

KR158 Spheres Harboring Slow-Cycling Cells Recapitulate High-Grade Glioma Features in an Immunocompetent System

 , , , ,
, , , , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Murine Glioma Cell Line
2.2. Gliomasphere Assay
2.3. Classical Adherent and Serum-Containing Cell Cultures
2.4. Scratch-Wound Assay and Time-Lapse Imaging
2.5. Intracranial Implant, Tumor Growth Monitoring and Survival Analysis
2.6. Isolation of Fast- and Slow-Cycling Cells
2.7. Short Tandem Repeat (STR) Analysis
2.8. Quantification of Secreted Lipocalin-2 Using ELISA
2.9. Immunocytochemistry
2.10. Flow Cytometric Analysis
2.11. Brain Tumor Tissue Processing
2.12. Live/Dead Assay
2.13. CyQUANT Direct Cell Proliferation Assay
2.14. Sphere-Forming Frequency (SFF) Assay
2.15. Bulk RNA-Seq Sample Preparation and Analysis
2.16. Single-Cell RNA Sequencing and Quality Control
2.17. Single-Cell RNA-Seq Data Analysis
2.18. Statistical Analyses
3. Results
3.1. Transcriptomic Difference between KR158, GL261, and CT-2A, with KR158 Overexpressing GBM-like Signatures
3.2. scRNA Sequencing Revealed Cellular Diversity in KR158 Tumors, Exhibiting Similarities with the Heterogeneity Observed in hGBM
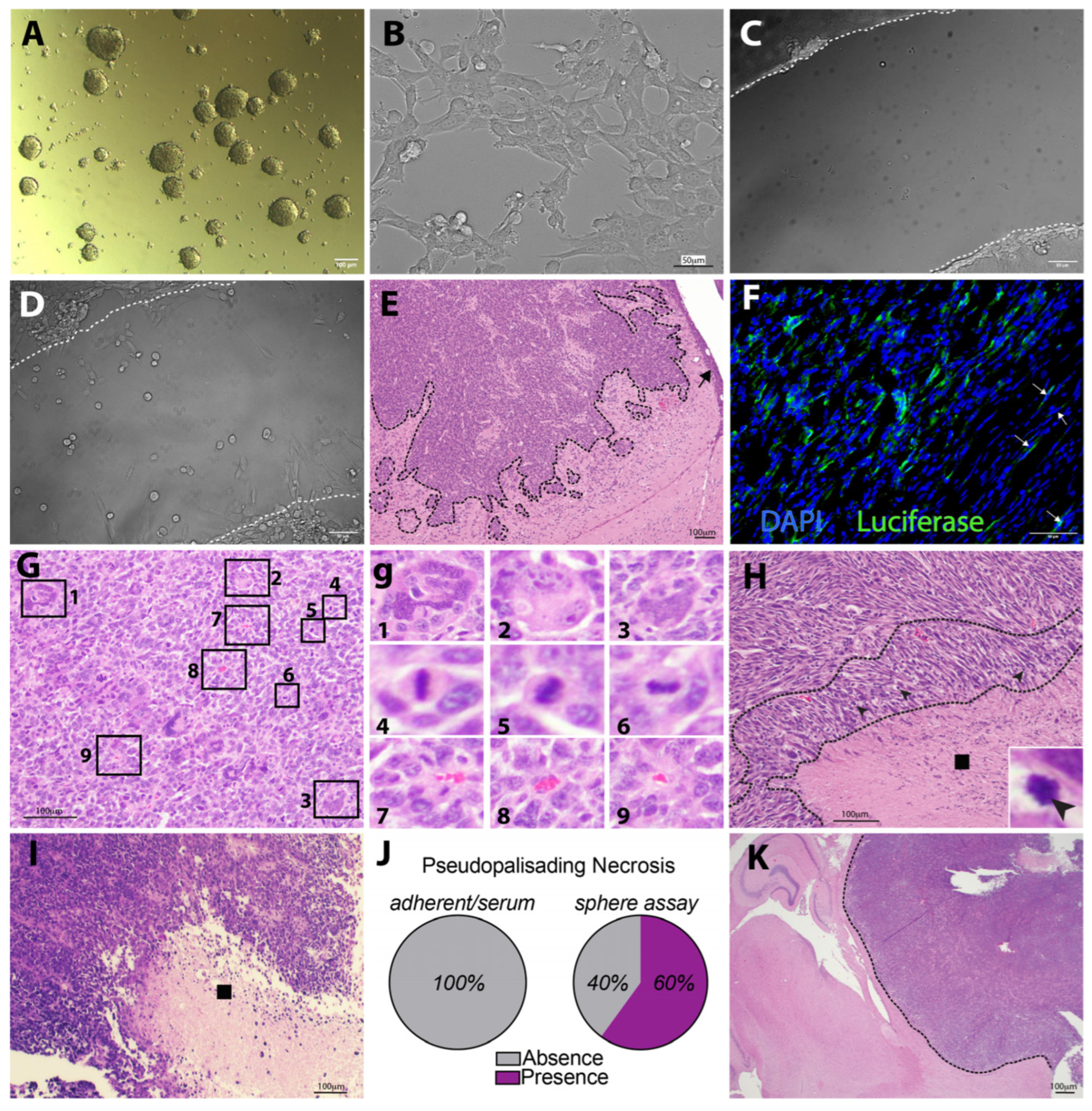
3.3. KR158 Cells Expanded Using the Gliomasphere Assay Generate Glioblastoma-like Tumors
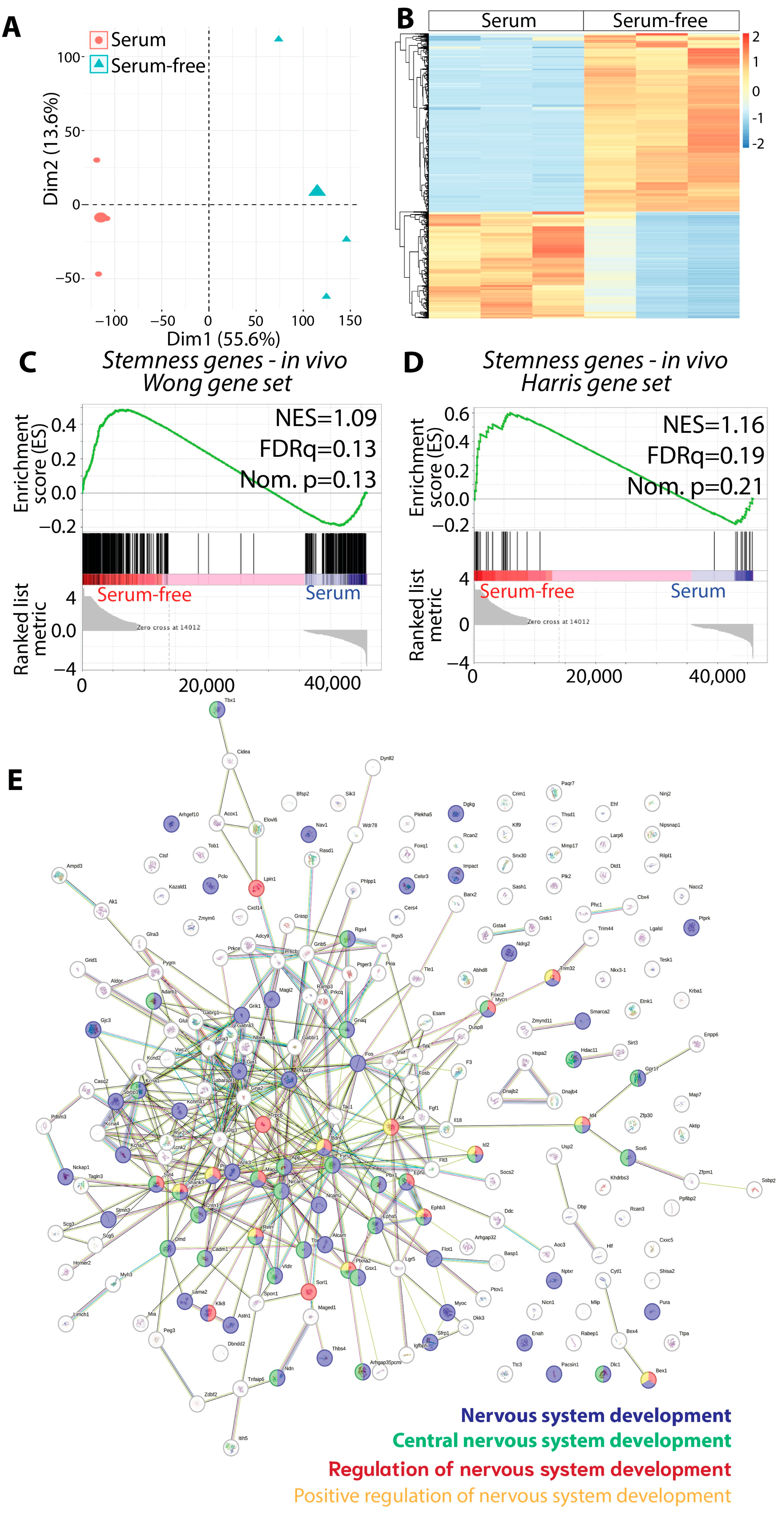
3.4. The Gliomasphere Assay Enhances Stemness Gene Signature in KR158 Cells
3.5. Evidence of SCCs in KR158 Gliomasphere Cultures Showing Similarities to SCCs in hGBM
3.6. Murine KR158 SCCs Form GBM-like Tumor with Heightened Stemness and Aggressive Phenotype
3.7. Murine KR158 SCCs Are More Tolerant to TMZ Compared to FCCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Badr, C.E.; Silver, D.J.; Siebzehnrubl, F.A.; Deleyrolle, L.P. Metabolic heterogeneity and adaptability in brain tumors. Cell. Mol. Life Sci. 2020, 77, 5101–5119. [Google Scholar] [CrossRef] [PubMed]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef]
- Nicholson, J.G.; Fine, H.A. Diffuse Glioma Heterogeneity and Its Therapeutic Implications. Cancer Discov. 2021, 11, 575–590. [Google Scholar] [CrossRef]
- Silver, A.; Feier, D.; Ghosh, T.; Rahman, M.; Huang, J.; Sarkisian, M.R.; Deleyrolle, L.P. Heterogeneity of glioblastoma stem cells in the context of the immune microenvironment and geospatial organization. Front. Oncol. 2022, 12, 1022716. [Google Scholar] [CrossRef]
- Graham, S.M.; Jorgensen, H.G.; Allan, E.; Pearson, C.; Alcorn, M.J.; Richmond, L.; Holyoake, T.L. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002, 99, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Dembinski, J.L.; Krauss, S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin. Exp. Metastasis 2009, 26, 611–623. [Google Scholar] [CrossRef]
