Abstract
Fungal contamination in rice and mycotoxins present significant challenges to both rice quality and food safety. However, there is a dearth of comprehensive research on the compositional and structural changes within fungal colonies in rice, particularly in typical rice-producing regions, as well as their underlying influencing factors. In this study, a comprehensive analysis of fungal taxa in rice grains was conducted using amplicon sequencing and bioinformatics methods on 99 rice samples collected in three major rice-producing regions in China: Northeast Plain (NP), Yangtze River Basin (YR), and Southeast Coastal Area (SC). A total of 6,019,722 fungal ITS sequences were obtained with an average sequence length of 235 base pairs, and effective ASVs (2014) accounted for approximately 97.58% of the total ASVs (2064). The fungal community diversity in rice grains exhibited significant variations across the three regions, with deterministic processes playing a predominant role in shaping the ecological dynamics of fungal taxa. Among the core microbiota (92 shared ASVs), the first five species (Alternaria, Fusarium, Curvularia, Epicoccum, and Ustilaginoidea) accounting for a proportion greater than 5% had been reported as potential pathogens for plants. Geographical variations in fungal community composition were evident, with a significantly higher number of shared populations observed between YR and CS regions compared to those in the NP region. Nutrient elements and climatic conditions were the internal and external driving factors of rice fungal community composition. Additionally, notable regional variations in fungal functionality were observed. The findings have significant implications for gaining a comprehensive understanding of the distribution patterns of fungal communities in the major rice-producing regions in China. Additionally, it provides valuable insights into controlling key influencing factors to effectively reduce the occurrence of toxin-producing fungi and mitigate the associated risks related to mycotoxin contamination, thereby contributing to improved risk management and assessment.
1. Introduction
Rice (Oryza sativa) is one of the most important food crops in China and even in the world. In China, more than 65% of the population depends on rice as a staple food [1,2]. Moreover, China’s rice production ranks first in the world (Food and Agriculture Organization of the United Nations (FAO), http://www.fao.org/faostat/zh/#data/QC (accessed on 2 March 2023)). The yield and quality of rice have a vital impact on food security and human and animal health [3].
However, rice is highly susceptible to fungal contamination throughout the production, harvesting, and storage processes. FAO estimates indicate that over 25% of global food crops are contaminated with mycotoxins [4]. The fungi possess the capability to produce toxic secondary metabolites, known as mycotoxins, which exhibit carcinogenic, mutagenic, teratogenic, and estrogenic effects under specific temperature and humidity conditions [5,6]. Previous studies have reported that at least 143 species of fungi and more than 400 kinds of mycotoxins existed in rice [7]. The main mycotoxins, including aflatoxins (AFs), ochratoxin A (OTA), deoxynivalenol (DON), and zearalenone (ZEN), are produced by Aspergillus, Penicillium, and Fusarium in rice [8,9,10,11,12]. In 1993, the International Agency for Research on Cancer (IARC) classified AFs and OTA as Group 1 and 2B carcinogens, respectively. (IARC, 1993, pp. 489–521). In the same year, DON was categorized as a Group 3 carcinogen by IARC, while ZEN received the same classification in 1999 [13]. At present, these four main mycotoxins are widely present in various cereals (such as rice, maize, barley and wheat). Consumption of these contaminated cereals by humans and animals can pose significant health risks [6].
The composition and distribution of grain-associated fungi can impact mycotoxin production and subsequently lead to food safety concerns [14]. The diversity and composition of microbial communities can be influenced by the interaction between microorganisms and environmental factors [15]. For instance, specific temperature conditions and optimal moisture content enhanced the activity of microorganisms, including pathogenic fungi [16], thereby leading to fungal toxin contamination and grain quality declining [17,18]. The structure and distribution of microbial communities are closely associated with climatic conditions in various geographical locations [19,20]. Due to diverse climate conditions, certain rice-growing regions in China, such as the Yangtze River basin, experience excessively humid rainy seasons. This results in rapid proliferation of mold-producing fungi in stored rice and subsequent mycotoxin production [13]. Moreover, interactions among different fungal species can also impact mycotoxin production [21]. The presence of Aspergillus flavus has been shown to significantly inhibit the toxin production capacity of Fusarium graminearum and Fusarium verticillioides [22].
Due to its vast land area, rice is widely cultivated in China under various climatic conditions and is extensively contaminated by multiple fungi. At present, there are many studies on mycotoxin contamination in rice in China, while research on the composition, distribution, and influencing factors of fungal communities on rice grains is comparatively scarce [1,3,13]. In this study, a total of 99 typical rice grain samples were collected from three major rice-producing regions in China to comprehensively analyze the fungal community using amplicon sequencing and bioinformatics analysis. The objectives were to (1) elucidate the diversity, composition, and distribution differences of fungal communities, particularly toxigenic fungi communities; (2) clarify the impact of intrinsic and extrinsic influencing factors on the distribution of fungal taxa; and (3) identify key influencing factors for risk management strategies aimed at reducing growth of toxigenic fungi and contamination by fungal mycotoxins.
2. Materials and Methods
2.1. Sample Collection
During the 2021 rice harvest season, a total of 99 rice grain samples with three biological duplications were systematically gathered from three major rice-producing regions in China—namely the Northeast Plain (NP), Yangtze River basin (YR), and Southeast Coastal region (SC) (Figure 1). These samples were collected based on considerations of geographical location, climatic conditions, and historical occurrences of fungal diseases. Detailed information such as geographic location and climate parameters are shown in the Supplementary Table S1. The weather data from 25 May to 10 June, and from 24 August to 10 September were downloaded from the website archive (https://www.timeanddate.com/, accessed on 2 March 2021). The samples (each about 500 g) collected from different sampling points were put in sterilized plastic-sealed bags separately, and quickly transported to the laboratory where they were stored at −80 °C.
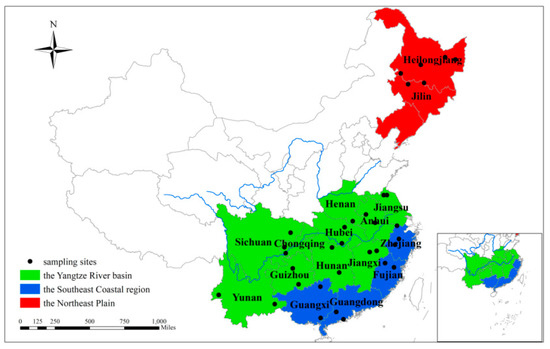
Figure 1.
Sampling locations in three major rice-producing regions of China (Table S1).
2.2. DNA Extraction, PCR Amplification, and High-Throughput Sequencing
Before genomic DNA extraction, rice grains’ surfaces need to be disinfected to prevent contamination by non-target microorganisms. The FastDNA Spin Kit (MP Biomedicals, Solon, OH, USA) was used to extract total fungal genomic DNA from rice grain samples following the manufacturer’s instructions. The quality and concentration of the DNA were assessed using 1% agarose gel electrophoresis and a NanoDrop® ND-2000 spectrophotometer (Thermo Scientific Inc., Waltham, MA, USA), respectively. The extracted DNA was stored at −80 °C until further use. Amplification of the fungal rDNA ITS1 region was performed using primer pairs ITS1F (5′-CTTGGTCATTTAGAGGAAGTAA-3′) and ITS2R (5′-GCTGCGTTCTTCATCGATGC-3′) [23] with an ABI GeneAmp® 9700 PCR thermocycler (ABI, Los Angeles, CA, USA).
The PCR reaction mixture contained 2 μL of a 10 × buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of each primer (5 μM), 0.2 μL of rTaq DNA polymerase, 10 ng of template DNA, 0.2 μL of BSA, and ddH2O to achieve a final volume of 20 µL. The PCR amplification program consisted of initial denaturation at 95 °C for 3 min, followed by denaturing at 95 °C for 30 s, annealing at 55 °C for 30 s and extension at 72 °C for 45 s in 35 cycles, with a final extension step at 72 °C for 10 min before being held at 4°C. To minimize PCR biases in PCR amplification results, all extracted genomic DNA from rice grain samples were amplified in triplicate. The resulting amplicons were visualized through gel electrophoresis on a 2% agarose gel. Subsequently, the AxyPrep DNA Gel Extraction Kit (Axygen-Biosciences, Union City, CA, USA) was employed to purify all amplicons according to the manufacturer’s protocol, and their quantification was determined utilizing the Quantus™ Fluorometer (Promega, Shanghai, China).
2.3. Illumina NovaSeq Sequencing
The NEXTFLEX Rapid DNA-Seq Kit was used for library construction of purified PCR products, which involved splicing linkers, magnetic bead screening to eliminate self-contiguous segments, enrichment of library templates through PCR amplification, and recovery of the final library via magnetic beads. Equimolar amounts of purified amplicons were combined and subjected to paired-end sequencing on an Illumina MiSeq PE300 platform at Shanghai Majorbio Bio-pharm Technology Co., Ltd. (Shanghai, China), following the standard protocols.
2.4. Determination of Nutrient Elements, Quality Index, and Mycotoxin Contents
The nutrient elements, quality index, and mycotoxin contents of 99 samples from three major rice production regions were analyzed to investigate the correlation between fungal distribution and influencing factors. Table 1 provides details about the detection indicators and methods used.

