.; Pathak, H.; Krupnik, T.J.; Six, J.; Kessel, C.V. Efficiency of fertilizer nitrogen in cereal production: Retrospects and prospects. Adv. Agron. 2005, 87, 85–156. [Google Scholar]
- Frink, C.R.; Waggoner, P.E.; Ausubel, J.H. Nitrogen fertilizer: Retrospect and prospect. Proc. Natl. Acad. Sci. USA 1999, 96, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Li, P.; Li, B.; Gong, J.; Wang, Y. Effects of fertigation scheme on N uptake and N use efficiency in cotton. Plant Soil 2007, 290, 115–126. [Google Scholar] [CrossRef]
- Ding, W.; Xu, X.; He, P.; Ullah, S.; Zhang, J.; Cui, Z.; Zhou, W. Improving yield and nitrogen use efficiency through alternative fertilization options for rice in China: A meta-analysis. Field Crops Res. 2018, 227, 11–18. [Google Scholar] [CrossRef]
- Li, H.; Hu, B.; Chu, C. Nitrogen use efficiency in crops: Lessons from Arabidopsis and rice. J. Exp. Bot. 2017, 68, 2477–2488. [Google Scholar] [CrossRef] [PubMed]
- Święcick, W.K.; Surma, M. The big five in the world of plants–the species that have changed the course of history. Pol. J. Agron. 2021, 47, 68–77. [Google Scholar]
- Różewicz, M. Production, use and efficiency of utilising grains of various cereal species as feed resources for poultry production. Pol. J. Agron. 2019, 38, 66–74. [Google Scholar]
- Simons, M.; Saha, R.; Guillard, L.; Clément, G.; Armengaud, P.; Cañas, R.; Maranas, C.D.; Lea, P.J.; Hirel, B. Nitrogen-use efficiency in maize (Zea mays L.): From ‘omics’ studies to metabolic modelling. J. Exp. Bot. 2014, 65, 5657–5671. [Google Scholar] [CrossRef]
- Kant, S. Understanding nitrate uptake, signaling and remobilisation for improving plant nitrogen use efficiency. Semin. Cell Dev. Biol. 2018, 74, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ji, M.; Liu, F.; Zhu, M.; Yang, Y.; Zhang, W.; Liu, S.; Wang, Y.; Lv, W.; Qi, S. NRG2 family members of Arabidopsis and maize regulate nitrate signalling and promote nitrogen use efficiency. Physiol. Plant. 2024, 176, e14251. [Google Scholar] [CrossRef] [PubMed]
- Kiba, T.; Krapp, A. Plant nitrogen acquisition under low availability: Regulation of uptake and root architecture. Plant Cell Physiol. 2016, 57, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Crawford, N.M. Nitrate: Nutrient and signal for plant growth. Plant Cell 1995, 7, 859–868. [Google Scholar]
- Garnett, T.; Conn, V.; Kaiser, B.N. Root based approaches to improving nitrogen use efficiency in plants. Plant Cell Environ. 2009, 32, 1272–1283. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Naz, M.; Fan, X.; Xuan, W.; Miller, A.J.; Xu, G. Plant nitrate transporters: From gene function to application. J. Exp. Bot. 2017, 68, 2463–2475. [Google Scholar] [CrossRef] [PubMed]
- Gojon, A.; Nacry, P.; Davidian, J.C. Root uptake regulation: A central process for NPS homeostasis in plants. Curr. Opin. Plant Biol. 2009, 12, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, M.; Abiko, T.; Yamaya, T. Assimilation of ammonium ions and reutilization of nitrogen in rice (Oryza sativa L.). J. Exp. Bot. 2007, 58, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Fan, X.; Miller, A.J. Plant nitrogen assimilation and use efficiency. Annu. Rev. Plant Biol. 2012, 63, 153–182. [Google Scholar] [CrossRef]
- Liu, K.H.; Tsay, Y.F. Switching between the two action modes of the dual-affinity nitrate transporter CHL1 by phosphorylation. EMBO J. 2003, 22, 1005–1013. [Google Scholar] [CrossRef]