- Gao, M.Q.; Choi, Y.P.; Kang, S.; Youn, J.H.; Cho, N.H. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene 2010, 29, 2672–2680. [Google Scholar] [CrossRef]
- Pece, S.; Tosoni, D.; Confalonieri, S.; Mazzarol, G.; Vecchi, M.; Ronzoni, S.; Bernard, L.; Viale, G.; Pelicci, P.G.; Di Fiore, P.P. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell 2010, 140, 62–73. [Google Scholar] [CrossRef]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef]
- Moore, N.; Houghton, J.; Lyle, S. Slow-cycling therapy-resistant cancer cells. Stem Cells Dev. 2012, 21, 1822–1830. [Google Scholar] [CrossRef] [PubMed]
- Moore, N.; Lyle, S. Quiescent, slow-cycling stem cell populations in cancer: A review of the evidence and discussion of significance. J. Oncol. 2011, 2011, 396076. [Google Scholar] [CrossRef] [PubMed]
- Zeuner, A.; Francescangeli, F.; Contavalli, P.; Zapparelli, G.; Apuzzo, T.; Eramo, A.; Baiocchi, M.; De Angelis, M.L.; Biffoni, M.; Sette, G.; et al. Elimination of quiescent/slow-proliferating cancer stem cells by Bcl-XL inhibition in non-small cell lung cancer. Cell Death Differ. 2014, 21, 1877–1888. [Google Scholar] [CrossRef]
- Shiokawa, D.; Sakai, H.; Ohata, H.; Miyazaki, T.; Kanda, Y.; Sekine, S.; Narushima, D.; Hosokawa, M.; Kato, M.; Suzuki, Y.; et al. Slow-Cycling Cancer Stem Cells Regulate Progression and Chemoresistance in Colon Cancer. Cancer Res. 2020, 80, 4451–4464. [Google Scholar] [CrossRef] [PubMed]
- Campos, B.; Gal, Z.; Baader, A.; Schneider, T.; Sliwinski, C.; Gassel, K.; Bageritz, J.; Grabe, N.; von Deimling, A.; Beckhove, P.; et al. Aberrant self-renewal and quiescence contribute to the aggressiveness of glioblastoma. J. Pathol. 2014, 234, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Deleyrolle, L.P.; Harding, A.; Cato, K.; Siebzehnrubl, F.A.; Rahman, M.; Azari, H.; Olson, S.; Gabrielli, B.; Osborne, G.; Vescovi, A.; et al. Evidence for label-retaining tumour-initiating cells in human glioblastoma. Brain J. Neurol. 2011, 134, 1331–1343. [Google Scholar] [CrossRef] [PubMed]
- Deleyrolle, L.P.; Rohaus, M.R.; Fortin, J.M.; Reynolds, B.A.; Azari, H. Identification and isolation of slow-dividing cells in human glioblastoma using carboxy fluorescein succinimidyl ester (CFSE). J. Vis. Exp. 2012. [Google Scholar] [CrossRef] [PubMed]
- Azari, H.; Deleyrolle, L.P.; Reynolds, B.A. Using Carboxy Fluorescein Succinimidyl Ester (CFSE) to Identify Quiescent Glioblastoma Stem-Like Cells. Methods Mol. Biol. 2018, 1686, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Hoang-Minh, L.B.; Siebzehnrubl, F.A.; Yang, C.; Suzuki-Hatano, S.; Dajac, K.; Loche, T.; Andrews, N.; Schmoll Massari, M.; Patel, J.; Amin, K.; et al. Infiltrative and drug-resistant slow-cycling cells support metabolic heterogeneity in glioblastoma. EMBO J. 2018, 37, e98772. [Google Scholar] [CrossRef]