Table 1.
Detection indices and methods used.
2.5. Amplicon Sequence Processing and Analysis
After demultiplexing, the raw sequences were quality-filtered with fastp (https://github.com/OpenGene/fastp, version 0.19.6 accessed on 2 March 2023) [35] and merged with FLASH (http://www.cbcb.umd.edu/software/flash, version 1.2.11 accessed on 2 March 2023) [36]. Then, the high-quality sequences were de-noised using the DADA2 [37] plugin in the Qiime2 [38] (version 2020.2) pipeline with recommended parameters to obtain representative sequences and abundance information of amplicon sequence variants (ASVs). To minimize the effects of sequencing depth on alpha and beta diversity measure, the number of sequences from each sample was rarefied to 34,731, which still yielded an average Good’s coverage of 99.91%. Species taxonomic analysis of ASVs was performed using Naive Bayes taxonomy classifiers from Qiime2 based on the unite8.0/its_fungi database.
2.6. Statistical Analysis
Bioinformatic analysis was performed on the free online platform of Majorbio Cloud platform (https://cloud.majorbio.com accessed on 2 March 2023) [39] and most analysis were carried out by the vegan package in R (version 3.3.1), unless otherwise noted [40]. Alpha diversity indices were calculated with Mothur v1.30.1 (http://www.mothur.org/wiki/Calculators accessed on 2 March 2023) [41]. The Kruskal–Wallis rank sum test was used to analyze the inter-group differences in Alpha diversity. The similarity among the fungal communities in different samples (beta-diversity) was conducted by principal coordinate analysis (PCoA) based on Bray–Curtis dissimilarities. The analysis of similarities (ANOSIM) was used to analyze the differences in community composition. The linear discriminant analysis (LDA) effect size (LEfSe) [42] (http://huttenhower.sph.harvard.edu/LEfSe accessed on 2 March 2023) was performed to identify the significantly abundant taxa (from phylum to genera) among the different groups (LDA score > 3.5, p < 0.05). The variable factors with variance inflation factor (VIF) values less than 10 were selected to build the final model [40]. An RDA/CCA (redundancy analysis/canonical correspondence analysis) was performed to investigate the effect of physicochemical properties on fungal community structures [40,43]. The correlation heatmap was used to analyze the correlation between fungal classification and environmental variables. The relative importance of geographic location, climatic condition, and rice grain physicochemical properties, etc., were measured by variation-partitioning analysis (VPA). Furthermore, to gain a deeper understanding of the mechanism behind fungal community formation in rice samples from various regions, the neutral community model (NCM) was employed to assess the significance of neutral processes in shaping these communities [44]. Based on the zero model, the picant package (version 1.8.2) was used to calculate the beta net relatedness index (βNTI) and the Raup Crick index (RCbray). The contribution of ecological processes to community assembly was further quantified by βNTI and RCbray indices [45]. MEGA software (version 11.0.10) was utilized to construct a phylogenetic tree of representative sequences from fungal ASVs using the maximum likelihood (ML) method. The fungal communities’ trophic modes (pathotroph, symbiotroph, or saprotroph) were classified and analyzed by comparing the ASVs against the Fungi Functional Guild (FUNGuild) database (https://github.com/UMNFuN/FUNGuild accessed on 2 March 2023) [46].
3. Results
3.1. Diversity Analysis of Fungal Community Structures
The fungal diversity of the 99 rice samples collected from three major rice producing regions in China (NP, YR, and SC) was investigated. A total of 6,019,722 fungal ITS sequences were obtained with an average sequence length of 235 base pairs, raw reads 6891703. The species annotation analysis resulted in the following statistics: domain: 1, kingdom: 1, phylum: 6, class: 27, order: 75, family: 198, genus: 437, species: 767. For post-analysis purposes, effective ASVs (2014) accounted for approximately 97.58% of the total ASVs (2064). At the phylum level, the dominant five taxa identified were Ascomycota, Basidiomycota, unclassified_k__Fungi, Mortierellomycota, and Rozellomycota; whereas at the genus level, Alternaria, Microdochium, unclassified_f__Phaeosphaeriaceae, Dendryphiella, and Phaeosphaeria emerged as the most prevalent taxon. To ensure downstream diversity and composition analyses could be conducted effectively, the sequences were rarefied to match the lowest number of sequences per sample (n = 34,731 sequences). The rarefaction curve (Figure S1) constructed based on the observed ASV (sobs index) indicated that it approached the asymptote, suggesting sufficient sequencing depth and comprehensive coverage of fungal communities in all samples.
Notable disparities in alpha diversity indices are depicted in Figure 2. The community richness (Figure 2A) within YR exhibited a significantly higher level compared to that of NP (p < 0.01). Furthermore, both community diversity and evenness (Figure 2B,C) within YR were significantly greater than those observed in SC (p < 0.05). Additionally, the phylogenetic diversity of fungal communities across these three regions mirrored the patterns observed for community richness, with YR displaying notably higher levels compared to NP (Figure 2D). Through the application of PCoA analysis based on the Bray–Curtis dissimilarity algorithm, it was observed that the 99 rice grain samples exhibited distinct clustering into three different groups (Figure 2E,F). The results of ANOSIM analyses further support the finding that there was a significant dissimilarity in fungal community composition between the NP group and both the SC and YR groups (r = 0.4604, p = 0.001). Therefore, it was concluded that there exist notable disparities in fungal community composition among rice grain samples from the NP region compared to those from the other two regions. While some differences were found between samples from the SC and YR regions, there were also numerous similarities. Additionally, MRPP analysis revealed that inter-regional variation exceeded intra-regional variation (A > 0), with a statistically significant difference between groups (p = 0.001) (Table S2).
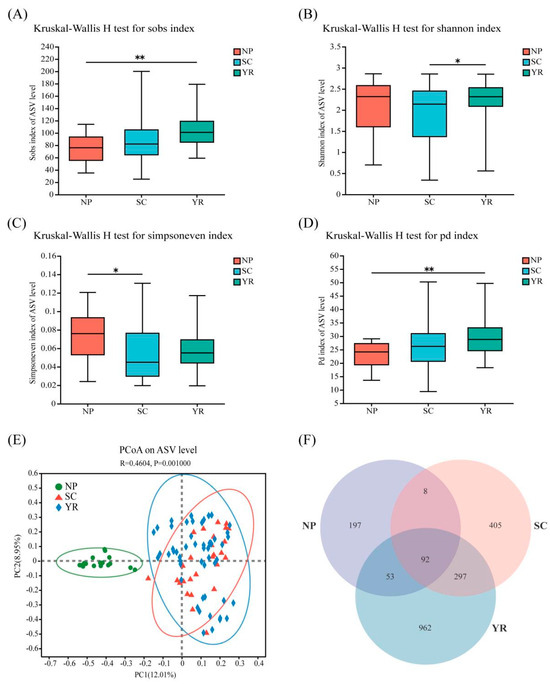
Figure 2.
Alpha diversity indices of the fungal communities among three major rice-producing regions of China (A–D): Sob index (A); Shannon index (B); Shannoneven index (C); Pd index (D) (*, p < 0.05; **, p < 0.05). Principal co-ordinates analysis (PCoA) of rice grain fungal communities based on Bray–Curtis distance algorithm (E), and Venn diagram of fungal ASV (F). NP, Northeast Plain; SC, Southeast Coastal region; YR, Yangtze River basin.
The neutral community model (NCM) was employed to evaluate the contribution of neutral processes in shaping fungal communities within rice grain samples, as depicted in Figure 3A–C. The overall goodness of fit for the NCM was represented by R2. The R2 values for the three regions were observed as follows: YR (36.81%), NP (24.00%), and SC (13.09%), respectively (Figure 3A–C), indicating that stochastic processes contribute to some extent to the community assembly of fungi in these regions, although their impact is not significant. In other words, deterministic processes exerted a stronger influence on community assembly than stochastic processes. Furthermore, the migration rate of fungi in the NP region (m = 0.0045) was found to be higher compared to that observed in the SC (m = 0.0007) and YR regions (m = 0.0005), indicating a relatively lower level of population restriction within samples in the NP region than in the other two regions. The community structure analysis using βNTI/RCbray (Figure 3D) revealed that fungal groups exhibited the aggregation primarily driven by homogeneous selection, suggesting a dominant role of deterministic processes in the ecological dynamics.
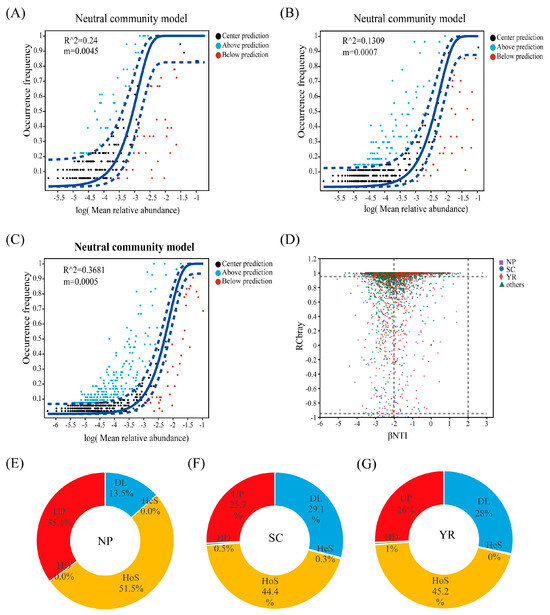
Figure 3.
