
Antiplatelet Agents Affecting GPCR Signaling Implicated in Tumor Metastasis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
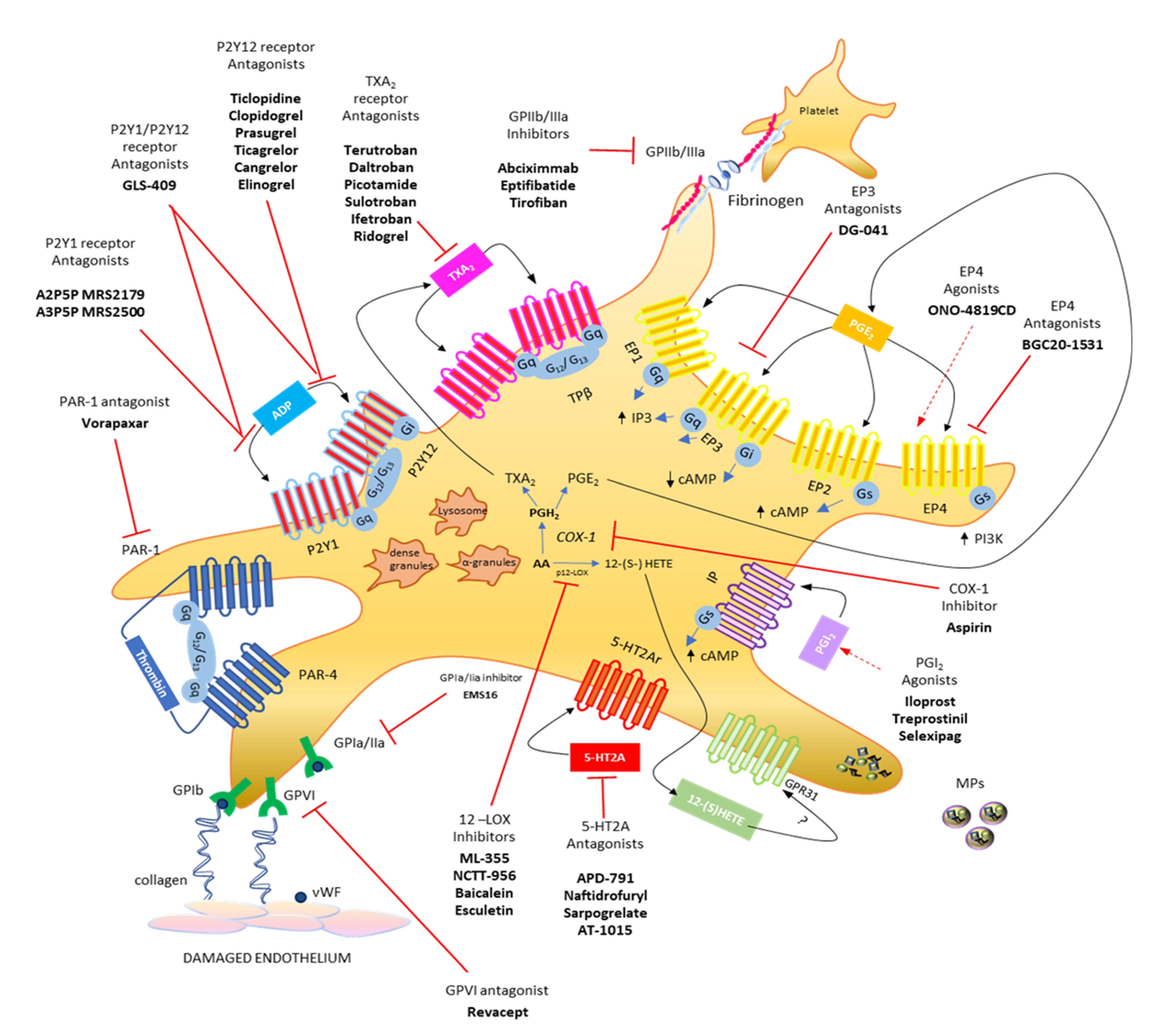
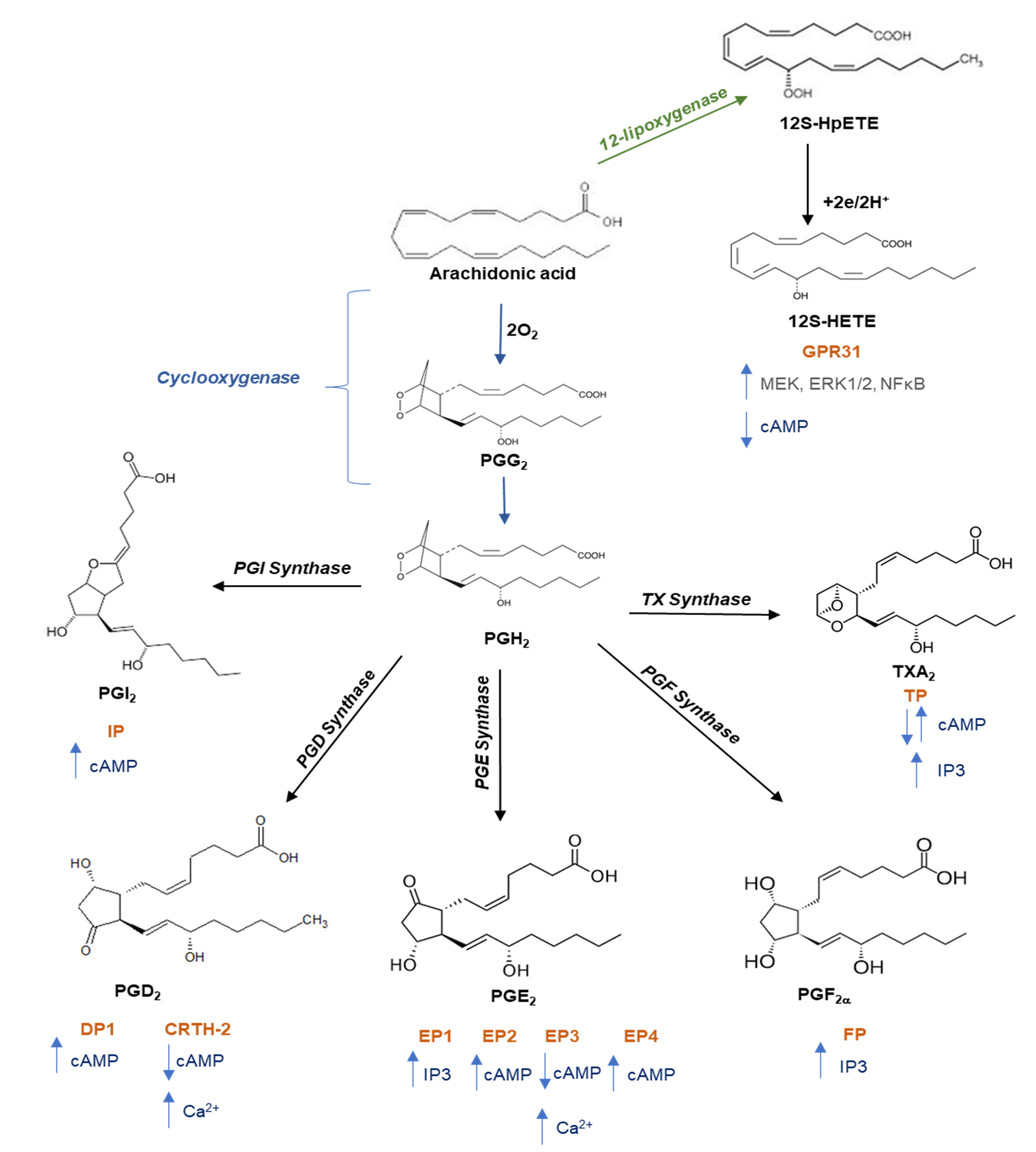
2. Major G Protein-Mediated Signaling during Platelet Activation
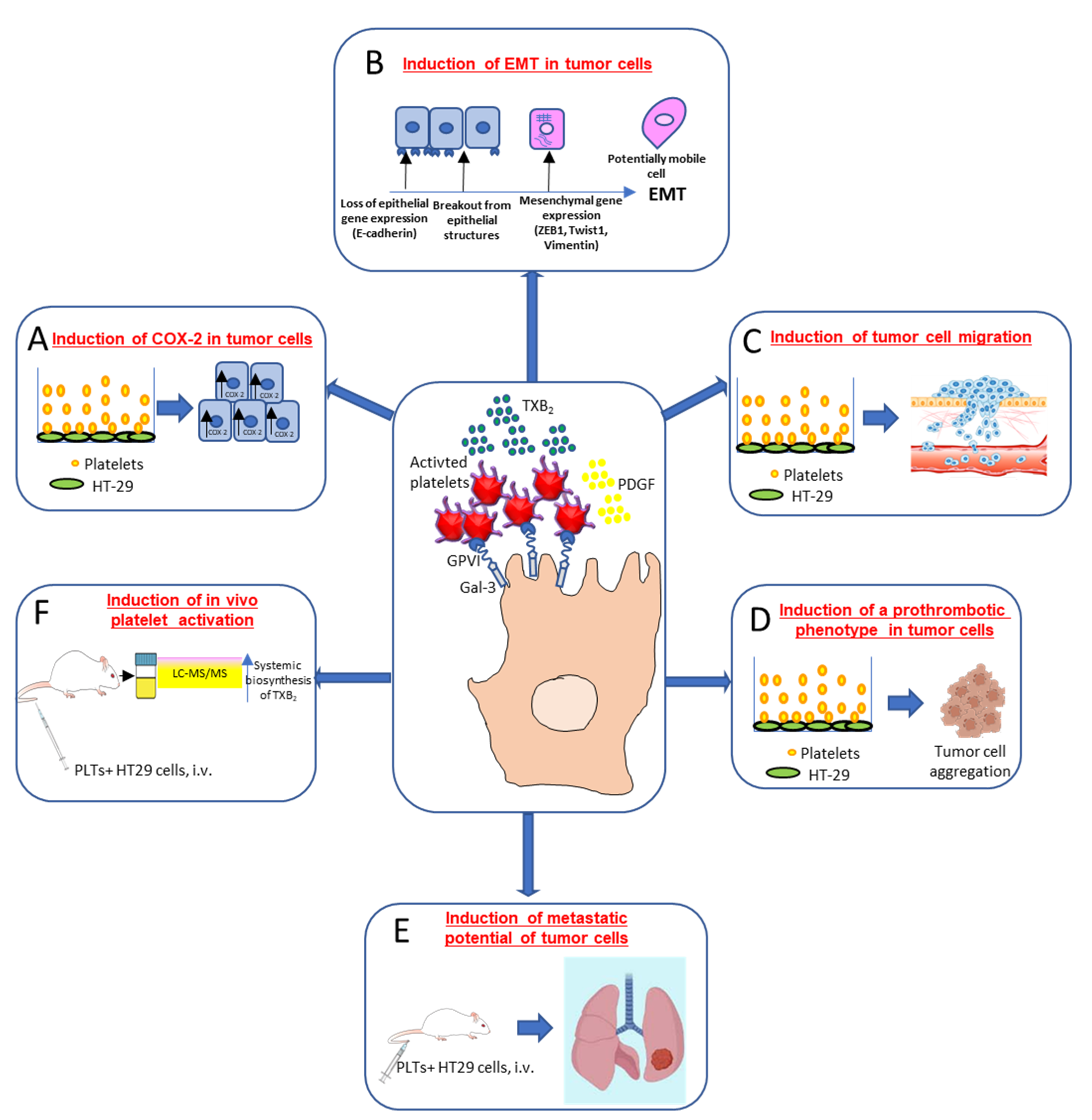
3. Role of Platelets in Tumorigenesis and Metastasis
4. Role of EP Receptors in Cancer and the Effect of Antagonists
5. Role of TP Receptors in Cancer and the Effect of Antagonists
6. Role of Thrombin in Cancer and the Effect of Antagonists
7. Adenine Nucleotides and Purinergic Receptors
8. Role of the P2Y12 Receptor in Cancer and the Effect of Antagonists
9. Development of Novel Antiplatelet Agents Targeting Intracellular Signaling Pathways
9.1. PI3Kβ Inhibitors
9.2. Gq Inhibitors
10. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Khorana, A.A.; Connolly, G.C. Assessing risk of venous thromboembolism in cancer patients. J. Clin. Oncol. 2009, 27, 4839–4847. [Google Scholar] [CrossRef] [PubMed]
- Lugassy, G.; Falanga, A.; Kakkar, A.; Rickles, F. Thrombosis and Cancer; Lugassy, G., Falanga, A., Kakkar, A., Rickles, F., Eds.; Taylor & Francis e-Library: London, UK, 2004. [Google Scholar]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Contursi, A.; Sacco, A.; Grande, R.; Dovizio, M.; Patrignani, P. Platelets as crucial partners for tumor metastasis: From mechanistic aspects to pharmacological targeting. Cell. Mol. Life Sci. 2017, 74, 3491–3507. [Google Scholar] [CrossRef] [PubMed]
- Dovizio, M.; Sacco, A.; Patrignani, P. Curbing tumorigenesis and malignant progression through the pharmacological control of the wound healing process. Vascul. Pharmacol. 2017, 89, 1–11. [Google Scholar] [CrossRef]
- Patrignani, P.; Patrono, C. Aspirin and Cancer. J. Am. Coll. Cardiol. 2016, 68, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Patrignani, P.; Patrono, C. Aspirin, platelet inhibition and cancer prevention. Platelets 2018, 29, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Wingler, L.M.; Lefkowitz, R.J. Conformational Basis of G Protein-Coupled Receptor Signaling Versatility. Trends Cell Biol. 2020, 30, 736–747. [Google Scholar] [CrossRef]
- Arang, N.; Gutkind, J.S. G Protein-Coupled receptors and heterotrimeric G proteins as cancer drivers. FEBS Lett. 2020, 594, 4201–4232. [Google Scholar] [CrossRef]
- Chaudhary, P.K.; Kim, S. An Insight into GPCR and G-Proteins as Cancer Drivers. Cells 2021, 10, 3288. [Google Scholar] [CrossRef]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [Green Version]
- Khalil, B.D.; Hsueh, C.; Cao, Y.; Abi Saab, W.F.; Wang, Y.; Condeelis, J.S.; Bresnick, A.R.; Backer, J.M. GPCR Signaling Mediates Tumor Metastasis via PI3Kβ. Cancer Res. 2016, 76, 2944–2953. [Google Scholar] [CrossRef] [Green Version]
- Stalker, T.J.; Newman, D.K.; Ma, P.; Wannemacher, K.M.; Brass, L.F. Platelet signaling. In Antiplatelet Agents. Handbook of Experimental Pharmacology; Gresele, P., Born, G., Patrono, C., Page, C., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; p. 210. [Google Scholar]
- Degrauwe, S.; Pilgrim, T.; Aminian, A.; Noble, S.; Meier, P.; Iglesias, J.F. Dual antiplatelet therapy for secondary prevention of coronary artery disease. Open Heart 2017, 4, e000651. [Google Scholar] [CrossRef] [Green Version]
- Gurbel, P.A.; Kuliopulos, A.; Tantry, U.S. G-protein-coupled receptors signaling pathways in new antiplatelet drug development. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 500–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggeri, Z.M. Platelets in atherothrombosis. Nat. Med. 2002, 8, 1227–1234. [Google Scholar] [CrossRef]
- Jackson, S.P.; Nesbitt, W.S.; Kulkarni, S. Signaling events underlying thrombus formation. J. Thromb. Haemost. 2003, 1, 1602–1612. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, B.; Watson, S.P. Platelet-collagen interaction: Is GPVI the central receptor? Blood 2003, 102, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, B.; Offermanns, S. Pharmacology of platelet adhesion and aggregation. In Cell Adhesion. Handbook of Experimental Pharmacology; Behrens, J., Nelson, W.J., Eds.; Springer: Berlin/Heidelberg, Germany, 2004; p. 165. [Google Scholar] [CrossRef]
- Yeung, J.; Adili, R.; Stringham, E.N.; Luo, R.; Vizurraga, A.; Rosselli-Murai, L.K.; Stoveken, H.M.; Yu, M.; Piao, X.; Holinstat, M.; et al. GPR56/ADGRG1 is a platelet collagen-responsive GPCR and hemostatic sensor of shear force. Proc. Natl. Acad. Sci. USA 2020, 117, 28275–28286. [Google Scholar] [CrossRef]
- Cattaneo, M. P2Y12 receptors: Structure and function. J. Thromb. Haemost. 2015, 13, S10–S16. [Google Scholar] [CrossRef]
- Patrignani, P.; Patrono, C. Cyclooxygenase inhibitors: From pharmacology to clinical read-outs. Biochim. Biophys. Acta 2015, 1851, 422–432. [Google Scholar] [CrossRef]
- Tourdot, B.E.; Holinstat, M. Targeting 12-Lipoxygenase as a Potential Novel Antiplatelet Therapy. Trends Pharmacol. Sci. 2017, 38, 1006–1015. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Kunapuli, S.P. Coactivation of two different G protein-coupled receptors is essential for ADP-induced platelet aggregation. Proc. Natl. Acad. Sci. USA 1998, 95, 8070–8074. [Google Scholar] [CrossRef] [Green Version]
- Offermanns, S.; Laugwitz, K.L.; Spicher, K.; Schultz, G. G proteins of the G12 family are activated via thromboxane A2 and thrombin receptors in human platelets. Proc. Natl. Acad. Sci. USA 1994, 91, 504–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knezevic, I.; Borg, C.; Le Breton, G.C. Identification of Gq as one of the G-proteins which copurify with human platelet thromboxane A2/prostaglandin H2 receptors. J. Biol. Chem. 1993, 268, 26011–26017. [Google Scholar] [CrossRef]
- Thomas, D.W.; Mannon, R.B.; Mannon, P.J.; Latour, A.; Oliver, J.A.; Hoffman, M.; Smithies, O.; Koller, B.H.; Coffman, T.M. Coagulation defects and altered hemodynamic responses in mice lacking receptors for thromboxane A2. J. Clin. Investig. 1998, 102, 1994–2001. [Google Scholar] [CrossRef] [Green Version]