- Hu, B.; Wang, W.; Ou, S.; Tang, J.; Li, H.; Che, R.; Zhang, Z.; Chai, X.; Wang, H.; Wang, Y. Variation in NRT1.1B contributes to nitrate-use divergence between rice subspecies. Nat. Genet. 2015, 47, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Tyerman, S.D.; Dechorgnat, J.; Ovchinnikova, E.; Dhugga, K.S.; Kaiser, B.N. Maize npf6 proteins are homologs of Arabidopsis chl1 that are selective for both nitrate and chloride. Plant Cell 2017, 29, 2581–2596. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Liu, Z.; Guo, J.; Jia, Z.; Shi, Y.; Kang, K.; Peng, W.; Wang, Z.; Chen, L.; Neuhaeuser, B.; et al. ZmNRT1.1B (ZmNPF6.6) determines nitrogen use efficiency via regulation of nitrate transport and signalling in maize. Plant Biotechnol. J. 2023, 22, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Marchive, C.; Roudier, F.; Castaings, L.; Brehaut, V.; Blondet, E.; Colot, V.; Meyer, C.; Krapp, A. Nuclear retention of the transcription factor NLP7 orchestrates the early response to nitrate in plants. Nat. Commun. 2013, 4, 1713. [Google Scholar] [CrossRef] [PubMed]
- Guan, P.; Wang, R.; Nacry, P.; Breton, G.; Kay, S.A.; Pruneda-Paz, J.L.; Davani, A.; Crawford, N.M. Nitrate foraging by Arabidopsis roots is mediated by the transcription factor TCP20 through the systemic signaling pathway. Proc. Natl. Acad. Sci. USA 2014, 111, 15267–15272. [Google Scholar] [CrossRef] [PubMed]
- Rubin, G.; Tohge, T.; Matsuda, F.; Saito, K.; Scheible, W.R. Members of the LBD family of transcription factors repress anthocyanin synthesis and affect additional nitrogen responses in Arabidopsis. Plant Cell 2009, 21, 3567–3584. [Google Scholar] [CrossRef] [PubMed]
- Vidal, E.A.; Moyano, T.C.; Riveras, E.; Contreras-Lopez, O.; Gutierrez, R.A. Systems approaches map regulatory networks downstream of the auxin receptor AFB3 in the nitrate response of Arabidopsis thaliana roots. Proc. Natl. Acad. Sci. USA 2013, 110, 12840–12845. [Google Scholar] [CrossRef] [PubMed]
- Vidal, E.A.; Alvarez, J.M.; Gutierrez, R.A. Nitrate regulation of AFB3 and NAC4 gene expression in Arabidopsis roots depends on NRT1.1 nitrate transport function. Plant Signal. Behav. 2014, 9, e28501. [Google Scholar] [CrossRef] [PubMed]
- Medici, A.; Marshall-Colon, A.; Ronzier, E.; Szponarski, W.; Wang, R.; Gojon, A.; Crawford, N.M.; Ruffel, S.; Coruzzi, G.M.; Krouk, G. AtNIGT1/HRS1 integrates nitrate and phosphate signals at the Arabidopsis root tip. Nat. Commun. 2015, 6, 6274. [Google Scholar] [CrossRef]
- Krouk, G.; Mirowski, P.; LeCun, Y.; Shasha, D.E.; Coruzzi, G.M. Predictive network modeling of the high-resolution dynamic plant transcriptome in response to nitrate. Genome Biol. 2010, 11, R123. [Google Scholar] [CrossRef]
- Xu, N.; Wang, R.; Zhao, L.; Zhang, C.; Li, Z.; Lei, Z.; Liu, F.; Guan, P.; Chu, Z.; Crawford, N.M.; et al. The Arabidopsis NRG2 protein mediates nitrate signaling and interacts with and regulates key nitrate regulators. Plant Cell 2016, 28, 485–504. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Wang, Y.; Liu, Y.; Jiang, L.; He, B.; Ning, L.; Du, H.; Lv, Y.; Zhou, L.; Lin, F.; et al. The NIN-like protein 5 (ZmNLP5) transcription factor is involved in modulating the nitrogen response in maize. Plant J. 2020, 102, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jia, Z.; Li, X.; Wang, Z.; Chen, F.; Mi, G.; Forde, B.; Takahashi, H.; Yuan, L. Involvement of a truncated MADS-box transcription factor ZmTMM1 in root nitrate foraging. J. Exp. Bot. 2020, 71, 4547–4561. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhong, Y.; Han, J.; Huang, L.; Wang, Y.; Shi, X.; Li, M.; Zhuang, Y.; Ren, W.; Liu, X. NIN-LIKE PROTEIN3.2 inhibits repressor Aux/IAA14 expression and enhances root biomass in maize seedlings under low nitrogen. Plant Cell 2024, koae184. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Chen, F.; Cai, H.; Liu, J.; Pan, Q.; Liu, Z.; Gu, R.; Mi, G.; Zhang, F.; Yuan, L. A genetic relationship between nitrogen use efficiency and seedling root traits in maize as revealed by QTL analysis. J. Exp. Bot. 2015, 66, 3175–3188. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhuang, Z.; Cai, H.; Cheng, S.; Soomro, A.A.; Liu, Z.; Gu, R.; Mi, G.; Yuan, L.; Chen, F. Use of genotype-environment interactions to elucidate the pattern of maize root plasticity to nitrogen deficiency. J. Integr. Plant Biol. 2016, 58, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Dong, L.; Lü, W.; Lü, J.; Meng, Q.; Liu, P. Transcriptome analysis of maize seedling roots in response to nitrogen-, phosphorus-, and potassium deficiency. Plant Soil 2020, 447, 637–658. [Google Scholar] [CrossRef]
- Trevisan, S.; Manoli, A.; Ravazzolo, L.; Botton, A.; Pivato, M.; Masi, A.; Quaggiotti, S. Nitrate sensing by the maize root apex transition zone: A merged transcriptomic and proteomic survey. J. Exp. Bot. 2015, 66, 3699–3715. [Google Scholar] [CrossRef] [PubMed]
- Para, A.; Li, Y.; Marshall-Colón, A.; Varala, K.; Francoeur, N.J.; Moran, T.M.; Edwards, M.B.; Hackley, C.; Bargmann, B.O.R.; Birnbaum, K.D.; et al. Hit-and-run transcriptional control by bZIP1 mediates rapid nutrient signaling in Arabidopsis. Proc. Natl. Acad. Sci. USA 2014, 111, 10371–10376. [Google Scholar] [CrossRef]
- Qi, J.; Yu, L.; Ding, J.; Ji, C.; Wang, S.; Wang, C.; Ding, G.; Shi, L.; Xu, F.; Cai, H. Transcription factor OsSNAC1 positively regulates nitrate transporter gene expression in rice. Plant Physiol. 2024, 192, 2923–2942. [Google Scholar]
- Konishi, M.; Yanagisawa, S. Arabidopsis NIN-like transcription factors have a central role in nitrate signalling. Nat. Commun. 2013, 4, 1617. [Google Scholar] [CrossRef] [PubMed]
- Kenchanmane Raju, S.K. Comparative profiling examines roles of dna regulatory sequences and accessible chromatin during cold stress response in grasses. Plant Cell 2020, 32, 2451–2452. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zheng, T.; Li, L.; Liu, W.; Qiu, L.; Ahmad, S.; Wang, J.; Cheng, T.; Zhang, Q.; Beckles, D. Integration of chromatin accessibility and gene expression reveals new regulators of cold hardening to enhance freezing tolerance in Prunus mume. J. Exp. Bot. 2023, 74, 2173–2187. [Google Scholar] [CrossRef] [PubMed]
- Pei, H.; Teng, W.; Gao, L.; Gao, H.; Ren, X.; Liu, Y.; Jia, J.; Tong, Y.; Wang, Y.; Lu, Z. Low-affinity SPL binding sites contribute to subgenome expression divergence in allohexaploid wheat. Sci. China Life Sci. 2022, 66, 819–834. [Google Scholar] [CrossRef]
- Wu, Z.; Jiang, Z.; Li, Z.; Jiao, P.; Zhai, J.; Liu, S.; Han, X.; Zhang, S.; Sun, J.; Gai, Z.; et al. Multi-omics analysis reveals spatiotemporal regulation and function of heteromorphic leaves in Populus. Plant Physiol. 2023, 192, 188–204. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T.; Ghaemmaghami, S.; Newman, J.R.S.; Weissman, J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 2009, 324, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Shi, J.; Chen, J.; Zhang, M.; Sun, S.; Xie, S.; Li, X.; Zeng, B.; Peng, L.; Hauck, A.; et al. Ribosome profiling reveals dynamic translational landscape in maize seedlings under drought stress. Plant J. 2015, 84, 1206–1218. [Google Scholar] [CrossRef]