Ecological processes of fungal communities in rice grains (A–D): neutral community model (NCM) of fungi in (A) NP, Northeast Plain; (B) SC, Southeast Coastal region; (C) YR, Yangtze River basin (The solid line represents the fit of the neutral model and the upper and lower dashed lines represent the 95% confidence level of the model predictions); βNTI/RCbray community structure (D). The relative importance of ecological processes in rice grain fungal community assembly (E,F): the importance of ecological processes was indicated by inferring community assembly mechanisms via phylogenetic bin-based null model analysis (iCAMP): (E) NP; (F) SC; (G) YR. HeS, Heterogeneous selection, HoS, Homogeneous selection, HD, Homogenizing dispersal, DL, Dispersal limitation, UP, Undominated processes.
The ecological processes of fungi were further quantified using the phylogenetic bin-based null model (iCAMP) to infer community assembly mechanisms (Figure 3E–G). Homogeneous selection (HoS), undominated processes (Up), and dispersal limitation (DL) were identified as the most significant factors determining the composition of fungal communities across all three regions. HoS played a dominant role, contributing to 51.5%, 44.4%, and 45.2%. While the importance of ecological processes in shaping rice fungal communities was similar between SC and YR regions (Figure 3F,G), it differed in NP region (Figure 3E). This difference could be attributed to regional variations, which will be thoroughly investigated and discussed in subsequent sections. Notably, SC and YR exhibited closer proximity in terms of geographical location and climatic conditions (Table S1).
3.2. Dominant Taxonomic Groups in Rice Grains
Venn diagram analysis of species revealed both shared and unique ASVs in the composition of fungal communities across different regions (Figure 2F). The YR region exhibited the highest total number of species (1404 ASVs), followed by the SC region (802 ASVs), while the NP region had the lowest count (350 ASVs). Amongst the 92 shared ASVs, referred to the core microorganisms, seven dominant ones accounted for a proportion greater than 5%: ASV37 (9.29%), ASV2 (7.85%), ASV14 (6.66%), ASV98 (5.84%), ASV1 (5.81%), ASV26 (5.24%), and ASV4 (5.07%). All these species were identified as pathogenic fungi causing plant diseases (Table S3). Geographical variations in fungal community composition on rice grains were evident, with a significantly higher number of shared populations observed between the YR and CS regions compared to those in the NP region. SourceTracker analysis indicated that approximately 70.97% of species in the NP region originated from the YR region, while 21.76% came from the SC region, and 7.27% were derived from other unidentified regions (Figure S2).
According to the results of taxonomic analysis, the community structure composition of different groups at the phylum and genus levels were determined (Figure 4). At the phylum level, Ascomycota, Basidiomycota, and unclassified fungi were found to be most dominant (Figure 4A). The relative abundance of all samples belonging to the Ascomycota phylum was overwhelmingly dominant. The primary genera across all samples included Alternaria, Microdochium, Epicoccum, unclassified-f-Phaeosphaeriaceae, Dendryphiella, Fusarium, Phaeosphaeria, Cladosporium, Gibberella, Aspergilus, and Ustilaginoidea (Figure 4B). Most of these strains were pathogenic to plants. For instance, Fusarium sp., Alternaria sp., Bipolaris sp., Curvularia sp., etc., were identified as pathogens causing rice ear rot. Among them, Alternaria alternata (Fr.) Keissl of Alternaria genus could lead to brown ears of grain, resulting in brown spots on rice grains [47]. In severe cases, rice grains became thick brown or dark brown affecting both yield and quality. In addition, rice grain samples from four regions, ZJH and ZLQ in SC, and HDEBT and HHC in NP, were tested for several major mycotoxins to validate our results (Figure S6). However, the relative abundance of dominant microbiome exhibited some differences at the genus level among samples from different regions. The microbial composition in SC and YR regions showed closer similarity, potentially due to similar prevailing climatic conditions in these two regions (Table S1). Comparative analysis across multiple groups also revealed significant differences in species composition among all three regions (Figure S3).
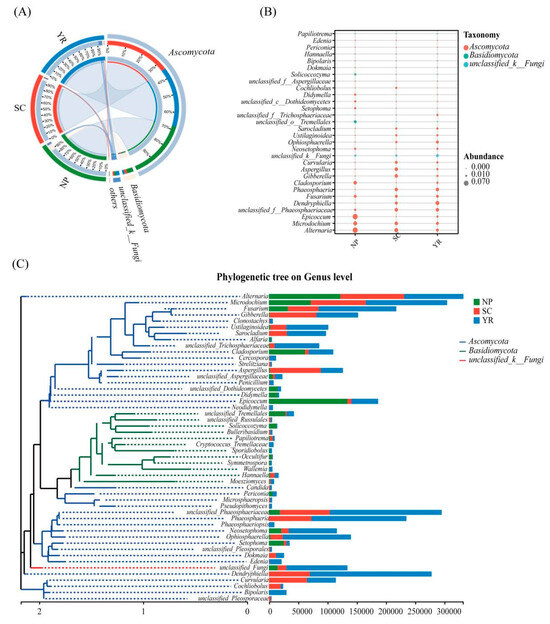
Figure 4.
The relative abundances of the most predominant fungal taxa associated with Chinese rice grains at the (A) phylum and (B) genus levels; (C) phylogenetic analysis on genus level. Notes: NP, Northeast Plain; SC, Southeast Coastal region; YR, Yangtze River basin.
The genera exhibiting distinct differences primarily included Alternaria, Epicoccum, Dendryphiella, Phaeosphaeria, Cladosporium, Gibberella, Aspergilus, Curvularia, Neosetophoma, Ophiosphaerella, Ustilaginoidea, Sarocladium, Setophoma, etc. Among these genera, most strains were pathogenic fungi or fungi capable of producing biotoxins. For instance, speciese of Alternaria [48], Epicoccum [49,50], Gibberella [51,52], Aspergillus [53], Curvularia [54,55], Ustilaginoidea [56,57,58], etc., were identified as pathogenic bacteria on rice corps. Furthermore, phylogenetic analysis (Figure 4C) also revealed that these pathogens belonged to the phylum Ascomycota, rather than Basidiomycota. The genera belonging to Basidiomycota posed relatively lower risks for rice cultivation.
3.3. Correlation Analysis between Fungal Distribution and the Influence Factors
3.3.1. The Impact of Intrinsic Influence Factors on Fungal Taxa
In the correlation analysis of environmental factors, nutrient elements, rice quality index, and mycotoxins as the intrinsic variables were analyzed. Nutrient elements comprised N, K, P, Ca, Fe, Mn, Mg, Na, and Zn, while the quality index included reducing sugar (RS), fat (FAT), protein (PRO), starch (STA), and water content (W). Through variance inflation factor (VIF) analysis, environmental factors with strong collinearity (VIF > 10) such as N, P, and PRO were excluded from further analysis. The remaining variables were considered as important environmental variables for subsequent CCA analysis and correlation heatmap analysis (Figure 5). As depicted in Figure 5A, seven elements displayed distinct correlations. Specifically, Mg was positively correlated with Na; Mn showed a positive correlation with Zn; and Fe exhibited a positive correlation with K, respectively, whereas Ca exhibited significant negative correlations with all other elements. Among these elements, Na, Mn, Zn, and Mg (R2 > 0.1, p = 0.001) exerted the greatest influence on species distribution (Figure 5A, Table S4). Furthermore, Na and Mg had the most pronounced impact on species distribution in NP region samples, while Mn and Zn played a major role in the SC and YR regions, followed by K and Fe. These mineral elements were essential nutrients for microbial growth and played crucial roles in participating in amino acids and enzymes composition in microorganisms, regulating the colloidal state of microbial protoplasm, and maintaining cell penetration and balance [59]. Although the content of Mn, Zn, and other trace elements were minuscule, their roles in microorganism growth was pivotal. The composition of fungal colonies varied across different regions due to variations in the types of main mineral elements present.
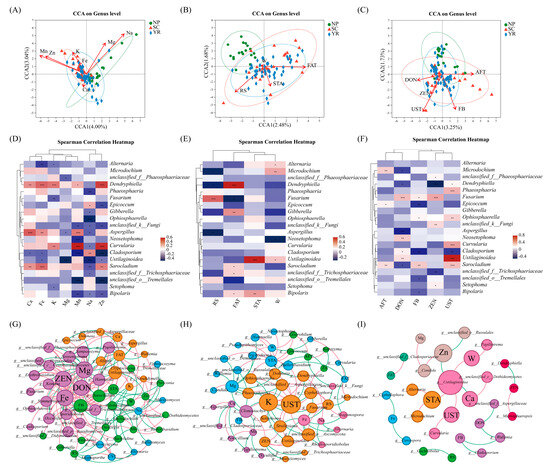
Figure 5.