- Friedman, E.A.; Ogletree, M.L.; Haddad, E.V.; Boutaud, O. Understanding the role of prostaglandin E2 in regulating human platelet activity in health and disease. Thromb. Res. 2015, 136, 493–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabre, J.E.; Nguyen, M.; Athirakul, K.; Coggins, K.; McNeish, J.D.; Austin, S.; Parise, L.K.; FitzGerald, G.A.; Coffman, T.M.; Koller, B.H. Activation of the murine EP3 receptor for PGE2 inhibits cAMP production and promotes platelet aggregation. J. Clin. Investig. 2001, 107, 603–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Hara, A.; Xiao, C.Y.; Okada, Y.; Takahata, O.; Nakaya, K.; Sugimoto, Y.; Ichikawa, A.; Narumiya, S.; Ushikubi, F. Increased bleeding tendency and decreased susceptibility to thromboembolism in mice lacking the prostaglandin E receptor subtype EP(3). Circulation 2001, 104, 1176–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coughlin, S.R. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J. Thromb. Haemost. 2005, 3, 1800–1814. [Google Scholar] [CrossRef]
- Kahn, M.L.; Zheng, Y.W.; Huang, W.; Bigornia, V.; Zeng, D.; Moff, S.; Farese, R.V., Jr.; Tam, C.; Coughlin, S.R. A dual thrombin receptor system for platelet activation. Nature 1998, 394, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Offermanns, S. Activation of platelet function through G protein-coupled receptors. Circ. Res. 2006, 99, 1293–1304. [Google Scholar] [CrossRef] [Green Version]
- Grosser, T.; Fries, S.; Fitzgerald, G.A. Biological basis for the cardiovascular consequences of COX-2 inhibition: Therapeutic challenges and opportunities. J. Clin. Investig. 2006, 116, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhang, W.; Giroux, C.; Cai, Y.; Ekambaram, P.; Dilly, A.K.; Hsu, A.; Zhou, S.; Maddipati, K.R.; Liu, J.; et al. Identification of the orphan G protein-coupled receptor GPR31 as a receptor for 12-(S)-hydroxyeicosatetraenoic acid. J. Biol. Chem. 2011, 286, 33832–33840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Doren, L.; Nguyen, N.; Garzia, C.; Fletcher, E.K.; Stevenson, R.; Jaramillo, D.; Kuliopulos, A.; Covic, L. Lipid Receptor GPR31 (G-Protein-Coupled Receptor 31) Regulates Platelet Reactivity and Thrombosis Without Affecting Hemostasis. Arterioscler. Thromb. Vasc. Biol. 2021, 41, e33–e45. [Google Scholar] [CrossRef]
- Adili, R.; Tourdot, B.E.; Mast, K.; Yeung, J.; Freedman, J.C.; Green, A.; Luci, D.K.; Jadhav, A.; Simeonov, A.; Maloney, D.J.; et al. First Selective 12-LOX Inhibitor, ML355, Impairs Thrombus Formation and Vessel Occlusion In Vivo With Minimal Effects on Hemostasis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1828–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Adili, R.; Liu, L.; Hong, K.; Holinstat, M.; Schwendeman, A. Synthetic high-density lipoproteins loaded with an antiplatelet drug for efficient inhibition of thrombosis in mice. Sci. Adv. 2020, 6, eabd0130. [Google Scholar] [CrossRef]
- Gay, L.J.; Felding-Habermann, B. Platelets alter tumor cell attributes to propel metastasis: Programming in transit. Cancer Cell 2011, 20, 553–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Läubli, H.; Stevenson, J.L.; Varki, A.; Varki, N.M.; Borsig, L. L-selectin facilitation of metastasis involves temporal induction of Fut7-dependent ligands at sites of tumor cell arrest. Cancer Res. 2006, 66, 1536–1542. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, J.S.; Talmage, K.E.; Massari, J.V.; La Jeunesse, C.M.; Flick, M.J.; Kombrinck, K.W.; Jirousková, M.; Degen, J.L. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood 2005, 105, 178–185. [Google Scholar] [CrossRef] [Green Version]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dovizio, M.; Maier, T.J.; Alberti, S.; Di Francesco, L.; Marcantoni, E.; Mu¨nch, G.; John, C.M.; Suess, B.; Sgambato, A.; Steinhilber, D.; et al. Pharmacological inhibition of platelet–tumor cell cross-talk prevents platelet-induced overexpression of cyclooxygenase-2 in HT29 human colon carcinoma cells. Mol. Pharmacol. 2013, 84, 25–40. [Google Scholar] [CrossRef]
- Guillem-Llobat, P.; Dovizio, M.; Bruno, A.; Ricciotti, E.; Cufino, V.; Sacco, A.; Grande, R.; Alberti, S.; Arena, V.; Cirillo, M.; et al. Aspirin prevents colorectal cancer metastasis in mice by splitting the crosstalk between platelets and tumor cells. Oncotarget 2016, 7, 32462–32477. [Google Scholar] [CrossRef] [PubMed]
- Menter, D.G.; Dubois, R.N. Prostaglandins in cancer cell adhesion, migration, and invasion. Int. J. Cell Biol. 2012, 2012, 723419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciulli, M.G.; Filabozzi, P.; Tacconelli, S.; Padovano, R.; Ricciotti, E.; Capone, M.L.; Grana, M.; Carnevale, V.; Patrignani, P. Platelet activation in patients with colorectal cancer. Prostaglandins Leukot. Essent. Fat. Acids 2005, 72, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Dovizio, M.; Tacconelli, S.; Ricciotti, E.; Bruno, A.; Maier, T.J.; Anzellotti, P.; Di Francesco, L.; Sala, P.; Signoroni, S.; Bertario, L.; et al. Effects of celecoxib on prostanoid biosynthesis and circulating angiogenesis proteins in familial adenomatous polyposis. J. Pharmacol Exp. Ther. 2012, 341, 242–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charman, W.N.; Charman, S.A.; Monkhouse, D.C.; Frisbee, S.E.; Lockhart, E.A.; Weisman, S.; Fitzgerald, G.A. Biopharmaceutical characterization of a low-dose (75 mg) controlled-release aspirin formulation. Br. J. Clin. Pharmacol. 1993, 36, 470–473. [Google Scholar] [CrossRef] [Green Version]
- Rothwell, P.M.; Fowkes, F.G.; Belch, J.F.; Ogawa, H.; Warlow, C.P.; Meade, T.W. Effect of daily aspirin on long-term risk of death due to cancer: Analysis of individual patient data from randomised trials. Lancet 2011, 377, 31–41. [Google Scholar] [CrossRef]
- Jurasz, P.; Alonso-Escolano, D.; Radomski, M.W. Platelet-cancer interactions: Mechanisms and pharmacology of tumour cell-induced platelet aggregation. Br. J. Pharmacol. 2004, 143, 819–826. [Google Scholar] [CrossRef] [Green Version]
- Honn, K.V. Inhibition of tumor cell metastasis by modulation of the vascular prostacyclin/thromboxane A2 system. Clin. Exp. Metastasis 1983, 1, 103–114. [Google Scholar] [CrossRef]
- Wang, D.; Dubois, R.N. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 2010, 29, 781–788. [Google Scholar] [CrossRef] [Green Version]