- Yang, C.; Tian, G.; Dajac, M.; Doty, A.; Wang, S.; Lee, J.H.; Rahman, M.; Huang, J.; Reynolds, B.A.; Sarkisian, M.R.; et al. Slow-Cycling Cells in Glioblastoma: A Specific Population in the Cellular Mosaic of Cancer Stem Cells. Cancers 2022, 14, 1126. [Google Scholar] [CrossRef]
- Sabelstrom, H.; Quigley, D.A.; Fenster, T.; Foster, D.J.; Fuchshuber, C.A.M.; Saxena, S.; Yuan, E.; Li, N.; Paterno, F.; Phillips, J.J.; et al. High density is a property of slow-cycling and treatment-resistant human glioblastoma cells. Exp. Cell Res. 2019, 378, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Jorg, D.J.; Cavalli, F.M.G.; Richards, L.M.; Nguyen, L.V.; Vanner, R.J.; Guilhamon, P.; Lee, L.; Kushida, M.M.; Pellacani, D.; et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 2017, 549, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Antonica, F.; Santomaso, L.; Pernici, D.; Petrucci, L.; Aiello, G.; Cutarelli, A.; Conti, L.; Romanel, A.; Miele, E.; Tebaldi, T.; et al. A slow-cycling/quiescent cells subpopulation is involved in glioma invasiveness. Nat. Commun. 2022, 13, 4767. [Google Scholar] [CrossRef] [PubMed]
- Atkins, R.J.; Stylli, S.S.; Kurganovs, N.; Mangiola, S.; Nowell, C.J.; Ware, T.M.; Corcoran, N.M.; Brown, D.V.; Kaye, A.H.; Morokoff, A.; et al. Cell quiescence correlates with enhanced glioblastoma cell invasion and cytotoxic resistance. Exp. Cell Res. 2019, 374, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Reilly, K.M.; Loisel, D.A.; Bronson, R.T.; McLaughlin, M.E.; Jacks, T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat. Genet. 2000, 26, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.; Fakurnejad, S.; Sayegh, E.T.; Clark, A.J.; Ivan, M.E.; Sun, M.Z.; Safaee, M.; Bloch, O.; James, C.D.; Parsa, A.T. Immunocompetent murine models for the study of glioblastoma immunotherapy. J. Transl. Med. 2014, 12, 107. [Google Scholar] [CrossRef] [PubMed]
- Binello, E.; Qadeer, Z.A.; Kothari, H.P.; Emdad, L.; Germano, I.M. Stemness of the CT-2A Immunocompetent Mouse Brain Tumor Model: Characterization In Vitro. J. Cancer 2012, 3, 166–174. [Google Scholar] [CrossRef]
- Zimmerman, H.M.; Arnold, H. Experimental Brain Tumors. I. Tumors Produced with Methylcholanthrene. Cancer Res. 1941, 1, 919–938. [Google Scholar]