CCA analysis of different factors’ effects on fungal taxa at the genus level: (A) nutrient elements; (B) quality index; (C) mycotoxins. Note: points of different colors or shapes represent different sample groups; the red arrow represents quantitative environmental factors, and the length of the arrow means the influence of nutrient elements on fungal distributions in different groups. The angles between the arrows represent positive and negative correlations (acute angles: positive correlations; obtuse angle: negative correlation; right angle: no correlation). Heat map of fungal populations and environmental factors at the genus level: (D) nutrient elements, (E) quality index, and (F) mycotoxin contents. Note: *, 0.01 < p ≤ 0.05, **, 0.001 < p ≤ 0.01, ***, p ≤ 0.001. The correlation network diagram of fungal species and environmental factors in rice samples from three regions at the genus level: (G) NP, Northeast Plain; (H) SC, Southeast Coastal region; (I) YR, Yangtze River basin. Note: node color classification is based on modularization; node size is based on eigenvector centrality; edge color classification is based on correlation. Red represents positive correlation and green represents negative correlation.
As can be seen from Figure 5B, the degree of influence of several quality indices on species distribution was RS (r2 = 0.3048, p = 0.001) > FAT (r2 = 0.262, p = 0.001) > STA (r2 = 0.112, p < 0.01). RS and STA, and STA and FAT were positively correlated, while RS and FAT were negatively correlated. RS had a positive correlation with the distribution of the sample community in the NP region, while STA and FAT had a greater negative correlation with the distribution of the sample community in the NP region. It can be seen from the Figure 5C the degree of influence of several mycotoxins on species distribution in the three regions was UST (r2 = 0.3132, p = 0.001) > AFT (r2 = 0.2041, p = 0.001) > FB (r2 = 0.1930, p = 0.001) > DON (r2 = 0.1405, p = 0.001) > ZEN (r2 = 0.0918, p < 0.05). UST was positively correlated with FB, DON, and ZEN, while UST was negatively correlated with AFT, and DON was negatively correlated with AFT. There was a negative correlation between the samples and mycotoxins in the NP region.
Variation partitioning approach (VPA) analysis was employed to assess the predominant environmental factors influencing fungal communities. This analysis further revealed the relative significance of nutrient elements, quality indicators, and mycotoxin contents in driving variations in fungal community structures (Figure S4). Nutrient elements accounted for the largest proportion of variation (7.57%), followed by quality index (5.71%) and mycotoxin contents (5.13%). Notably, 83.73% of microbial community changes remained unexplained by these aforementioned environmental factors. We hypothesize that this phenomenon may be related to the fact that changes in microbial communities are influenced by a variety of factors, including the complexity of environmental conditions, ecosystem diversity, and other environmental factors that might play key roles in the variations, suggesting the involvement of other key determinants that warrant further investigation.
The relative abundances of the dominant genera (the top 20 genera) among all samples were depicted in Figure 5D. Furthermore, the Spearman correlation heatmap revealed strong associations between certain microorganisms with environmental factors, suggesting that these microorganisms might serve as key species influencing environmental variables. Thus, they could potentially be utilized as microbial biomarkers for distinguishing control and treatment groups. Notably, the pathogenic genus Dendryphiella exhibited significant positive correlations with Fe, K, and Zn (p < 0.001), while being associated with various elements except Na and Mg. Conversely, Fusarium displayed a negative correlation solely with K (p < 0.01). The fungal species Aspergillus, Curvularia, and Sarochadium exhibited predominantly positive correlations with elements, and specifically significantly correlated with Ca and Mn, Mn, Zn, and Fe, respectively (p < 0.001). Conversely, other fungi such as Alternaria, Fusarium, Cladosporium, Ustilaginoidea, and unclassified_o__Tremellales displayed primarily negative associations with elements factors. These fungi could be considered as key indicators influencing environmental variables. From the element point of view, Mn and Zn showed a strong correlation with several genera (p < 0.001). Na, on the other hand, was the most correlated element with the top 20 species in relative abundance. The above analysis also revealed that different fungal populations have different requirements for mineral elements.
The correlation between the quality indicators of rice and fungal populations appeared to be less significant compared to the correlation between nutrient elements and fungal populations (Figure 5E). However, each quality indicator still exhibited a significant association with specific species. For instance, RS showed a significantly positive correlation with Fusarium, FAT demonstrated a significantly positive correlation with Dendryphiella, and STA displayed a significantly positive correlation with Ustilaginoidea (p < 0.001). Microdochium, Dendryphiella, and Aspergillus exhibited negative correlations with RS (0.01 < p ≤ 0.05), while Fusarium was negatively correlated with FAT and Neosetophoma was negatively associated with W (0.01 < p ≤ 0.05). There was a certain correlation between mycotoxin levels and fungal population distribution; however, the degree of correlation varied for different mycotoxins and fungal populations (Figure 5F). For instance, UST exhibited not only a significant positive correlation with Ustilaginoidea (p < 0.001), but also with some rice pathogens such as Curvularia, Phaeosphaeria, and Sarocladium (p < 0.001) [54,55]. Conversely, it demonstrated a significant negative correlation with Cladosporium (0.001 < p ≤ 0.01). This result suggested that in the presence of the rice smut fungus—Ustilaginoidea—UST might facilitate the growth of fungi like Curvularia, Phaeosphaeria, and Sarocladium, leading to plant diseases such as rice grain rot and sheath blight. Meanwhile, Cladosporium might exert some inhibitory effect on UST. Both DON and ZEN were significantly positively correlated with Fusarium (p < 0.001), while DON showed a significant negative correlation with Aspergillus (p < 0.001). There was a competitive relationship between Fusarium and Aspergillus, therefore the likelihood of both fungal mycotoxins coexisting and causing harm was extremely low. No significant association was observed between Alternaria and Gibberella among the five common mycotoxins.
Meanwhile, the correlation between the top 50 species and their intrinsic environmental factors in the three regions was investigated by constructing a microbial ecological network to examine the relationship among species with environmental factors (Figure 5G–I). The microbial symbiosis network of the NP and environmental factors exhibited distinct topological characteristics, while the microbial network of the YR and environmental factors showed low connectivity and complexity. NP, SC, and YR had 59, 53, and 29 nodes, respectively, with 181, 96, and 23 edges, respectively. Compared to the other two groups, the NP-environmental factors network displayed a lower modularity value (0.309) as well as fewer communities (4). Additionally, key nodes crucial for shaping the network structure were identified based on intra-module connectivity and inter-module connectivity of each member in the network: Mg, ZEN and DON were mainly observed in the NP-environmental factors network; K, UST were primarily found in the SC-environmental factors network; Ustilaginoidea dominated in the YR-environmental factors network. These findings collectively suggested that both connectivity and hub nodes within the microbial networks of the NP were significantly stronger compared to those of the SC or YR which might indicate that under influence from environmental factors, the structure of a rice grain’s microbial community was more organized.
3.3.2. The Impact of Extrinsic Influence Factors on Fungal Taxa
LEfSe can be used to identify the multi-level species characteristics that best account for differences between two or more groups of samples, as well as the degree to which these characteristics contribute to group dissimilarities. By employing LDA values to quantify the impact of species on effect size, LEfSe suggests key roles played by certain species in environmental change processes. This analysis also serves as an effective approach for discovering biomarkers distinguishing different groups. In this study, LEfSe was employed to analyze the interspecific variations among three major rice-producing regions from phylum to genus level (Figure 6A,B). LEfSe revealed that each of the three regions exhibited its own significantly enriched fungal taxa. In the NP region, Basidiomycota and Ascomycota, which were the two main phyla, displayed significant enrichment at a species level. Notably, enriched taxa included Cladosporium, Cladosporiaceae, Capnodiales, Basidiomycota, unclassified_Tremellales, unclassified_Dothideomycetes, Didymella, Cystobasidiaceae, Cystobasidiales, Occultifur, unclassified_Hypocreales (LDA > 3.5). In the SC region, the key microbial populations were predominantly concentrated within the Ascomycota phylum. Fungal taxa that exhibited significant enrichment included Aspergillus, Aspergillaceae, Ascomycota, Eurotiales, Curvularia, Massarinaceae, Stagonospora, etc. (LDA > 3.5). Similarly, in the YR region, the species composition shared similarities with those observed in the SC region and also belonged to the Ascomycota phylum. Notable constituents encompassed Dendryphiella, Phaeosphaeria, Ophiosphaerella, Trichosphaeriaceae, Trichosphaeriales, unclassified_f_Trichosphaeriaceae, Sarocladium, Edenia, Corynespora, and Corynesporascaceae (LDA > 3.5). These distinctive species mentioned above hold potential as core species or biomarkers specific to their respective regions. A Kruskal–Wallis H test revealed clearly that there were significant differences in the abundance of some biotoxin-producing species in different regions, while no significant regional differences existed in others. Some results were shown in Figure 6C–H and Figure S3. In addition, two genera—Epicoccum and Dendryphiella—showed obvious regional specificity in Figure 6G,H [60].