- Pradono, P.; Tazawa, R.; Maemondo, M.; Tanaka, M.; Usui, K.; Saijo, Y.; Hagiwara, K.; Nukiwa, T. Gene transfer of thromboxane A(2) synthase and prostaglandin I(2) synthase antithetically altered tumor angiogenesis and tumor growth. Cancer Res. 2002, 62, 63–66. [Google Scholar]
- Matsui, Y.; Amano, H.; Ito, Y.; Eshima, K.; Suzuki, T.; Ogawa, F.; Iyoda, A.; Satoh, Y.; Kato, S.; Nakamura, M.; et al. Thromboxane A₂ receptor signaling facilitates tumor colonization through P-selectin-mediated interaction of tumor cells with platelets and endothelial cells. Cancer Sci. 2012, 103, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; DuBois, R.N. Role of prostanoids in gastrointestinal cancer. J. Clin. Investig. 2018, 128, 2732–2742. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Zeller, W.; Zhou, N.; Hategen, G.; Mishra, R.; Polozov, A.; Yu, P.; Onua, E.; Zhang, J.; Zembower, D.; et al. Antagonists of the EP3 receptor for prostaglandin E2 are novel antiplatelet agents that do not prolong bleeding. ACS Chem. Biol. 2009, 4, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Fulton, A.M.; Ma, X.; Kundu, N. Targeting prostaglandin E EP receptors to inhibit metastasis. Cancer Res. 2006, 66, 9794–9797. [Google Scholar] [CrossRef] [Green Version]
- Take, Y.; Koizumi, S.; Nagahisa, A. Prostaglandin E Receptor 4 Antagonist in CancerImmunotherapy: Mechanisms of Action. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Parikh, A.; Shapiro, G.I.; Varga, A.; Naing, A.; Meric-Bernstam, F.; Ataman, Ö.; Reyderman, L.; Binder, T.A.; Ren, M.; et al. First-in-human phase I study of immunomodulatory E7046, an antagonist of PGE2-receptor E-type 4 (EP4), in patients with advanced cancers. J. Immunother. Cancer 2020, 8, e000222. [Google Scholar] [CrossRef]
- O’Callaghan, G.; Houston, A. Prostaglandin E2 and the EP receptors in malignancy: Possible therapeutic targets? Br. J. Pharmacol. 2015, 172, 5239–5250. [Google Scholar] [CrossRef] [Green Version]
- af Forselles, K.J.; Root, J.; Clarke, T.; Davey, D.; Aughton, K.; Dack, K.; Pullen, N. In vitro and in vivo characterization of PF-04418948, a novel, potent and selective prostaglandin EP₂ receptor antagonist. Br. J. Pharmacol. 2011, 164, 1847–1856. [Google Scholar] [CrossRef] [Green Version]
- Sales, K.J.; Maudsley, S.; Jabbour, H.N. Elevated prostaglandin EP2 receptor in endometrial adenocarcinoma cells promotes vascular endothelial growth factor expression via cyclic 3’,5’-adenosine monophosphate-mediated transactivation of the epidermal growth factor receptor and extracellular signal-regulated kinase 1/2 signaling pathways. Mol. Endocrinol. 2004, 18, 1533–1545. [Google Scholar] [CrossRef]
- Chang, S.H.; Liu, C.H.; Wu, M.T.; Hla, T. Regulation of vascular endothelial cell growth factor expression in mouse mammary tumor cells by the EP2 subtype of the prostaglandin E2 receptor. Prostaglandins Other Lipid Mediat. 2005, 76, 48–58. [Google Scholar] [CrossRef]
- Kamiyama, M.; Pozzi, A.; Yang, L.; DeBusk, L.M.; Breyer, R.M.; Lin, P.C. EP2, a receptor for PGE2, regulates tumor angiogenesis through direct effects on endothelial cell motility and survival. Oncogene 2006, 25, 7019–7028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinski, P. Regulation of immune responses by prostaglandin E2. J. Immunol. 2012, 188, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.Y.; Zhang, H.; Zhang, M.; Xia, S.K.; Bai, X.M.; Zhang, L.; Ma, J.; Rong, R.; Wang, Y.P.; Du, M.Z.; et al. Prostaglandin E2 receptor EP2 mediates Snail expression in hepatocellular carcinoma cells. Oncol. Rep. 2014, 31, 2099–2106. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Schiemann, W.P. PGE2 receptor EP2 mediates the antagonistic effect of COX-2 on TGF-beta signaling during mammary tumorigenesis. FASEB J. 2010, 24, 1105–1116. [Google Scholar] [CrossRef] [Green Version]
- Namba, T.; Sugimoto, Y.; Negishi, M.; Irie, A.; Ushikubi, F.; Kakizuka, A.; Ito, S.; Ichikawa, A.; Narumiya, S. Alternative splicing of C-terminal tail of prostaglandin E receptor subtype EP3 determines G-protein specificity. Nature 1993, 365, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Woodward, D.F.; Jones, R.L.; Narumiya, S. International Union of Basic and Clinical Pharmacology. LXXXIII: Classification of prostanoid receptors, updating 15 years of progress. Pharmacol. Rev. 2011, 63, 471–538. [Google Scholar] [CrossRef] [Green Version]
- Dey, I.; Lejeune, M.; Chadee, K. Prostaglandin E2 receptor distribution and function in the gastrointestinal tract. Br. J. Pharmacol. 2006, 149, 611–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, K.; Kawamori, T.; Nakatsugi, S.; Ohta, T.; Ohuchida, S.; Yamamoto, H.; Maruyama, T.; Kondo, K.; Narumiya, S.; Sugimura, T.; et al. Inhibitory effect of a prostaglandin E receptor subtype EP(1) selective antagonist, ONO-8713, on development of azoxymethane-induced aberrant crypt foci in mice. Cancer Lett. 2000, 156, 57–61. [Google Scholar] [CrossRef]
- Watanabe, K.; Kawamori, T.; Nakatsugi, S.; Ohta, T.; Ohuchida, S.; Yamamoto, H.; Maruyama, T.; Kondo, K.; Ushikubi, F.; Narumiya, S.; et al. Role of the prostaglandin E receptor subtype EP1 in colon carcinogenesis. Cancer Res. 1999, 59, 5093–5096. [Google Scholar]
- Kawamori, T.; Uchiya, N.; Nakatsugi, S.; Watanabe, K.; Ohuchida, S.; Yamamoto, H.; Maruyama, T.; Kondo, K.; Sugimura, T.; Wakabayashi, K. Chemopreventive effects of ONO-8711, a selective prostaglandin E receptor EP(1) antagonist, on breast cancer development. Carcinogenesis 2001, 22, 2001–2004. [Google Scholar] [CrossRef] [Green Version]
- Tober, K.L.; Wilgus, T.A.; Kusewitt, D.F.; Thomas-Ahner, J.M.; Maruyama, T.; Oberyszyn, T.M. Importance of the EP(1) receptor in cutaneous UVB-induced inflammation and tumor development. J. Investig. Dermatol. 2006, 126, 205–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Kundu, N.; Ioffe, O.B.; Goloubeva, O.; Konger, R.; Baquet, C.; Gimotty, P.; Reader, J.; Fulton, A.M. Prostaglandin E receptor EP1 suppresses breast cancer metastasis and is linked to survival differences and cancer disparities. Mol. Cancer Res. 2010, 8, 1310–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorat, M.A.; Morimiya, A.; Mehrotra, S.; Konger, R.; Badve, S.S. Prostanoid receptor EP1 expression in breast cancer. Mod. Pathol. 2008, 21, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Iyú, D.; Glenn, J.R.; White, A.E.; Johnson, A.J.; Fox, S.C.; Heptinstall, S. The role of prostanoid receptors in mediating the effects of PGE(2) on human platelet function. Platelets 2010, 21, 329–342. [Google Scholar] [CrossRef]