- Ou, G.; Charles, L.; Matton, S.; Rodner, C.; Hurley, M.; Kuhn, L.; Gronowicz, G. Fibroblast growth factor-2 stimulates the proliferation of mesenchyme-derived progenitor cells from aging mouse and human bone. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Hebert, J.M.; Basilico, C.; Goldfarb, M.; Haub, O.; Martin, G.R. Isolation of cDNAs encoding four mouse FGF family members and characterization of their expression patterns during embryogenesis. Dev. Biol. 1990, 138, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Herr, A.B.; Ornitz, D.M.; Sasisekharan, R.; Venkataraman, G.; Waksman, G. Heparin-induced self-association of fibroblast growth factor-2. Evidence for two oligomerization processes. J. Biol. Chem. 1997, 272, 16382–16389. [Google Scholar] [CrossRef] [PubMed]
- Yayon, A.; Klagsbrun, M.; Esko, J.D.; Leder, P.; Ornitz, D.M. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell 1991, 64, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Goutam, R.S.; Kumar, V.; Lee, U.; Kim, J. Exploring the Structural and Functional Diversity among FGF Signals: A Comparative Study of Human, Mouse, and Xenopus FGF Ligands in Embryonic Development and Cancer Pathogenesis. Int. J. Mol. Sci. 2023, 24, 7556. [Google Scholar] [CrossRef] [PubMed]
- Siebzehnrubl, F.A.; Silver, D.J.; Tugertimur, B.; Deleyrolle, L.P.; Siebzehnrubl, D.; Sarkisian, M.R.; Devers, K.G.; Yachnis, A.T.; Kupper, M.D.; Neal, D.; et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol. Med. 2013, 5, 1196–1212. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Tao, H.; Karachi, A.; Grippin, A.J.; Jin, L.; Chang, Y.E.; Zhang, W.; Dyson, K.A.; Hou, A.Y.; Na, M.; et al. Dysregulation of Glutamate Transport Enhances Treg Function That Promotes VEGF Blockade Resistance in Glioblastoma. Cancer Res. 2020, 80, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.J.; Liu, H.; Ridky, T.W.; Cassarino, D.; Segal, E.; Chang, H.Y. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell 2008, 2, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.A.; Yang, H.; Low, B.E.; Mukherjee, J.; Guha, A.; Bronson, R.T.; Shultz, L.D.; Israel, M.A.; Yun, K. Cancer stem cells are enriched in the side population cells in a mouse model of glioma. Cancer Res. 2008, 68, 10051–10059. [Google Scholar] [CrossRef]
- Trivedi, V.; Yang, C.; Klippel, K.; Yegorov, O.; von Roemeling, C.; Hoang-Minh, L.; Fenton, G.; Ogando-Rivas, E.; Castillo, P.; Moore, G.; et al. mRNA-based precision targeting of neoantigens and tumor-associated antigens in malignant brain tumors. Genome Med. 2024, 16, 17. [Google Scholar] [CrossRef]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Berger, G.; Knelson, E.H.; Jimenez-Macias, J.L.; Nowicki, M.O.; Han, S.; Panagioti, E.; Lizotte, P.H.; Adu-Berchie, K.; Stafford, A.; Dimitrakakis, N.; et al. STING activation promotes robust immune response and NK cell-mediated tumor regression in glioblastoma models. Proc. Natl. Acad. Sci. USA 2022, 119, e2111003119. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhao, Y.; Huang, Y.; Zhu, K.; Zeng, F.; Zhao, J.; Zhang, H.; Zhu, X.; Kettenmann, H.; Xiang, X. TREM2 promotes glioma progression and angiogenesis mediated by microglia/brain macrophages. Glia 2023, 71, 2679–2695. [Google Scholar] [CrossRef] [PubMed]