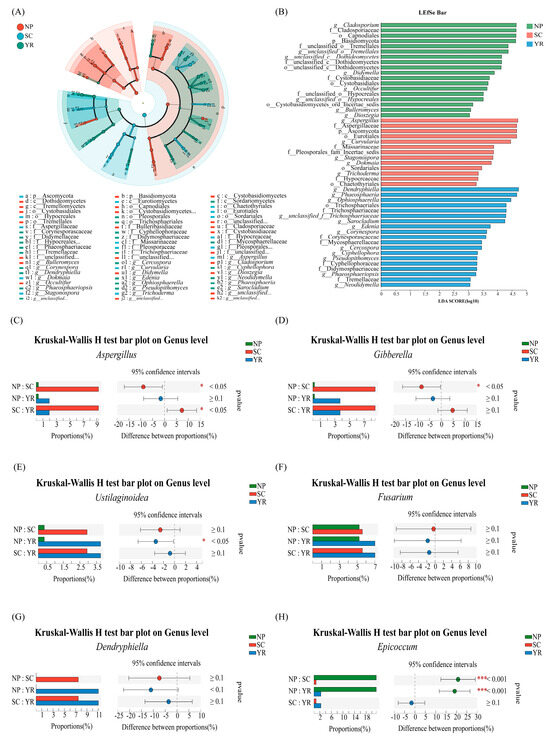
Figure 6.
Species difference analysis of samples among 3 major rice-producing regions. (A) LEfSe analysis (LDA > 3); (B) LEfSe bar (LDA > 3); (C–H), Kruskal–Wallis H test bar plot. Note: The horizontal coordinate of the bar chart on the left represents the percentage of the abundance of a certain species in each group of samples, and the vertical coordinate represents the group category of pair comparison. Different colors represent different groups. The figure on the right represents the proportion of species abundance differences within the set confidence interval, and the far right is the p value, * 0.01 < p ≤ 0.05, *** p ≤ 0.001.
Furthermore, the variation and distribution of fungal communities were primarily influenced by external influencing factors—geographic, environmental, farming and variety factors including latitude, longitude, altitude, mean annual temperature (MAT), mean annual precipitation (MAP), windspeed, summer mean temperature (SMT), winter mean temperature (WMT), harvest high temperature (HHT), harvest mean temperature (HMT), harvest mean precipitation (HMP), harvest mean wind speed (HMW), months of sowing, months of harvesting, and rice varieties (Var1-Oryza japonica/Oryza indica, and Var2-early rice/middle-late rice). Similarly, VIF analysis was employed to eliminate the factors with high autocorrelation among the aforementioned environmental variables and identify representative ones. Additionally, a combination of RDA/CCA analysis helped identify key environmental factors that significantly influenced the distribution of fungal communities as shown in Table 2. SMT (r2 = 0.4865, p = 0.001), harvesting period (r2 = 0.4721, p = 0.001), seeding (r2 = 0.4292, p = 0.001), and HMP (r2 = 0.4211, p = 0.001) emerged as the most influential factors associated with fungal community distribution, followed by agronomic habits—Var1 (r2 = 0.4180, p = 0.001) and Var2 (r2 = 0.4136, p = 0.001). Conversely, windspeed (r2 = 0.0481, p > 0.05) and altitude (r2 = 0.0025, p > 0.05) exerted minimal impact on differences in fungal community composition. Combined with the VPA analysis (Figure S5), it could be concluded that the fungal composition in rice grains was jointly affected by multi-dimensional environmental factors, and climate factors had the highest explanation for fungal community differences in samples (8.26%).
Table 2.
Table for RDA/CCA analysis.
Furthermore, the Mantel-test network heatmap analysis visually demonstrated the key factors influencing the variation in fungal community composition (Figure 7A, Table S5). The effects of various environmental factors on fungal community distribution varied across the three regions. Apart from latitude, altitude, MAT, HHT, and HMT, there was a significant positive correlation between other environmental factors and fungal communities in the NP region. These findings greatly contribute to our understanding of changes in fungal community structure (0.024 ≤ p ≤ 0.001). The distribution of fungi in the SC region showed no significant correlation with HMP, SMT, and Var1; however, it exhibited a significant correlation with other factors (0.043 ≤ p ≤ 0.001). In the YR region, the fungal community structure was positively correlated with environmental factors except for longitude, altitude, and WMT (0.015 ≤ p ≤ 0.001). Furthermore, the negative correlation depicted in Figure 7A lost its practical significance due to p ≥ 0.05 and therefore was not discussed here. Table S5 also revealed that, compared with samples from the SC (13 factors) and YR (12 factors), there were relatively few environmental factors (6 factors) influencing fungal community structure changes from the NP region.
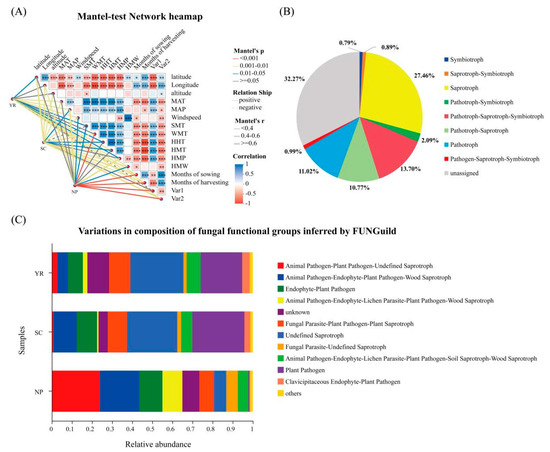
Figure 7.
Mantel-test network heatmap analyzing the key factors affecting the difference in fungal community composition in three regions (A). Note: Mantel-test heat map: The lines represent the correlation between community and environmental factors, and the heatmap represents the correlation between environmental factors. Line thickness: the correlation between community and environmental factors, and using Mantel’r (absolute value of R) to draw; relationship: Positive and Negative representing the positive and negative correlation between community and environmental factors; different colors in the heatmap represent positive and negative correlation, color depth represents the magnitude of positive and negative correlation, asterisk represents significance, * 0.01 < p ≤ 0.05, ** 0.001 < p ≤ 0.01, *** p ≤ 0.001. FunGuild-predicted fungal functional groups in rice grains: (B) nutrient classification and (C) functional groups.
In addition, significant positive or negative correlations were observed among several types of environmental factors (Figure 7A, Table S6). For instance, a significant negative correlation was found between farming factors (months of sowing and months of harvesting) and climate factors (SMT, WMT, HHT, HMT, and HMP), as well as between geographic factors (latitude and altitude) and climate factors (SMT, WMT, HHT, HMT, and HMP) (p < 0.001). Additionally, there was a significant negative correlation between climate factors (MAT and MAP) and climate factors (SMT, WMT, HHT, HMT, and HMP) (p < 0.001). The correlation between Var1 and the three types of factors—geographic factors (latitude, and longitude), climate factors (MAT, MAP, SMT, WMT, HHT, HMT, and HMP), and farming factors (months of sowing and months of harvesting)—exhibited an opposite pattern compared to that observed for Var2.
3.4. Fungal Functional Prediction
The FunGuild database was utilized to predict the functional characteristics of fungal communities [46]. FunGuild effectively grouped species based on their utilization of environmental resources, regardless of genetic relatedness. Fungi were classified into three main categories according to nutritional mode: pathotroph, symbiotroph, and saprotroph, which were further divided into 12 guilds based on their nutritional strategies. Figure 7B displayed the relative abundances of rice grain fungi categorized by nutrient patterns and functional groups. The fungal population primarily consisted of eight trophic types, with saprotroph accounting for the highest proportion (27.46%), obtaining nutrients through decomposing dead host cells—a process closely linked to organic matter decomposition and nutrient cycling [61]. Pathotroph–saprotroph–symbiotroph, accounted for 13.70%, followed by pathotroph (11.02%) and pathotroph–saprotroph (10.77%), respectively, in Figure 7C. Among them, pathotrophic fungi that damage host cells to acquire nutrients had a detrimental impact on plant growth [62].
The functional groups of the three regions exhibited certain differences, as depicted in Figure 7C. In the NP region, saprotrophic fungi—Animal Pathogen-Plant Pathogen-Undefined Saprotroph and Animal Pathogen-Endophyte-Plant Pathogen-Wood Saprotroph—were found to have the highest relative abundance, followed by Endophyte-Plant Pathogen; in the SC and YR, Plant Pathogen and Undefined Saprotroph accounted for the largest proportion. Additionally, relatively abundant fungi in the SC region included Animal Pathogen-Endophyte-Plant Pathogen-Wood Saprotroph, Endophyte-Plant Pathogen, and Fungal Parasite-Plant Pathogen-Plant Saprotroph. Notably, Fungal Parasite-Plant Pathogen-Plant Saprotroph exhibited a high relative abundance in the YR region.
4. Discussion
China is a vast country with abundant resources and a large population, where rice serves as the staple food for over 65% of the population [1,2]. Ensuring the quality and safety of rice production in the three major rice-producing regions holds paramount importance. Certain microorganisms and their toxins pose significant threats to the quality and safety of rice and its products. Therefore, it is of special significance for us to conduct a study on fungal communities, especially toxigenic fungi/plant pathogens, and their influencing factors in 99 samples of rice grains covering these three major rice-producing regions.