- Clapp, L.; Giembycz, M.; Heinemann, A.; Jones, R.L.; Narumiya, S.; Norel, X.; Sugimoto, Y.; Woodward, D.F.; Yao, C. Prostanoid receptors in GtoPdb v.2021.2. IUPHAR/BPS Guide Pharmacol. CITE, 2021; 2. [Google Scholar] [CrossRef]
- Nakahata, N. Thromboxane A2: Physiology/pathophysiology, cellular signal transduction and pharmacology. Pharmacol. Ther. 2008, 118, 18–35. [Google Scholar] [CrossRef]
- Needleman, P.; Minkes, M.; Raz, A. Thromboxanes: Selective biosynthesis and distinct biological properties. Science 1976, 193, 163–165. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tai, H.-H. Thromboxane A2 receptor-mediated release of matrix metalloproteinase-1 (MMP-1) induces expression of monocyte chemoattractant protein-1 (MCP-1) by activation of protease-activated receptor 2 (PAR2) in A549 human lung adenocarcinoma cells. Mol. Carcinog. 2014, 53, 659–666. [Google Scholar] [CrossRef]
- Mulvaney, E.P.; Shilling, C.; Eivers, S.B.; Perry, A.S.; Bjartell, A.; Kay, E.W.; Watson, R.W.; Kinsella, B.T. Expression of the TPα and TPβ isoforms of the thromboxane prostanoid receptor (TP) in prostate cancer: Clinical significance and diagnostic potential. Oncotarget 2016, 7, 73171–73187. [Google Scholar] [CrossRef] [Green Version]
- Sobolesky, P.M.; Halushka, P.V.; Garrett-Mayer, E.; Smith, M.T.; Moussa, O. Regulation of the tumor suppressor FOXO3 by the thromboxane-A2 receptors in urothelial cancer. PLoS ONE 2014, 9, e107530. [Google Scholar] [CrossRef] [Green Version]
- Orr, K.; Buckley, N.E.; Haddock, P.; James, C.; Parent, J.-L.; McQuaid, S.; Mullan, P.B. Thromboxane A2 receptor (TBXA2R) is a potent survival factor for triple negative breast cancers (TNBCs). Oncotarget 2016, 7, 55458–55472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Lee, M.-H.; Liu, K.; Wang, T.; Song, M.; Han, Y.; Yao, K.; Xie, H.; Zhu, F.; Grossmann, M.; et al. Inhibiting breast cancer by targeting the thromboxane A2 pathway. NPJ Precis. Oncol. 2017, 1, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Fujii, T.; Takahashi, Y.; Takahashi, Y.; Suzuki, T.; Ukai, M.; Tauchi, K.; Horikawa, N.; Tsukada, K.; Sakai, H. Up-regulation of Kv7.1 channels in thromboxane A2-induced colonic cancer cell proliferation. Pflugers Arch. 2014, 466, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Ekambaram, P.; Lambiv, W.; Cazzolli, R.; Ashton, A.W.; Honn, K. V The thromboxane synthase and receptor signaling pathway in cancer: An emerging paradigm in cancer progression and metastasis. Cancer Metastasis Rev. 2011, 30, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Werfel, T.A.; Hicks, D.J.; Rahman, B.; Bendeman, W.E.; Duvernay, M.T.; Maeng, J.G.; Hamm, H.; Lavieri, R.R.; Joly, M.M.; Pulley, J.M.; et al. Repurposing of a Thromboxane Receptor Inhibitor Based on a Novel Role in Metastasis Identified by Phenome-Wide Association Study. Mol. Cancer Ther. 2020, 19, 2454–2464. [Google Scholar] [CrossRef]
- Honn, K.V.; Meyer, J.; Neagos, G.; Henderson, T.; Westley, C.; Ratanatharathorn, V. Control of tumor growth and metastasis with prostacyclin and thromboxane synthetase inhibitors: Evidence for a new antitumor and antimetastatic agent (BAY g 6575). Prog. Clin. Biol. Res. 1982, 89, 295–331. [Google Scholar]
- Tesfamariam, B. Involvement of platelets in tumor cell metastasis. Pharmacol. Ther. 2016, 157, 112–119. [Google Scholar] [CrossRef]
- Mehta, P.; Lawson, D.; Ward, M.B.; Lee-Ambrose, L.; Kimura, A. Effects of thromboxane A2 inhibition on osteogenic sarcoma cell-induced platelet aggregation. Cancer Res. 1986, 46, 5061–5063. [Google Scholar]
- de Leval, X.; Benoit, V.; Delarge, J.; Julémont, F.; Masereel, B.; Pirotte, B.; Merville, M.P.; David, J.L.; Dogné, J.M. Pharmacological evaluation of the novel thromboxane modulator BM-567 (II/II). Effects of BM-567 on osteogenic sarcoma-cell-induced platelet aggregation. Prostaglandins Leukot. Essent. Fat. Acids 2003, 68, 55–59. [Google Scholar] [CrossRef]
- Unsworth, A.J.; Bye, A.P.; Sage, T.; Gaspar, R.S.; Eaton, N.; Drew, C.; Stainer, A.; Kriek, N.; Volberding, P.J.; Hutchinson, J.L.; et al. Antiplatelet properties of Pim kinase inhibition are mediated through disruption of thromboxane A2 receptor signaling. Haematologica 2021, 106, 1968–1978. [Google Scholar] [CrossRef]
- Allison, S.E.; Petrovic, N.; Mackenzie, P.I.; Murray, M. Pro-migratory actions of the prostacyclin receptor in human breast cancer cells that over-express cyclooxygenase-2. Biochem. Pharmacol. 2015, 96, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Mitrugno, A.; Williams, D.; Kerrigan, S.W.; Moran, N. A novel and essential role for FcγRIIa in cancer cell-induced platelet activation. Blood 2014, 123, 249–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso-Escolano, D.; Strongin, A.Y.; Chung, A.W.; Deryugina, E.I.; Radomski, M.W. Membrane type-1 matrix metalloproteinase stimulates tumour cell-induced platelet aggregation: Role of receptor glycoproteins. Br. J. Pharmacol. 2004, 141, 241–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, C.; Jurasz, P.; Santos-Martinez, M.J.; Jeong, S.S.; Mitsky, T.; Chen, R.; Radomski, M.W. Platelet aggregation-induced by caco-2 cells: Regulation by matrix metalloproteinase-2 and adenosine diphosphate. J. Pharmacol. Exp. Ther. 2006, 317, 739–745. [Google Scholar] [CrossRef]
- Lian, L.; Li, W.; Li, Z.-Y.; Mao, Y.-X.; Zhang, Y.-T.; Zhao, Y.-M.; Chen, K.; Duan, W.-M.; Tao, M. Inhibition of MCF-7 breast cancer cell-induced platelet aggregation using a combination of antiplatelet drugs. Oncol. Lett. 2013, 5, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Zarà, M.; Canobbio, I.; Visconte, C.; Canino, J.; Torti, M.; Guidetti, G.F. Molecular mechanisms of platelet activation and aggregation induced by breast cancer cells. Cell. Signal. 2018, 48, 45–53. [Google Scholar] [CrossRef]
- Krishna Priya, S.; Nagare, R.P.; Sneha, V.S.; Sidhanth, C.; Bindhya, S.; Manasa, P.; Ganesan, T.S. Tumour angiogenesis-Origin of blood vessels. Int. J. Cancer 2016, 139, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Daniel, T.O.; Liu, H.; Morrow, J.D.; Crews, B.C.; Marnett, L.J. Thromboxane A2 is a mediator of cyclooxygenase-2-dependent endothelial migration and angiogenesis. Cancer Res. 1999, 59, 4574–4577. [Google Scholar]