- Zamler, D.B.; Shingu, T.; Kahn, L.M.; Huntoon, K.; Kassab, C.; Ott, M.; Tomczak, K.; Liu, J.; Li, Y.; Lai, I.; et al. Immune landscape of a genetically engineered murine model of glioma compared with human glioma. JCI Insight 2022, 7, e148990. [Google Scholar] [CrossRef] [PubMed]
- Johanns, T.M.; Ward, J.P.; Miller, C.A.; Wilson, C.; Kobayashi, D.K.; Bender, D.; Fu, Y.; Alexandrov, A.; Mardis, E.R.; Artyomov, M.N.; et al. Endogenous Neoantigen-Specific CD8 T Cells Identified in Two Glioblastoma Models Using a Cancer Immunogenomics Approach. Cancer Immunol. Res. 2016, 4, 1007–1015. [Google Scholar] [CrossRef] [PubMed]
- Karachi, A.; Yang, C.; Dastmalchi, F.; Sayour, E.J.; Huang, J.; Azari, H.; Long, Y.; Flores, C.; Mitchell, D.A.; Rahman, M. Modulation of temozolomide dose differentially affects T-cell response to immune checkpoint inhibition. Neuro-Oncol. 2019, 21, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Altieri, R.; Zenga, F.; Fontanella, M.M.; Cofano, F.; Agnoletti, A.; Spena, G.; Crobeddu, E.; Fornaro, R.; Ducati, A.; Garbossa, D. Glioma Surgery: Technological Advances to Achieve a Maximal Safe Resection. Surg. Technol. Int. 2015, 27, 297–302. [Google Scholar] [PubMed]
- Lopez-Bertoni, H.; Johnson, A.; Rui, Y.; Lal, B.; Sall, S.; Malloy, M.; Coulter, J.B.; Lugo-Fagundo, M.; Shudir, S.; Khela, H.; et al. Sox2 induces glioblastoma cell stemness and tumor propagation by repressing TET2 and deregulating 5hmC and 5mC DNA modifications. Signal Transduct. Target. Ther. 2022, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Blanco, J.; Sanz-Arriazu, L.; Lorenzoni, R.; Blanco-Prieto, M.J. Glioblastoma chemotherapeutic agents used in the clinical setting and in clinical trials: Nanomedicine approaches to improve their efficacy. Int. J. Pharm. 2020, 581, 119283. [Google Scholar] [CrossRef]
- Patterson, J.D.; Wongsurawat, T.; Rodriguez, A. A Glioblastoma Genomics Primer for Clinicians. Med. Res. Arch. 2020, 8, 1–17. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2011, 59, 1169–1180. [Google Scholar] [CrossRef]
- Mair, D.B.; Ames, H.M.; Li, R. Mechanisms of invasion and motility of high-grade gliomas in the brain. Mol. Biol. Cell 2018, 29, 2509–2515. [Google Scholar] [CrossRef]
- Saurty-Seerunghen, M.S.; Daubon, T.; Bellenger, L.; Delaunay, V.; Castro, G.; Guyon, J.; Rezk, A.; Fabrega, S.; Idbaih, A.; Almairac, F.; et al. Glioblastoma cell motility depends on enhanced oxidative stress coupled with mobilization of a sulfurtransferase. Cell Death Dis. 2022, 13, 913. [Google Scholar] [CrossRef]