According to the PCA analysis, the fungal communities in the three regions were distinctly divided into three clusters based on their geographical locations (Figure 2E). There were significant differences in fungal community structure between different regions, with the NP region exhibiting a much greater dissimilarity compared to the CS and YR regions (Figure S3), which might be attributed to the climatic conditions (for example, the extreme cold temperatures in winter in the NP region) of these regions [63]. This finding was consistent with the study by Qi et al. [1] on rice microbiomes in northern (LN) and southern China (CQ). Zhou et al. [64] and Katsurayama et al. [65] also suggested that geographical location plays an important role in influencing the composition and structure of rice-associated bacterial communities. Furthermore, our analysis revealed a noteworthy discovery: dominant fungal taxa in each region were predominantly plant pathogens such as Alternaria [48], Epicoccum [49,50], Gibberella [51,52], Aspergillus [53], Curvularia [54,55], Ustilaginoidea [56,57,58], etc. (Table S3), but their distribution varied significantly across different regions (Figure 4 and Figure S3). These pathogens not only impacted rice yield but also produced biotoxins that had significant effects on rice quality and safety (Table S3). Additionally, there were variations in the distribution patterns of different genera among the three studied regions. Alternaria, Microdochium, and Fusarium showed relatively balanced distributions across all three regions. Gibberella and Ustilaginoidea were predominantly found in the SC and YR, respectively. Epicoccum was mainly distributed in the NP; however, its population abundance was very low in the SC. Curvularia and Dendryphiella were exclusively detected in the SC and YR. The factors contributing to regional variations in strain composition were determined through subsequent analyses.
The previous studies have demonstrated that both environmental and climatic factors played an equally significant role in determining the composition of fungal communities associated with rice grains [3,64,66,67]. Variations in these factors among samples from different regions might result in disparities in fungal species diversity and population distribution. This study not only comprehensively investigated the influence of external factors including environment and climate on the distribution and structure of fungal colonies, but also investigated the influence of internal factors, such as nutritional quality and elements, on this aspect, which was rarely covered by previous studies. Through VPA assessment, key influencing factors determining the distribution of toxigenic fungi/pathogens populations in rice grains were identified. The nutrient elements had the greatest influence on the difference in fungal communities (Figure S4). There was a close relationship between the variations of element content within rice grains and the toxigenic fungi/plant pathogens. For instance, biomarkers Aspergillus and Curvularia, which respond to intrinsic environmental variables, exhibited positive correlations with Ca-Mn and Mn-Zn, respectively (p < 0.001), while Alternaria, Fusarium, and Ustilaginoidea primarily displayed negative correlations with the aforementioned elements. From an elemental perspective, Mn and Zn showed strong associations with several genera (p < 0.001), whereas Na was linked to most species among the top 20 relative abundances—all of which warrant special attention.
Differently, the correlation between quality indicators and fungal populations seemed to be less closely related than that between nutrient elements and fungi (Figure 5E), but each indicator still had a close association with specific fungal genera. This was consistent with the conclusions of previous studies [68]. The levels of different toxins also showed varying degrees of correlation with the distribution of fungal populations (Figure 5F). However, an interesting finding was that each toxin not only correlated with its own toxin-producing fungi but also exhibited positive or negative correlations with other rice pathogens, such as UST and DON. This suggested that there might be some connections among various pathogenic microorganisms, and there could potentially be certain correlations between the diseases they cause. The occurrence of one disease might lead to the simultaneous occurrence of another; in other words, if one associated disease was effectively controlled or prevented, the likelihood of other diseases occurring would correspondingly decrease. This finding was consistent with previous reports [21,22].
Analysis of the correlation between microbial population distribution and external environmental factors revealed significant regional differences in plant pathogens, such as Epicoccum and Dendryphiella (Figure 6) [64]. These core species with significant differences could serve as biomarkers for determining the occurrence of plant diseases in their respective regions. Among the four categories of external environmental factors, the influence of climate conditions was most pronounced (Figure S5). Overall, SMT in climate conditions had the greatest impact on the distribution of fungal communities in rice grains and was a key influencing factor (Table 2). Previous reports have suggested that wind speed played an important role in fungal spore dispersal and was a key environmental factor affecting rice microbiome communities [15,69]. In our study, windspeed was one of the main contributing factors but not the most important one (Table 2). A Mantel-test network heatmap analysis revealed certain correlations among various environmental factors. The synergistic effects between these external and internal environmental factors contributed to microbial population differences among the three major rice-producing regions. In other words, the distribution of pathogenic fungal taxa was a result of multidimensional interactions among different environmental factors. The early prevention and control measures of rice pathogens should consider many factors to achieve accurate prediction and evaluation, and formulate targeted control strategies
5. Conclusions
The present study employed high-throughput sequencing technology in conjunction with bioinformatics analysis to comprehensively assess and predict the fungal community composition, distribution, and intrinsic and extrinsic influencing factors, as well as community functional characteristics in 99 representative rice grain samples collected from the three major rice-producing regions in China. There were significant differences in the diversity and composition of fungal communities among the three regions, and the differences between the NP and the other two regions were even greater. Deterministic processes exert a predominant influence on shaping the ecological dynamics of fungal taxa, such as SMT, harvesting period, seeding, and HMP. The core fungal species are mainly potential plant pathogens. The factors influencing variations in fungal community structure are multifaceted, with nutrient elements and climatic conditions serving as key internal and external driving forces for alterations in fungal community composition. Additionally, notable regional variations in fungal functionality within rice grains were investigated across the three regions. This study has important reference significance for the government supervision departments to fully grasp the distribution of fungal populations in the main rice producing areas of China and to deal with and control rice fungal pollution and mycotoxin production.
The following recommendations can address and control fungal contamination of rice and its mycotoxins production:
- Regional management measures. As studies have found significant differences in the diversity and composition of fungal communities in different regions, management strategies should be developed in each region according to specific ecological conditions and fungal characteristics. For example, more targeted control measures can be taken for the core plant pathogenic fungi in their respective regions.
- Strengthen field management. Optimize planting and harvesting time (e.g., timely sowing and rational harvesting) to reduce environmental conditions conducive to fungal growth. Select disease-resistant varieties suited to local climate and soil conditions to improve the crop’s ability to resist fungal infection.
- Nutrient element management. According to the effect of nutrient elements on fungal community structure mentioned in the study, rational fertilizer application and balanced soil nutrients will improve the growth and health of the crop, thus reducing the production of fungi and mycotoxins.
- Climate adaptation measures. Given that climatic conditions are important factors affecting fungal communities, adaptive agricultural measures should be taken to cope with the effects of climate change on rice growth and fungal growth. For example, selecting moisture-tolerant rice varieties to cope with changes in rainfall.
- Monitoring and testing system. Establish a long-term monitoring program to regularly assess fungal contamination and mycotoxin levels in rice from different regions and take timely countermeasures. Such monitoring can also help to better understand the dynamics of fungal communities.
- Public and farmer education. Enhance farmers’ and public awareness of fungal contamination and its potential hazards, and improve their knowledge of good agricultural practices (GAP) and food safety so as to reduce the risk of fungal contamination at the source.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/agronomy14081681/s1. Figure S1 Rarefaction curve; Figure S2 Source tracker species source analysis was based on a Bayesian approach. (A) SC, Southeast Coastal region; (B) YR, Yangtze River basin; Figure S3 Comparative analysis of different species in rice samples from three regions. Abbreviations: (A) NP, Northeast Plain; (B) SC, Southeast Coastal region; (C) YR, Yangtze River basin. *, 0.01 < p ≤ 0.05; **, 0.001 < p ≤ 0.01; ***, p ≤ 0.001; Figure S4 Variation partitioning approach (VPA). Abbreviations: A, quality index; B, mycotoxin contents; C, nutrient elements; Figure S5 Variation partitioning approach (VPA). Notes: climate, MAT, MAP, windspeed, SMT, WMT, HHT, HMT, HMP and HMW; farming, months of sowing and months of harvesting; Var, Var1 (Oryza indica/Oryza japonica) and Var2 (early rice/middle-late rice); geographic, latitude, longitude and altitude; Figure S6; Table S1 Environmental factors; Table S2 MRPP Analysis; Table S3 Pathogenic fungi causing plant diseases; Table S4 CCA Analysis of Envfit; Table S5 Mantel Test results; Table S6 Environmental factor heatmap correlation results table.
Author Contributions
F.Z.: Conceptualization, methodology, visualization, investigation, writing—original draft. Z.C.: Methodology, investigation, writing—review and editing. X.Z.: Writing—review and editing. Q.Y.: Writing—review and editing. M.G.: Writing—review and editing. M.C.: Conceptualization, supervision, funding acquisition, validation. X.L.: Conceptualization, methodology, writing—review and editing, supervision, funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This project has been supported by “Pioneer” and “Leading Goose” R&D Program of Zhejiang Province, China (No. 2023C02014), Zhejiang Provincial Natural Science Foundation of China (No. LY24C030002), Central Public Interest Scientific Institution Basal Research Fund for China National Rice Research Institute (No. CPSIBRF-CNRRI-202407), the China Agriculture Research System (No. CARS-01), the Agricultural Science and Technology Innovation Program, China (ASTIP), and National Natural Science Foundation of China (No. 32301766).