- Wei, J.; Yan, W.; Li, X.; Ding, Y.; Tai, H.H. Thromboxane receptor alpha mediates tumor growth and angiogenesis via induction of vascular endothelial growth factor expression in human lung cancer cells. Lung Cancer 2010, 69, 26–32. [Google Scholar] [CrossRef]
- Nie, D.; Lamberti, M.; Zacharek, A.; Li, L.; Szekeres, K.; Tang, K.; Chen, Y.; Honn, K.V. Thromboxane A(2) regulation of endothelial cell migration, angiogenesis, and tumor metastasis. Biochem. Biophys. Res. Commun. 2000, 267, 245–251. [Google Scholar] [CrossRef]
- Kuwano, T.; Nakao, S.; Yamamoto, H.; Tsuneyoshi, M.; Yamamoto, T.; Kuwano, M.; Ono, M. Cyclooxygenase 2 is a key enzyme for inflammatory cytokine-induced angiogenesis. FASEB J. 2004, 18, 300–310. [Google Scholar] [CrossRef]
- Kim, S.-R.; Jung, Y.-H.; Park, H.-J.; Kim, M.-K.; Jeong, J.-W.; Jang, H.-O.; Yun, I.; Bae, S.-K.; Bae, M.-K. Upregulation of thromboxane synthase mediates visfatin-induced interleukin-8 expression and angiogenic activity in endothelial cells. Biochem. Biophys. Res. Commun. 2012, 418, 662–668. [Google Scholar] [CrossRef]
- Ashton, A.W.; Yokota, R.; John, G.; Zhao, S.; Suadicani, S.O.; Spray, D.C.; Ware, J.A. Inhibition of endothelial cell migration, intercellular communication, and vascular tube formation by thromboxane A(2). J. Biol. Chem. 1999, 274, 35562–35570. [Google Scholar] [CrossRef] [Green Version]
- Ashton, A.W.; Ware, J.A. Thromboxane A2 receptor signaling inhibits vascular endothelial growth factor-induced endothelial cell differentiation and migration. Circ. Res. 2004, 95, 372–379. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.; Wu, J.; Murray, J.K.; Gellman, S.H.; Wozniak, M.A.; Keely, P.J.; Boyer, M.E.; Gomez, T.M.; Hasso, S.M.; Fallon, J.F.; et al. An antiangiogenic neurokinin-B/thromboxane A2 regulatory axis. J. Cell Biol. 2006, 174, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Ashton, A.W.; Cheng, Y.; Helisch, A.; Ware, J.A. Thromboxane A2 receptor agonists antagonize the proangiogenic effects of fibroblast growth factor-2: Role of receptor internalization, thrombospondin-1, and alpha(v)beta3. Circ. Res. 2004, 94, 735–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Yokota, R.; Tang, S.; Ashton, A.W.; Ware, J.A. Reversal of angiogenesis in vitro, induction of apoptosis, and inhibition of AKT phosphorylation in endothelial cells by thromboxane A(2). Circ. Res. 2000, 87, 739–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Cruz, J.P.; Moreno, A.; Ruiz-Ruiz, M.I.; Sánchez De La Cuesta, F. Effect of DT-TX 30, a combined thromboxane synthase inhibitor and thromboxane receptor antagonist, on retinal vascularity in experimental diabetes mellitus. Thromb. Res. 2000, 97, 125–131. [Google Scholar] [CrossRef]
- Beauchamp, M.H.; Martinez-Bermudez, A.K.; Gobeil, F.; Marrache, A.M.; Hou, X.; Speranza, G.; Abran, D.; Quiniou, C.; Lachapelle, P.; Roberts, J.; et al. Role of thromboxane in retinal microvascular degeneration in oxygen-induced retinopathy. J. Appl. Physiol. 2001, 90, 2279–2288. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.-H.; Shi, C.; Cohen, R.A. High glucose via peroxynitrite causes tyrosine nitration and inactivation of prostacyclin synthase that is associated with thromboxane/prostaglandin H(2) receptor-mediated apoptosis and adhesion molecule expression in cultured human aortic endothelial cells. Diabetes 2002, 51, 198–203. [Google Scholar] [CrossRef] [Green Version]
- Benndorf, R.A.; Schwedhelm, E.; Gnann, A.; Taheri, R.; Kom, G.; Didié, M.; Steenpass, A.; Ergün, S.; Böger, R.H. Isoprostanes inhibit vascular endothelial growth factor-induced endothelial cell migration, tube formation, and cardiac vessel sprouting in vitro, as well as angiogenesis in vivo via activation of the thromboxane A(2) receptor: A potential link between oxidative stress and impaired angiogenesis. Circ. Res. 2008, 103, 1037–1046. [Google Scholar] [CrossRef] [Green Version]
- Wilson, S.J.; McGinley, K.; Huang, A.J.; Smyth, E.M. Heterodimerization of the alpha and beta isoforms of the human thromboxane receptor enhances isoprostane signaling. Biochem. Biophys. Res. Commun. 2007, 352, 397–403. [Google Scholar] [CrossRef] [Green Version]
- Wojtukiewicz, M.Z.; Tang, D.G.; Ciarelli, J.J.; Nelson, K.K.; Walz, D.A.; Diglio, C.A.; Mammen, E.F.; Honn, K.V. Thrombin increases the metastatic potential of tumor cells. Int. J. Cancer 1993, 54, 793–806. [Google Scholar] [CrossRef]
- Nierodzik, M.; Kajumo, F.; Karpatkin, S. Effect of thrombin treatment of tumor cells on adhesion of tumor cells to platelets in vitro and metastasis in vivo. Cancer Res. 1992, 52, 3267–3272. [Google Scholar] [PubMed]
- Huang, Z.; Miao, X.; Luan, Y.; Zhu, L.; Kong, F.; Lu, Q.; Pernow, J.; Nilsson, G.; Li, N. PAR1-stimulated platelet releasate promotes angiogenic activities of endothelial progenitor cells more potently than PAR4-stimulated platelet releasate. Thromb. Haemostasis 2015, 13, 465–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufmann, R.; Junker, U.; Junker, K.; Nuske, K.; Ranke, C.; Zieger, M.; Scheele, J. The serine proteinase thrombin promotes migration of human renal carcinoma cells by a PKA-dependent mechanism. Cancer Lett. 2002, 180, 183–190. [Google Scholar] [CrossRef]
- Radjabi, A.R.; Sawada, K.; Jagadeeswaran, S.; Eichbichler, A.; Kenny, H.A.; Montag, A.; Bruno, K.; Lengyel, E. Thrombin induces tumor invasion through the induction and association of matrix metalloproteinase-9 and beta1-integrin on the cell surface. J. Biol. Chem. 2008, 283, 2822–2834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nierodzik, M.; Plotkin, A.; Kajumo, F.; Karpatkin, S. Thrombin stimulates tumor-platelet adhesion in vitro and metastasis in vivo. J. Clin. Investig. 1991, 87, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Zigler, M.; Kamiya, T.; Brantley, E.C.; Villares, G.J.; Bar-Eli, M. PAR-1 and thrombin: The ties that bind the microenvironment to melanoma metastasis. Cancer Res. 2011, 71, 6561–6566. [Google Scholar] [CrossRef] [Green Version]