- Sambraus, H.H. Remarkable domestic animal breeds in California. Tierarztl. Prax. 1990, 18, 547–556. [Google Scholar]
- Varlakhanova, N.V.; Cotterman, R.F.; deVries, W.N.; Morgan, J.; Donahue, L.R.; Murray, S.; Knowles, B.B.; Knoepfler, P.S. myc maintains embryonic stem cell pluripotency and self-renewal. Differentiation 2010, 80, 9–19. [Google Scholar] [CrossRef]
- Laurenti, E.; Varnum-Finney, B.; Wilson, A.; Ferrero, I.; Blanco-Bose, W.E.; Ehninger, A.; Knoepfler, P.S.; Cheng, P.F.; MacDonald, H.R.; Eisenman, R.N.; et al. Hematopoietic stem cell function and survival depend on c-Myc and N-Myc activity. Cell Stem Cell 2008, 3, 611–624. [Google Scholar] [CrossRef]
- Cotterman, R.; Knoepfler, P.S. N-Myc regulates expression of pluripotency genes in neuroblastoma including lif, klf2, klf4, and lin28b. PLoS ONE 2009, 4, e5799. [Google Scholar] [CrossRef]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Medina-Menendez, C.; Garcia-Corzo, L.; Cordoba-Beldad, C.M.; Quiroga, A.C.; Calleja Barca, E.; Zinchuk, V.; Munoz-Lopez, S.; Rodriguez-Martin, P.; Ciorraga, M.; et al. SoxD genes are required for adult neural stem cell activation. Cell Rep. 2022, 38, 110313. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Gasco, A.; Parcerisas, A.; Hino, K.; Herranz-Perez, V.; Ulloa, F.; Elias-Tersa, A.; Bosch, M.; Garcia-Verdugo, J.M.; Simo, S.; Pujadas, L.; et al. Regulation of young-adult neurogenesis and neuronal differentiation by neural cell adhesion molecule 2 (NCAM2). Cereb. Cortex 2023, 33, 10931–10948. [Google Scholar] [CrossRef]
- Mossahebi-Mohammadi, M.; Quan, M.; Zhang, J.S.; Li, X. FGF Signaling Pathway: A Key Regulator of Stem Cell Pluripotency. Front. Cell Dev. Biol. 2020, 8, 79. [Google Scholar] [CrossRef]
- Gouaze-Andersson, V.; Gherardi, M.J.; Lemarie, A.; Gilhodes, J.; Lubrano, V.; Arnauduc, F.; Cohen-Jonathan Moyal, E.; Toulas, C. FGFR1/FOXM1 pathway: A key regulator of glioblastoma stem cells radioresistance and a prognosis biomarker. Oncotarget 2018, 9, 31637–31649. [Google Scholar] [CrossRef]
- Hsu, Y.C.; Kao, C.Y.; Chung, Y.F.; Lee, D.C.; Liu, J.W.; Chiu, I.M. Activation of Aurora A kinase through the FGF1/FGFR signaling axis sustains the stem cell characteristics of glioblastoma cells. Exp. Cell Res. 2016, 344, 153–166. [Google Scholar] [CrossRef]
- Yarden, Y.; Kuang, W.J.; Yang-Feng, T.; Coussens, L.; Munemitsu, S.; Dull, T.J.; Chen, E.; Schlessinger, J.; Francke, U.; Ullrich, A. Human proto-oncogene c-kit: A new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 1987, 6, 3341–3351. [Google Scholar] [CrossRef]