- Shalgi, R.; Hurt, J.A.; Krykbaeva, I.; Taipale, M.; Lindquist, S.; Burge, C.B. Widespread regulation of translation by elongation pausing in heat shock. Mol. Cell 2013, 49, 439–452. [Google Scholar] [CrossRef]
- Wang, P.; Jin, S.; Chen, X.; Wu, L.; Zheng, Y.; Yue, C.; Guo, Y.; Zhang, X.; Yang, J.; Ye, N. Chromatin accessibility and translational landscapes of tea plants under chilling stress. Hortic. Res. 2021, 8, 96. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y.; Li, M.; Zhang, W.; Lu, Y.; Hua, K.; Ling, X.; Chen, T.; Guo, D.; Yang, Y. Translatome and transcriptome analyses reveal the mechanism that underlies the enhancement of salt stress by the small peptide Ospep5 in plants. J. Agric. Food Chem. 2024, 72, 4277–4291. [Google Scholar] [CrossRef]
- Qanmber, G.; You, Q.; Yang, Z.; Fan, L.; Zhang, Z.; Chai, M.; Gao, B.; Li, F.; Yang, Z. Transcriptional and translational landscape fine-tune genome annotation and explores translation control in cotton. J. Adv. Res. 2024, 58, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, H.; Wang, H.; Yang, X.; Xu, Y.; Yang, Z.; Xu, C.; Li, P. Integrating transcriptome, co-expression and QTL-seq analysis reveals that primary root growth in maize is regulated via flavonoid biosynthesis and auxin signal transduction. J. Exp. Bot. 2021, 72, 4773–4795. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, T.; Yang, J.; Wang, H.; Ji, W.; Xu, Y.; Yang, Z.; Xu, C.; Li, P. GWAS and transcriptome analysis reveal key genes affecting root growth under low nitrogen supply in maize. Genes 2022, 13, 1632. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Karaletsos, T.; Drewe, P.; Sreedharan, V.T.; Kuo, D.; Singh, K.; Wendel, H.G.; Ratsch, G. RiboDiff: Detecting changes of mRNA translation efficiency from ribosome footprints. Bioinformatics 2017, 33, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Bajic, M.; Maher, K.A.; Deal, R.B. Identification of open chromatin regions in plant genomes using atac-seq. Methods Mol. Biol. 2018, 1675, 183–201. [Google Scholar] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring regulatory networks from expression data using tree-based methods. PLoS ONE 2010, 5, e12776. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. Proc. Int. AAAI Conf. Web Soc. Media 2009, 3, 361–362. [Google Scholar] [CrossRef]
- Gaudinier, A.; Rodriguez-Medina, J.; Zhang, L.; Olson, A.; Liseron-Monfils, C.; Bagman, A.-M.; Foret, J.; Abbitt, S.; Tang, M.; Li, B.; et al. Transcriptional regulation of nitrogen-associated metabolism and growth. Nature 2018, 563, 259–264. [Google Scholar] [CrossRef]
- Ivanov, I.P.; Loughran, G.; Atkins, J.F. uORFs with unusual translational start codons autoregulate expression of eukaryotic ornithine decarboxylase homologs. Proc. Natl. Acad. Sci. USA 2008, 105, 10079–10084. [Google Scholar] [CrossRef] [PubMed]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Juntawong, P.; Girke, T.; Bazin, J.; Bailey-Serres, J. Translational dynamics revealed by genome-wide profiling of ribosome footprints in Arabidopsis. Proc. Natl. Acad. Sci. USA 2014, 111, E203–E212. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, J.Q.; Crawford, N.M. Identification and characterization of a chlorate-resistant mutant of Arabidopsis thaliana with mutations in both nitrate reductase structural genes NIA1 and NIA2. Mol. Gen. Genet. 1993, 239, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Wang, Y.; Chen, G.; Zhang, A.; Yang, S.; Shang, L.; Wang, D.; Ruan, B.; Liu, C.; Jiang, H.; et al. The indica nitrate reductase gene OsNR2 allele enhances rice yield potential and nitrogen use efficiency. Nat. Commun. 2019, 10, 5207. [Google Scholar] [CrossRef] [PubMed]