- Cisowski, J.; O’Callaghan, K.; Kuliopulos, A.; Yang, J.; Nguyen, N.; Deng, Q.; Yang, E.; Fogel, M.; Tressel, S.; Foley, C.; et al. Targeting protease-activated receptor-1 with cell-penetrating pepducins in lung cancer. Am. J. Pathol. 2011, 179, 513–523. [Google Scholar] [CrossRef]
- Chen, Y.; Yuan, Y.; Li, W. Sorting machineries: How platelet-dense granules differ from α-granules. Biosci. Rep. 2018, 38, BSR20180458. [Google Scholar] [CrossRef] [Green Version]
- Enjyoji, K.; Sévigny, J.; Lin, Y.; Frenette, P.S.; Christie, P.D.; Esch, J.S., 2nd; Imai, M.; Edelberg, J.M.; Rayburn, H.; Lech, M.; et al. Targeted disruption of cd39/ATP diphosphohydrolase results in disordered hemostasis and thromboregulation. Nat. Med. 1999, 5, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic Signaling in the Cardiovascular System. Circ. Res. 2017, 120, 207–228. [Google Scholar] [CrossRef] [Green Version]
- Ballerini, P.; Dovizio, M.; Bruno, A.; Tacconelli, S.; Patrignani, P. P2Y12 Receptors in Tumorigenesis and Metastasis. Front. Pharmacol. 2018, 9, 66. [Google Scholar] [CrossRef] [Green Version]
- Gachet, C. P2Y(12) receptors in platelets and other hematopoietic and non-hematopoietic cells. Purinergic Signal. 2012, 8, 609–619. [Google Scholar] [CrossRef]
- Lee, N.T.; Ong, L.K.; Gyawali, P.; Nassir, C.M.N.C.M.; Mustapha, M.; Nandurkar, H.H.; Sashindranath, M. Role of Purinergic Signalling in Endothelial Dysfunction and Thrombo-Inflammation in Ischaemic Stroke and Cerebral Small Vessel Disease. Biomolecules 2021, 11, 994. [Google Scholar] [CrossRef] [PubMed]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317. [Google Scholar] [CrossRef] [Green Version]
- Yegutkin, G.G. Nucleotide- and nucleoside-converting ectoenzymes: Important modulators of purinergic signalling cascade. Biochim. Biophys. Acta 2008, 1783, 673–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegatti, P.; Falzoni, S.; Pinton, P.; Rizzuto, R.; Di Virgilio, F. A novel recombinant plasma membrane-targeted luciferase reveals a new pathway for ATP secretion. Mol. Biol. Cell 2005, 16, 3659–3665. [Google Scholar] [CrossRef] [Green Version]
- De Marchi, E.; Orioli, E.; Pegoraro, A.; Adinolfi, E.; Di Virgilio, F. Detection of Extracellular ATP in the Tumor Microenvironment, Using the pmeLUC Biosensor. Methods Mol. Biol. 2020, 2041, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Vultaggio-Poma, V.; Sarti, A.C.; Di Virgilio, F. Extracellular ATP: A Feasible Target for Cancer Therapy. Cells 2020, 9, 2496. [Google Scholar] [CrossRef] [PubMed]
- Salmaso, V.; Jacobson, K.A. Purinergic Signaling: Impact of GPCR Structures on Rational Drug Design. ChemMedChem 2020, 15, 1958–1973. [Google Scholar] [CrossRef] [PubMed]
- Hechler, B.; Gachet, C. P2 receptors and platelet function. Purinergic Signal. 2011, 7, 293–303. [Google Scholar] [CrossRef] [Green Version]
- Cattaneo, M. New P2Y(12) inhibitors. Circulation 2010, 121, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Schrör, K.; Siller-Matula, J.M.; Huber, K. Pharmacokinetic basis of the antiplatelet action of prasugrel. Fundam. Clin. Pharmacol. 2012, 26, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Kügelgen, I. Pharmacological profiles of cloned mammalian P2Y-receptor subtypes. Pharmacol. Ther. 2006, 110, 415–432. [Google Scholar] [CrossRef]
- Abbracchio, M.P.; Burnstock, G.; Boeynaems, J.M.; Barnard, E.A.; Boyer, J.L.; Kennedy, C.; Knight, G.E.; Fumagalli, M.; Gachet, C.; Jacobson, K.A.; et al. International Union of Pharmacology LVIII: Update on the P2Y G protein-coupled nucleotide receptors: From molecular mechanisms and pathophysiology to therapy. Pharmacol. Rev. 2006, 58, 281–341. [Google Scholar] [CrossRef]
- Ohlmann, P.; Lecchi, A.; El-Tayeb, A.; Müller, C.E.; Cattaneo, M.; Gachet, C. The platelet P2Y(12) receptor under normal and pathological conditions. Assessment with the radiolabeled selective antagonist [(3)H]PSB-0413. Purinergic Signal. 2013, 9, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef]
- Suzuki, T.; Kohyama, K.; Moriyama, K.; Ozaki, M.; Hasegawa, S.; Ueno, T.; Saitoe, M.; Morio, T.; Hayashi, M.; Sakuma, H. Extracellular ADP augments microglial inflammasome and NF-κB activation via the P2Y12 receptor. Eur. J. Immunol. 2020, 50, 205–219. [Google Scholar] [CrossRef]
- Su, X.; Floyd, D.H.; Hughes, A.; Xiang, J.; Schneider, J.G.; Uluckan, O.; Heller, E.; Deng, H.; Zou, W.; Craft, C.S.; et al. The ADP receptor P2RY12 regulates osteoclast function and pathologic bone remodeling. J. Clin. Investig. 2012, 122, 3579–3592. [Google Scholar] [CrossRef] [PubMed]
- Diehl, P.; Olivier, C.; Halscheid, C.; Helbing, T.; Bode, C.; Moser, M. Clopidogrel affects leukocyte dependent platelet aggregation by P2Y12 expressing leukocytes. Basic Res. Cardiol. 2010, 105, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Muniz, V.S.; Baptista-Dos-Reis, R.; Benjamim, C.F.; Mata-Santos, H.A.; Pyrrho, A.S.; Strauch, M.A.; Melo, P.A.; Vicentino, A.R.; Silva-Paiva, J.; Bandeira-Melo, C.; et al. Purinergic P2Y12 Receptor Activation in Eosinophils and the Schistosomal Host Response. PLoS ONE 2015, 10, e0139805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kronlage, M.; Song, J.; Sorokin, L.; Isfort, K.; Schwerdtle, T.; Leipziger, J.; Robaye, B.; Conley, P.B.; Kim, H.C.; Sargin, S.; et al. Autocrine purinergic receptor signaling is essential for macrophage chemotaxis. Sci. Signal. 2010, 3, ra55. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Guo, Y.; Zhuang, X.; Li, W.; Zou, J.; Su, Q.; Zhao, J.; Liu, Y.; Liao, X.; Du, Z.; et al. Immunosuppressive Effect of Ticagrelor on Dendritic Cell Function: A New Therapeutic Target of Antiplatelet Agents in Cardiovascular Disease. J. Biomed. Nanotechnol. 2018, 14, 1665–1673. [Google Scholar] [CrossRef] [PubMed]
- Wihlborg, A.K.; Wang, L.; Braun, O.O.; Eyjolfsson, A.; Gustafsson, R.; Gudbjartsson, T.; Erlinge, D. ADP receptor P2Y12 is expressed in vascular smooth muscle cells and stimulates contraction in human blood vessels. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1810–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, X.; Pi, S.L.; Baral, S.; Xia, Y.P.; He, Q.W.; Li, Y.N.; Jin, H.J.; Li, M.; Wang, M.D.; Mao, L.; et al. P2Y12 Promotes Migration of Vascular Smooth Muscle Cells Through Cofilin Dephosphorylation During Atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 515–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Kunapuli, S.P. P2Y12 receptor in platelet activation. Platelets 2011, 22, 56–60. [Google Scholar] [CrossRef]