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Qi, Z.; Zhou, X.; Tian, L.; Zhang, H.; Cai, L.; Tang, F. Temporal and Spatial Variation of Microbial Communities in Stored Rice Grains from Two Major Depots in China. Food Res. Int. 2022, 152, 110876. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.D.; Su, P.; Shan, H. Mycotoxin Contamination of Rice in China. J. Food Sci. 2017, 82, 573–584. [Google Scholar] [CrossRef]
- Qi, Z.; Zhou, X.; Tian, L.; Zhang, H.; Cai, L.; Tang, F. Distribution of Mycotoxin-Producing Fungi across Major Rice Production Areas of China. Food Control 2022, 134, 108572. [Google Scholar] [CrossRef]
- Eskola, M.; Kos, G.; Elliott, C.T.; Hajšlová, J.; Mayar, S.; Krska, R. Worldwide Contamination of Food-Crops with Mycotoxins: Validity of the Widely Cited ‘Fao Estimate’ of 25%. Crit. Rev. Food Sci. Nutr. 2020, 60, 2773–2789. [Google Scholar] [CrossRef]
- Glenn, A.E. Mycotoxigenic Fusarium Species in Animal Feed. Anim. Feed Sci. Technol. 2007, 137, 213–240. [Google Scholar] [CrossRef]
- Nielsen, K.F.; Mogensen, J.M.; Johansen, M.; Larsen, T.O.; Frisvad, J.C. Review of Secondary Metabolites and Mycotoxins from the Aspergillus niger Group. Anal. Bioanal. Chem. 2009, 395, 1225–1242. [Google Scholar] [CrossRef] [PubMed]
- Reddy, O.R.; Sathyanarayana, N. Seed-Borne Fungi of Rice and Quarantine Significance. In Major Fungal Diseases of Rice, Recent Advances; Sreenivasaprasad, S., Johnson, R., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2001; p. 367. [Google Scholar]
- Hoeltz, M.; Fagundes, C.A.; Alcayata, E.A.L.; Noll, I.B. Mycobiota and Mycotoxins in Rice Samples Collected during the Stationary Drying and Storage System. Cienc. Rural 2009, 39, 803–808. [Google Scholar] [CrossRef]
- Frisvad, J.C.; Hubka, V.; Ezekiel, C.N.; Hong, S.B.; Nováková, A.; Chen, A.J.; Arzanlou, M.; Larsen, T.O.; Sklenář, F.; Mahakarnchanakul, W.; et al. Taxonomy of Aspergillus section Flavi and Their Production of Aflatoxins, Ochratoxins and Other Mycotoxins. Stud. Mycol. 2019, 93, 1–63. [Google Scholar] [CrossRef]
- Juan, C.; Zinedine, A.; Idrissi, L.; Mañes, J. Ochratoxin A in Rice on the Moroccan Retail Market. Int. J. Food Microbiol. 2008, 126, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.R.N.; Reddy, C.S.; Muralidharan, K. Potential of Botanicals and Biocontrol Agents on Growth and Aflatoxin Production by Aspergillus flavus Infecting Rice Grains. Food Control 2009, 20, 173–178. [Google Scholar] [CrossRef]
- Ferre, F.S.; Santamarina, M.P. Efficacy of Trichoderma harzianum in Suppression of Fusarium culmorum. Ann. Microbiol. 2010, 60, 335–340. [Google Scholar] [CrossRef]
- Li, R.; Wang, X.; Zhou, T.; Yang, D.; Wang, Q.; Zhou, Y. Occurrence of Four Mycotoxins in Cereal and Oil Products in Yangtze Delta Region of China and Their Food Safety Risks. Food Control 2014, 35, 117–122. [Google Scholar] [CrossRef]
- Mannaa, M.; Kim, K.D. Influence of Temperature and Water Activity on Deleterious Fungi and Mycotoxin Production during Grain Storage. Mycobiology 2017, 45, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Lessard, F.F. Integrated Management of the Risks of Stored Grain Spoilage by Seedborne Fungi and Contamination by Storage Mould Mycotoxins—An Update. J. Stored Prod. Res. 2017, 71, 22–40. [Google Scholar] [CrossRef]
- Laca, A.; Mousia, Z.; Díaz, M.; Webb, C.; Pandiella, S.S. Distribution of Microbial Contamination within Cereal Grains. J. Food Eng. 2006, 72, 332–338. [Google Scholar] [CrossRef]
- Ehrlich, K.; Ciegler, A.; Klich, M.; Lee, L. Fungal Competition and Mycotoxin Production on Corn. Experientia 1985, 41, 691–693. [Google Scholar] [CrossRef]
- Hoffmann, A.; Lischeid, G.; Koch, M.; Lentzsch, P.; Sommerfeld, T.; Müller, M.E.H. Co-cultivation of Fusarium, Alternaria, and Pseudomonas on Wheat-Ears Affects Microbial Growth and Mycotoxin Production. Microorganisms 2021, 9, 443. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.; Johnson, C.; Santos-Medellín, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Sundaresan, V. Structure, Variation, and Assembly of the Root-Associated Microbiomes of Rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911–E920. [Google Scholar] [CrossRef]
- Edwards, S.G. Impact of Agronomic and Climatic Factors on the Mycotoxin Content of Harvested Oats in the United Kingdom. Food Addit. Contam. Part A-Chem. Anal. Control Expo. Risk Assess. 2017, 34, 2230–2241. [Google Scholar] [CrossRef]
- Ding, N.; Xing, F.; Liu, X.; Selvaraj, J.N.; Wang, L.; Zhao, Y.; Wang, Y.; Guo, W.; Dai, X.; Liu, Y. Variation in Fungal Microbiome (Mycobiome) and Aflatoxin in Stored in-Shell Peanuts at Four Different Areas of China. Front. Microbiol. 2015, 6, 1055. [Google Scholar] [CrossRef]
- Giorni, P.; Magan, N.; Battilani, P. Environmental Factors Modify Carbon Nutritional Patterns and Niche Overlap between Aspergillus flavus and Fusarium verticillioides Strains from Maize. Int. J. Food Microbiol. 2009, 130, 213–218. [Google Scholar] [CrossRef]
- Adams, R.I.; Miletto, M.; Taylor, J.W.; Bruns, T.D. Dispersal in Microbes: Fungi in Indoor Air are Dominated by Outdoor Air and Show Dispersal Limitation at Short Distances. ISME J. 2013, 7, 1262–1273. [Google Scholar] [CrossRef]
- da Silva, I.J.S.; Lavorante, A.F.; Paim, A.P.S.; da Silva, M.J. Microwave-Assisted Digestion Employing Diluted Nitric Acid for Mineral Determination in Rice by ICP OES. Food Chem. 2020, 319, 126435. [Google Scholar] [CrossRef]
- Pei, T.; Jiang, J.; Gan, D.S.; Li, P.W. Dumas Combustion Method for Determination of Crude Protein Content in Oilseeds and Products. Chin. J. Oil Crop Sci. 2012, 34, 650–654. [Google Scholar]
- Wu, H.M.; Wang, W.T.; Ren, X.M.; Fan, L.; Fu, J.Q.; Tian, H.Y. Comparison of Determination of Fat in Egg Products by Chloroform Methanol Method and Acid Hydrolysis Method. J. Food Saf. Qual. 2020, 11, 7472–7475. [Google Scholar] [CrossRef]
- Lu, L.; Duan, B.W. Improvement of Determination of Starch Content in Rice by Polarimetry. China Rice 2011, 17, 25–27. [Google Scholar] [CrossRef]
- Lee, K.H.; Woo, K.S.; Yong, H.I.; Jo, C.; Lee, S.K.; Lee, B.W.; Oh, S.K.; Lee, Y.Y.; Lee, B.; Kim, H.J. Assessment of Microbial Safety and Quality Changes of Brown and White Cooked Rice Treated with Atmospheric Pressure Plasma. Food Sci. Biotechnol. 2018, 27, 661–667. [Google Scholar] [CrossRef]
- Kaur, M.; Asthir, B. Characterization of Biochemical and Proximate Composition in Rice Grains as Influenced by Germination. Cereal Res. Commun. 2021, 49, 291–299. [Google Scholar] [CrossRef]
- Cao, K.; Xie, C.; Wang, M.; Wang, P.; Gu, Z.; Yang, R. Effects of Soaking and Germination on Deoxynivalenol Content, Nutrition and Functional Quality of Fusarium Naturally Contaminated Wheat. LWT 2022, 160, 113324. [Google Scholar] [CrossRef]
- Schneweis, I.; Meyer, K.; Engelhardt, G.; Bauer, J. Occurrence of Zearalenone-4-β -D—Glucopyranoside in Wheat. J. Agric. Food Chem. 2002, 50, 1736–1738. [Google Scholar] [CrossRef]
- Liu, J.; Yu, D.N.; Xiong, N.; Liu, Y.; Li, D. Study on Aflatoxin in Paddy and in Whole Paddy Seed by Immunoaffinity Column-Photochemical Derivatization and HPLC. J. Chin. Cereals Oils Assoc. 2011, 26, 107–111. [Google Scholar]
- Natalia, A.M.; Huertas-Pérez, J.F.; García-Campaña, A.M.; Gámiz-Gracia, L. Simple Methodology for the Determination of Mycotoxins in Pseudocereals, Spelt and Rice. Food Control. 2014, 36, 94–101. [Google Scholar] [CrossRef]
- Fang, J.; Cao, H.A.; Xu, J.H.; Yin, X.C.; Shi, J.R. Simultaneous Quantitative Determination of Ustiloxin A and Ustiloxin D in Rice Grains by High Performance Liquid Chromatography-Tandem Mass Spectrometry. Chin. J. Rice Sci. 2012, 26, 246–250. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast Length Adjustment of Short Reads to Improve Genome Assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, J.C.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Yu, G.; Shi, C.; Liu, L.; Guo, Q.; Han, C.; Zhang, D.; Zhang, L.; Liu, B.; Gao, H.; et al. Majorbio Cloud: A One-Stop, Comprehensive Bioinformatic Platform for Multi-Omics Analyses. iMeta 2022, 1, e12. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; O’hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.; Wagner, H. Vegan: Community Ecology Package. R Package Version 2.4-2. 2017. Available online: https://cran.r-project.org/package=vegan (accessed on 17 January 2017).