- Maisel, M.; Herr, A.; Milosevic, J.; Hermann, A.; Habisch, H.J.; Schwarz, S.; Kirsch, M.; Antoniadis, G.; Brenner, R.; Hallmeyer-Elgner, S.; et al. Transcription profiling of adult and fetal human neuroprogenitors identifies divergent paths to maintain the neuroprogenitor cell state. Stem Cells 2007, 25, 1231–1240. [Google Scholar] [CrossRef]
- Dermit, M.; Dodel, M.; Lee, F.C.Y.; Azman, M.S.; Schwenzer, H.; Jones, J.L.; Blagden, S.P.; Ule, J.; Mardakheh, F.K. Subcellular mRNA Localization Regulates Ribosome Biogenesis in Migrating Cells. Dev. Cell 2020, 55, 298–313 e210. [Google Scholar] [CrossRef]
- Shao, R.; Scully, S.J., Jr.; Yan, W.; Bentley, B.; Mueller, J.; Brown, C.; Bigelow, C.; Schwartz, L.M. The novel lupus antigen related protein acheron enhances the development of human breast cancer. Int. J. Cancer 2012, 130, 544–554. [Google Scholar] [CrossRef]
- Santiago-Sanchez, G.S.; Pita-Grisanti, V.; Quinones-Diaz, B.; Gumpper, K.; Cruz-Monserrate, Z.; Vivas-Mejia, P.E. Biological Functions and Therapeutic Potential of Lipocalin 2 in Cancer. Int. J. Mol. Sci. 2020, 21, 4365. [Google Scholar] [CrossRef] [PubMed]
- Deis, J.; Lin, T.Y.; Bushman, T.; Chen, X. Lipocalin 2 Deficiency Alters Prostaglandin Biosynthesis and mTOR Signaling Regulation of Thermogenesis and Lipid Metabolism in Adipocytes. Cells 2022, 11, 1535. [Google Scholar] [CrossRef] [PubMed]
- Uhrbom, L.; Hesselager, G.; Nister, M.; Westermark, B. Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res. 1998, 58, 5275–5279. [Google Scholar]
- Zhu, H.; Acquaviva, J.; Ramachandran, P.; Boskovitz, A.; Woolfenden, S.; Pfannl, R.; Bronson, R.T.; Chen, J.W.; Weissleder, R.; Housman, D.E.; et al. Oncogenic EGFR signaling cooperates with loss of tumor suppressor gene functions in gliomagenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 2712–2716. [Google Scholar] [CrossRef]
- Kwon, C.H.; Zhao, D.; Chen, J.; Alcantara, S.; Li, Y.; Burns, D.K.; Mason, R.P.; Lee, E.Y.; Wu, H.; Parada, L.F. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Res. 2008, 68, 3286–3294. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Amankulor, N.M.; Helmy, K.Y.; Becher, O.J.; Holland, E.C. Modeling Adult Gliomas Using RCAS/t-va Technology. Transl. Oncol. 2009, 2, 89–95. [Google Scholar] [CrossRef]
- Uhrbom, L.; Dai, C.; Celestino, J.C.; Rosenblum, M.K.; Fuller, G.N.; Holland, E.C. Ink4a-Arf loss cooperates with KRas activation in astrocytes and neural progenitors to generate glioblastomas of various morphologies depending on activated Akt. Cancer Res. 2002, 62, 5551–5558. [Google Scholar]
- Munoz, D.M.; Tung, T.; Agnihotri, S.; Singh, S.; Guha, A.; Zadeh, G.; Hawkins, C. Loss of p53 cooperates with K-ras activation to induce glioma formation in a region-independent manner. Glia 2013, 61, 1862–1872. [Google Scholar] [CrossRef]