- Siller-Matula, J.M.; Trenk, D.; Schrör, K.; Gawaz, M.; Kristensen, S.D.; Storey, R.F.; Huber, K.; EPA (European Platelet Academy). Response variability to P2Y12 receptor inhibitors: Expectations and reality. JACC Cardiovasc. Interv. 2013, 6, 1111–1128. [Google Scholar] [CrossRef]
- Fitzgerald, D.J.; Fitzgerald, G.A. Historical lessons in translational medicine: Cyclooxygenase inhibition and P2Y12 antagonism. Circ. Res. 2013, 112, 174–194. [Google Scholar] [CrossRef] [PubMed]
- von Kügelgen, I. Molecular pharmacology of P2Y receptor subtypes. Biochem. Pharmacol. 2021, 187, 114361. [Google Scholar] [CrossRef] [PubMed]
- Guidetti, G.F.; Lova, P.; Bernardi, B.; Campus, F.; Baldanzi, G.; Graziani, A.; Balduini, C.; Torti, M. The Gi-coupled P2Y12 receptor regulates diacylglycerol-mediated signaling in human platelets. J. Biol. Chem. 2008, 283, 28795–28805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baig, A.; Bao, X.; Wolf, M.; Haslam, R.J. The platelet protein kinase C substrate pleckstrin binds directly to SDPR protein. Platelets 2009, 20, 446–457. [Google Scholar] [CrossRef]
- Garcia, A.; Kim, S.; Bhavaraju, K.; Schoenwaelder, S.M.; Kunapuli, S.P. Role of phosphoinositide 3-kinase beta in platelet aggregation and thromboxane A2 generation mediated by Gi signalling pathways. Biochem. J. 2010, 429, 369–377. [Google Scholar] [CrossRef] [Green Version]
- Cosemans, J.M.; Munnix, I.C.; Wetzker, R.; Heller, R.; Jackson, S.P.; Heemskerk, J.W. Continuous signaling via PI3K isoforms beta and gamma is required for platelet ADP receptor function in dynamic thrombus stabilization. Blood 2006, 108, 3045–3052. [Google Scholar] [CrossRef]
- Schoenwaelder, S.M.; Ono, A.; Sturgeon, S.; Chan, S.M.; Mangin, P.; Maxwell, M.J.; Turnbull, S.; Mulchandani, M.; Anderson, K.; Kauffenstein, G.; et al. Identification of a unique co-operative phosphoinositide 3-kinase signaling mechanism regulating integrin alpha IIb beta 3 adhesive function in platelets. J. Biol. Chem. 2007, 282, 28648–28658. [Google Scholar] [CrossRef] [Green Version]
- Kohga, S.; Kinjo, M.; Tanaka, K.; Ogawa, H.; Ishihara, M.; Tanaka, N. Effects of 5-(2-chlorobenzyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine hydrochloride (Ticlopidine), a platelet aggregation inhibitor, on blood-borne metastasis. Cancer Res. 1981, 41, 4710–4714. [Google Scholar]
- Wang, Y.; Sun, Y.; Li, D.; Zhang, L.; Wang, K.; Zuo, Y.; Gartner, T.K.; Liu, J. Platelet P2Y12 is involved in murine pulmonary metastasis. PLoS ONE 2013, 8, e80780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gareau, A.J.; Brien, C.; Gebremeskel, S.; Liwski, R.S.; Johnston, B.; Bezuhly, M. Ticagrelor inhibits platelet-tumor cell interactions and metastasis in human and murine breast cancer. Clin. Exp. Metastasis 2018, 35, 25–35. [Google Scholar] [CrossRef]
- Gebremeskel, S.; LeVatte, T.; Liwski, R.S.; Johnston, B.; Bezuhly, M. The reversible P2Y12 inhibitor ticagrelor inhibits metastasis and improves survival in mouse models of cancer. Int. J. Cancer 2015, 136, 234–240. [Google Scholar] [CrossRef]
- Cho, M.S.; Noh, K.; Haemmerle, M.; Li, D.; Park, H.; Hu, Q.; Hisamatsu, T.; Mitamura, T.; Mak, S.L.C.; Kunapuli, S.; et al. Role of ADP receptors on platelets in the growth of ovarian cancer. Blood 2017, 130, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Czajkowski, R.; Banachewicz, W.; Ilnytska, O.; Drobot, L.B.; Baranska, J. Differential effects of P2Y1 and P2Y12 nucleotide receptors on ERK1/ERK2 and phosphatidylinositol 3-kinase signalling and cell proliferation in serum-deprived and nonstarved glioma C6 cells. Br. J. Pharmacol. 2004, 141, 497–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shchors, K.; Massaras, A.; Hanahan, D. Dual Targeting of the Autophagic Regulatory Circuitry in Gliomas with Repurposed Drugs Elicits Cell-Lethal Autophagy and Therapeutic Benefit. Cancer Cell 2015, 28, 456–471. [Google Scholar] [CrossRef] [Green Version]
- Elaskalani, O.; Domenichini, A.; Abdol Razak, N.B.; Dye, D.; Falasca, M.; Metharom, P. Antiplatelet Drug Ticagrelor Enhances Chemotherapeutic Efficacy by Targeting the Novel P2Y12-AKT Pathway in Pancreatic Cancer Cells. Cancers 2020, 12, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawton, J.S.; Tamis-Holland, J.E.; Bangalore, S.; Bates, E.R.; Beckie, T.M.; Bischoff, J.M.; Bittl, J.A.; Cohen, M.G.; DiMaio, J.M.; Don, C.W.; et al. 2021 ACC/AHA/SCAI Guideline for Coronary Artery Revascularization: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2022, 79, e21–e129. [Google Scholar] [CrossRef]
- Levine, G.N.; Bates, E.R.; Bittl, J.A.; Brindis, R.G.; Fihn, S.D.; Fleisher, L.A.; Granger, C.B.; Lange, R.A.; Mack, M.J.; Mauri, L.; et al. 2016 ACC/AHA Guideline Focused Update on Duration of Dual Antiplatelet Therapy in Patients With Coronary Artery Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2016, 68, 1082–1115. [Google Scholar] [CrossRef] [PubMed]
- Collet, J.P.; Thiele, H.; Barbato, E.; Barthélémy, O.; Bauersachs, J.; Bhatt, D.L.; Dendale, P.; Dorobantu, M.; Edvardsen, T.; Folliguet, T.; et al. ESC Scientific Document Group. 2020 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur. Heart J. 2021, 42, 1289–1367. [Google Scholar] [CrossRef]
- Serebruany, V.L.; Tomek, A.; Kim, M.H. Survival After Solid Cancers in Antithrombotic Trials. Am. J. Cardiol. 2015, 116, 969–972. [Google Scholar] [CrossRef]
- Elmariah, S.; Mauri, L.; Doros, G.; Galper, B.Z.; O’Neill, K.E.; Steg, P.G.; Kereiakes, D.J.; Yeh, R.W. Extended duration dual antiplatelet therapy and mortality: A systematic review and meta-analysis. Lancet 2015, 385, 792–798. [Google Scholar] [CrossRef] [Green Version]
- Leader, A.; Zelikson-Saporta, R.; Pereg, D.; Spectre, G.; Rozovski, U.; Raanani, P.; Hermoni, D.; Lishner, M. The Effect of Combined Aspirin and Clopidogrel Treatment on Cancer Incidence. Am. J. Med. 2017, 130, 826–832. [Google Scholar] [CrossRef] [Green Version]
- Kotronias, R.A.; Kwok, C.S.; Wong, C.W.; Kinnaird, T.; Zaman, A.; Mamas, M.A. Cancer Event Rate and Mortality with Thienopyridines: A Systematic Review and Meta-Analysis. Drug Saf. 2017, 40, 229–240. [Google Scholar] [CrossRef]