- Prinsi, B.; Espen, L. Mineral nitrogen sources differently affect root glutamine synthetase isoforms and amino acid balance among organs in maize. BMC Plant Biol. 2015, 15, 96. [Google Scholar] [CrossRef] [PubMed]
- Xin, W.; Zhang, L.; Zhang, W.; Gao, J.; Yi, J.; Zhen, X.; Du, M.; Zhao, Y.; Chen, L. An integrated analysis of the rice transcriptome and metabolome reveals root growth regulation mechanisms in response to nitrogen availability. Int. J. Mol. Sci. 2019, 20, 5893. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ge, S.; Xu, X.; Xing, Y.; Du, X.; Zhang, X.; Lv, M.; Liu, J.; Zhu, Z.; Jiang, Y. Multiomics analysis reveals new insights into the apple fruit quality decline under high nitrogen conditions. J. Agric. Food Chem. 2021, 69, 5559–5572. [Google Scholar] [CrossRef]
- Potter, K.C.; Wang, J.; Schaller, G.E.; Kieber, J.J. Cytokinin modulates context-dependent chromatin accessibility through the type-B response regulators. Nat. Plants 2018, 4, 1102–1111. [Google Scholar] [CrossRef]
- Huang, M.K.; Zhang, L.; Zhou, L.M.; Yung, W.S.; Li, M.W.; Lam, H.M. Genomic features of open chromatin regions (OCRs) in wild soybean and their effects on gene expressions. Genes 2021, 12, 640. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Shen, W.; Li, M.W.; Huang, M.; Zhang, J.; Li, H. The chromatin accessibility landscape of pistils and anthers in rice. Plant Physiol. 2022, 190, 2797–2811. [Google Scholar] [CrossRef] [PubMed]
- Fredes, I.; Moreno, S.; Diaz, F.P.; Gutierrez, R.A. Nitrate signaling and the control of Arabidopsis growth and development. Curr. Opin. Plant Biol. 2019, 47, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Vidal, E.A.; Alvarez, J.M.; Araus, V.; Riveras, E.; Brooks, M.D.; Krouk, G.; Ruffel, S.; Lejay, L.; Crawford, N.M.; Coruzzi, G.M.; et al. Nitrate in 2020: Thirty years from transport to signaling networks. Plant Cell 2020, 32, 2094–2119. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, S.; Ji, M.; Zhu, T.; Wang, Y.; Tang, X.; Zhu, X.; Yang, Z.; Xu, C.; Wang, H.; Li, P. Multi-Omics Analysis Reveals the Transcriptional Regulatory Network of Maize Roots in Response to Nitrogen Availability. Agronomy 2024, 14, 1541. https://doi.org/10.3390/agronomy14071541
Fang S, Ji M, Zhu T, Wang Y, Tang X, Zhu X, Yang Z, Xu C, Wang H, Li P. Multi-Omics Analysis Reveals the Transcriptional Regulatory Network of Maize Roots in Response to Nitrogen Availability. Agronomy. 2024; 14(7):1541. https://doi.org/10.3390/agronomy14071541
Chicago/Turabian StyleFang, Shuai, Minggang Ji, Tianze Zhu, Yunyun Wang, Xiao Tang, Xinjie Zhu, Zefeng Yang, Chenwu Xu, Houmiao Wang, and Pengcheng Li. 2024. "Multi-Omics Analysis Reveals the Transcriptional Regulatory Network of Maize Roots in Response to Nitrogen Availability" Agronomy 14, no. 7: 1541. https://doi.org/10.3390/agronomy14071541
APA StyleFang, S., Ji, M., Zhu, T., Wang, Y., Tang, X., Zhu, X., Yang, Z., Xu, C., Wang, H., & Li, P. (2024). Multi-Omics Analysis Reveals the Transcriptional Regulatory Network of Maize Roots in Response to Nitrogen Availability. Agronomy, 14(7), 1541. https://doi.org/10.3390/agronomy14071541