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Justine, R.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; et al. Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic Biomarker Discovery and Explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Legendre, P.; Gallagher, E.D. Ecologically Meaningful Transformations for Ordination of Species Data. Oecologia 2001, 129, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Sloan, W.T.; Lunn, M.; Woodcock, S.; Head, I.M.; Nee, S.; Curtis, T.P. Quantifying the Roles of Immigration and Chance in Shaping Prokaryote Community Structure. Environ. Microbiol. 2006, 8, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Ning, D.; Yuan, M.; Wu, L.; Zhang, Y.; Guo, X.; Zhou, X.; Yang, Y.; Arkin, A.P.; Firestone, M.K.; Zhou, J. A Quantitative Framework Reveals Ecological Drivers of Grassland Microbial Community Assembly in Response to Warming. Nat. Commun. 2020, 11, 4717. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.H.; Song, Z.W.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An Open Annotation Tool for Parsing Fungal Community Datasets by Ecological Guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Mu, J.W.; Li, P.; Li, D.P.; Han, Y.C. The New Rice Disease-Browning Ear of Grain. Reclaiming Rice Cultiv. 2006, 5, 46–47. [Google Scholar]
- Lekagne, J.B.D.; Fouelefack, F.R.; Daboy, C.D.; Galani, Y.J.H.; Fotio, D.; Nguefack, J. Chemical Composition, Antifungal Properties and Seed Treatment Potential of Essential Oil Fractions of Callistemon citrinus Against Two Seed-Borne Fungi of Rice: Alternaria padwickii and Bipolaris oryzae. J. Plant Dis. Prot. 2023, 130, 833–841. [Google Scholar] [CrossRef]
- Deepika, Y.S.; Mahadevakumar, S.; Amruthesh, K.N.; Lakshmidevi, N. First Report of Epicoccum nigrum Associated with Leaf Spot Disease of Cowpea (Vigna unguiculata) from India. J. Plant Pathol. 2021, 103, 391–392. [Google Scholar] [CrossRef]
- Gai, X.T.; Jiang, N.; Lu, C.H.; Xia, Z.Y.; Qin, X.Y. First Report of Epicoccum latusicollum Causing Root Rot on Nicotiana tabacum in China. J. Plant Pathol. 2020, 102, 1291. [Google Scholar] [CrossRef]
- Hsuan, H.M.; Salleh, B.; Zakaria, L. Molecular Identification of Fusarium Species in Gibberella fujikuroi Species Complex from Rice, Sugarcane and Maize from Peninsular Malaysia. Int. J. Mol. Sci. 2011, 12, 6722–6732. [Google Scholar] [CrossRef]
- Jo, Y.-K.; Cromwell, W.; Jeong, H.-K.; Thorkelson, J.; Roh, J.-H.; Shin, D.-B. Use of Silver Nanoparticles for Managing Gibberella fujikuroi on Rice Seedlings. Crop Prot. 2015, 74, 65–69. [Google Scholar] [CrossRef]
- Schabo, D.C.; Martins, L.M.; Iamanaka, B.T.; Maciel, J.F.; Taniwaki, M.H.; Schaffner, D.W.; Magnani, M. Modeling Aflatoxin B1 Production by Aspergillus flavus During Wheat Malting for Craft Beer as a Function of Grains Steeping Degree, Temperature and Time of Germination. Int. J. Food Microbiol. 2020, 333, 108777. [Google Scholar] [CrossRef]
- Chen, X.; Tang, T.; Chen, C.; Wei, L.; Zhou, D. First Report of Curvularia Leaf Spot Caused by Curvularia muehlenbeckiae on Zizania latifolia in China. J. Plant Pathol. 2021, 103, 1073. [Google Scholar] [CrossRef]
- Palwasha, P.; Din, S.U.; Fahim, M. Eco-Friendly Strategies for the Management of Curvularia spicifera through Phytobiocides and Biological Antagonists. Eur. J. Plant Pathol. 2022, 164, 551–565. [Google Scholar] [CrossRef]
- Liu, X.; Matsumoto, H.; Lv, T.; Zhan, C.; Fang, H.; Pan, Q.; Xu, H.; Fan, X.; Chu, T.; Chen, S.; et al. Phyllosphere Microbiome Induces Host Metabolic Defence Against Rice False-Smut Disease. Nat. Microbiol. 2023, 8, 1419–1433. [Google Scholar] [CrossRef]
- Thomazella, D.P.d.T.; Teixeira, P.J.P.L. Microbiome-Mediated Metabolic Defence. Nat. Plants 2023, 9, 1174–1175. [Google Scholar] [CrossRef]
- Zheng, D.W.; Wang, Y.; Han, Y.; Xu, J.R.; Wang, C.F. UvHOG1 is Important for Hyphal Growth and Stress Responses in the Rice False Smut Fungus Ustilaginoidea virens. Sci. Rep. 2016, 6, 24824. [Google Scholar] [CrossRef]
- Dong, H.; Lu, A. Mineral-Microbe Interactions and Implications for Remediation. Elements 2012, 8, 95–100. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, T.Q.; Zhu, J.L. Diversity of Endophytic Fungi in Transgenic Rice Seeds from Different Planting Sites Based on PTN System. Sci. Agric. Sin. 2020, 53, 2305–2320. [Google Scholar] [CrossRef]
- Phillips, L.A.; Ward, V.; Jones, M.D. Ectomycorrhizal Fungi Contribute to Soil Organic Matter Cycling in Sub-Boreal Forests. ISME J. 2014, 8, 699–713. [Google Scholar] [CrossRef]
- Anthony, M.A.; Frey, S.D.; Stinson, K.A. Fungal Community Homogenization, Shift in Dominant Trophic Guild, and Appearance of Novel Taxa with Biotic Invasion. Ecosphere 2017, 8, e01951. [Google Scholar] [CrossRef]
- Valeria, G.; Elena, M.; Ileana, A.; Alexandra, D.A.; Irina, S.; Elena, C.M.; Alexandra, O.O.; Enuta, I.; Nastasia, B. Post-Harvest Contamination with Mycotoxins in the Context of the Geographic and Agroclimatic Conditions in Romania. Toxins 2018, 10, 533. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, J.T.; Wang, W.H.; Clement, K.M.T.; Lei, C. Changes in Bacterial and Fungal Microbiomes Associated with Tomatoes of Healthy and Infected by Fusarium oxysporum f. sp. Lycopersici. Microb. Ecol. 2020, 81, 1004–1017. [Google Scholar] [CrossRef] [PubMed]
- Katsurayama, A.M.; Martins, L.M.; Iamanaka, B.T.; Fungaro, M.H.P.; Silva, J.J.; Pitt, J.I.; Frisvad, J.C.; Taniwaki, M.H. Fungal Communities in Rice Cultivated in Different Brazilian Agroclimatic Zones: From Field to Market. Food Microbiol. 2020, 87, 103378. [Google Scholar] [CrossRef] [PubMed]
- Devin, C.D.; Damaris, D.; Citlali, F.G.; Stephen, G.; Scott, C.; Tanja, W.; Gretchen, N.; Axel, V.; Laila, P.P.M.; Susannah, G.T. Plant Compartment and Biogeography Affect Microbiome Composition in Cultivated and Native Agave Species. New Phytol. 2016, 209, 798–811. [Google Scholar] [CrossRef]
- Klaedtke, S.; Jacques, M.; Raggi, L.; Préveaux, A.; Bonneau, S.; Negri, V.; Chable, V.; Barret, M. Terroir is a Key Driver of Seed-Associated Microbial Assemblages. Environ. Microbiol. 2016, 18, 1792–1804. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pei, F.; Fang, Y.; Li, P.; Xia, J.; Sun, L.; Zou, Y.; Shen, F.; Hu, Q. Interactions among Fungal Community, Fusarium Mycotoxins, and Components of Harvested Wheat under Simulated Storage Conditions. J. Agric. Food Chem. 2019, 67, 8411–8418. [Google Scholar] [CrossRef] [PubMed]
- Doohan, F.M.; Brennan, J.; Cooke, B.M. Influence of Climatic Factors on Fusarium Species Pathogenic to Cereals. Eur. J. Plant Pathol. 2003, 109, 755–768. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).