- Rodríguez-Miguel, A.; García-Rodríguez, L.A.; Gil, M.; Montoya, H.; Rodríguez-Martín, S.; de Abajo, F.J. Clopidogrel and Low-Dose Aspirin, Alone or Together, Reduce Risk of Colorectal Cancer. Clin. Gastroenterol. Hepatol. 2019, 17, 2024–2033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, J.R.; Chauhan, M.; Shah, C.; Ring, A.; Thomas, A.L.; Goodall, A.H.; Adlam, D. The TICONC (Ticagrelor-Oncology) Study: Implications of P2Y12 Inhibition for Metastasis and Cancer-Associated Thrombosis. JACC CardioOncol. 2020, 2, 236–250. [Google Scholar] [CrossRef]
- Gremmel, T.; Yanachkov, I.B.; Yanachkova, M.I.; Wright, G.E.; Wider, J.; Undyala, V.V.; Michelson, A.D.; Frelinger, A.L. 3rd.; Przyklenk, K. Synergistic Inhibition of Both P2Y1 and P2Y12 Adenosine Diphosphate Receptors as Novel Approach to Rapidly Attenuate Platelet-Mediated Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 501–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolensky Koganov, E.; Michelson, A.D.; Yanachkov, I.B.; Yanachkova, M.I.; Wright, G.E.; Przyklenk, K.; Frelinger, A.L. 3rd. GLS-409, an Antagonist of Both P2Y1 and P2Y12, Potently Inhibits Canine Coronary Artery Thrombosis and Reversibly Inhibits Human Platelet Activation. Sci. Rep. 2018, 8, 14529. [Google Scholar] [CrossRef]
- Jackson, S.P.; Schoenwaelder, S.M.; Goncalves, I.; Nesbitt, W.S.; Yap, C.L.; Wright, C.E.; Kenche, V.; Anderson, K.E.; Dopheide, S.M.; Yuan, Y.; et al. PI 3-kinase p110beta: A new target for antithrombotic therapy. Nat. Med. 2005, 11, 507–514. [Google Scholar] [CrossRef]
- Martin, V.; Guillermet-Guibert, J.; Chicanne, G.; Cabou, C.; Jandrot-Perrus, M.; Plantavid, M.; Vanhaesebroeck, B.; Payrastre, B.; Gratacap, M.P. Deletion of the p110beta isoform of phosphoinositide 3-kinase in platelets reveals its central role in Akt activation and thrombus formation in vitro and in vivo. Blood 2010, 115, 2008–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nylander, S.; Kull, B.; Björkman, J.A.; Ulvinge, J.C.; Oakes, N.; Emanuelsson, B.M.; Andersson, M.; Skärby, T.; Inghardt, T.; Fjellström, O.; et al. Human target validation of phosphoinositide 3-kinase (PI3K)β: Effects on platelets and insulin sensitivity, using AZD6482 a novel PI3Kβ inhibitor. J. Thromb. Haemost. 2012, 10, 2127–2136. [Google Scholar] [CrossRef] [PubMed]
- Nylander, S.; Wågberg, F.; Andersson, M.; Skärby, T.; Gustafsson, D. Exploration of efficacy and bleeding with combined phosphoinositide 3-kinase β inhibition and aspirin in man. J. Thromb. Haemost. 2015, 13, 1494–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, S.P.; Schoenwaelder, S.M. Antithrombotic phosphoinositide 3-kinase β inhibitors in humans: A ‘shear’ delight! J. Thromb. Haemost. 2012, 10, 2123–2126. [Google Scholar] [CrossRef] [Green Version]
- Uemura, T.; Kawasaki, T.; Taniguchi, M.; Moritani, Y.; Hayashi, K.; Saito, T.; Takasaki, J.; Uchida, W.; Miyata, K. Biological properties of a specific Galpha q/11 inhibitor, YM-254890, on platelet functions and thrombus formation under high-shear stress. Br. J. Pharmacol. 2006, 148, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Peng, Q.; Alqahtani, S.; Nasrullah, M.Z.A.; Shen, J. Functional evidence for biased inhibition of G protein signaling by YM-254890 in human coronary artery endothelial cells. Eur. J. Pharmacol. 2021, 891, 173706. [Google Scholar] [CrossRef] [PubMed]
- Maree, A.O.; Fitzgerald, D.J. Variable platelet response to aspirin and clopidogrel in atherothrombotic disease. Circulation 2007, 115, 2196–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capone, M.L.; Tacconelli, S.; Sciulli, M.G.; Grana, M.; Ricciotti, E.; Minuz, P.; Di Gregorio, P.; Merciaro, G.; Patrono, C.; Patrignani, P. Clinical pharmacology of platelet, monocyte, and vascular cyclooxygenase inhibition by naproxen and low-dose aspirin in healthy subjects. Circulation 2004, 109, 1468–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audoly, L.P.; Rocca, B.; Fabre, J.E.; Koller, B.H.; Thomas, D.; Loeb, A.L.; Coffman, T.M.; FitzGerald, G.A. Cardiovascular responses to the isoprostanes iPF(2alpha)-III and iPE(2)-III are mediated via the thromboxane A(2) receptor in vivo. Circulation 2000, 101, 2833–2840. [Google Scholar] [CrossRef] [Green Version]
- van Kooten, F.; Ciabattoni, G.; Patrono, C.; Dippel, D.W.; Koudstaal, P.J. Platelet activation and lipid peroxidation in patients with acute ischemic stroke. Stroke 1997, 28, 1557–1563. [Google Scholar] [CrossRef] [Green Version]
- Bousser, M.G.; Amarenco, P.; Chamorro, A.; Fisher, M.; Ford, I.; Fox, K.M.; Hennerici, M.G.; Mattle, H.P.; Rothwell, P.M.; PERFORM Study Investigators; et al. Terutroban versus aspirin in patients with cerebral ischaemic events (PERFORM): A randomised, double-blind, parallel-group trial. Lancet 2011, 377, 2013–2022. [Google Scholar] [CrossRef]
- Fitzgerald, D.J.; Fragetta, J.; FitzGerald, G.A. Prostaglandin endoperoxides modulate the response to thromboxane synthase inhibition during coronary thrombosis. J. Clin. Investig. 1988, 82, 1708–1713. [Google Scholar] [CrossRef]
- Sacco, A.; Bruno, A.; Contursi, A.; Dovizio, M.; Tacconelli, S.; Ricciotti, E.; Guillem-Llobat, P.; Salvatore, T.; Di Francesco, L.; Fullone, R.; et al. Platelet-Specific Deletion of Cyclooxygenase-1 Ameliorates Dextran Sulfate Sodium-Induced Colitis in Mice. J. Pharmacol. Exp. Ther. 2019, 370, 416–426. [Google Scholar] [CrossRef]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rovati, G.; Contursi, A.; Bruno, A.; Tacconelli, S.; Ballerini, P.; Patrignani, P. Antiplatelet Agents Affecting GPCR Signaling Implicated in Tumor Metastasis. Cells 2022, 11, 725. https://doi.org/10.3390/cells11040725
Rovati G, Contursi A, Bruno A, Tacconelli S, Ballerini P, Patrignani P. Antiplatelet Agents Affecting GPCR Signaling Implicated in Tumor Metastasis. Cells. 2022; 11(4):725. https://doi.org/10.3390/cells11040725
Chicago/Turabian StyleRovati, Gianenrico, Annalisa Contursi, Annalisa Bruno, Stefania Tacconelli, Patrizia Ballerini, and Paola Patrignani. 2022. "Antiplatelet Agents Affecting GPCR Signaling Implicated in Tumor Metastasis" Cells 11, no. 4: 725. https://doi.org/10.3390/cells11040725