- Uhrbom, L.; Kastemar, M.; Johansson, F.K.; Westermark, B.; Holland, E.C. Cell type-specific tumor suppression by Ink4a and Arf in Kras-induced mouse gliomagenesis. Cancer Res. 2005, 65, 2065–2069. [Google Scholar] [CrossRef]
- Hede, S.M.; Hansson, I.; Afink, G.B.; Eriksson, A.; Nazarenko, I.; Andrae, J.; Genove, G.; Westermark, B.; Nister, M. GFAP promoter driven transgenic expression of PDGFB in the mouse brain leads to glioblastoma in a Trp53 null background. Glia 2009, 57, 1143–1153. [Google Scholar] [CrossRef]
- Hicks, W.H.; Bird, C.E.; Traylor, J.I.; Shi, D.D.; El Ahmadieh, T.Y.; Richardson, T.E.; McBrayer, S.K.; Abdullah, K.G. Contemporary Mouse Models in Glioma Research. Cells 2021, 10, 712. [Google Scholar] [CrossRef]
- Candolfi, M.; Curtin, J.F.; Nichols, W.S.; Muhammad, A.G.; King, G.D.; Pluhar, G.E.; McNiel, E.A.; Ohlfest, J.R.; Freese, A.B.; Moore, P.F.; et al. Intracranial glioblastoma models in preclinical neuro-oncology: Neuropathological characterization and tumor progression. J. Neuro-Oncol. 2007, 85, 133–148. [Google Scholar] [CrossRef]
- Szatmari, T.; Lumniczky, K.; Desaknai, S.; Trajcevski, S.; Hidvegi, E.J.; Hamada, H.; Safrany, G. Detailed characterization of the mouse glioma 261 tumor model for experimental glioblastoma therapy. Cancer Sci. 2006, 97, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Ausman, J.I.; Shapiro, W.R.; Rall, D.P. Studies on the chemotherapy of experimental brain tumors: Development of an experimental model. Cancer Res. 1970, 30, 2394–2400. [Google Scholar] [PubMed]
- Martinez-Murillo, R.; Martinez, A. Standardization of an orthotopic mouse brain tumor model following transplantation of CT-2A astrocytoma cells. Histol. Histopathol. 2007, 22, 1309–1326. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; el-Abbadi, M.; Roy, M.L. Ganglioside distribution in murine neural tumors. Mol. Chem. Neuropathol. 1992, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Miyai, M.; Tomita, H.; Soeda, A.; Yano, H.; Iwama, T.; Hara, A. Current trends in mouse models of glioblastoma. J. Neuro-Oncol. 2017, 135, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Akter, F.; Simon, B.; de Boer, N.L.; Redjal, N.; Wakimoto, H.; Shah, K. Pre-clinical tumor models of primary brain tumors: Challenges and opportunities. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188458. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Singh, R.K.; Fidler, I.J.; Raz, A. Murine models to evaluate novel and conventional therapeutic strategies for cancer. Am. J. Pathol. 2007, 170, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Nakashima, H.; Alayo, Q.A.; Penaloza-MacMaster, P.; Freeman, G.J.; Kuchroo, V.K.; Reardon, D.A.; Fernandez, S.; Caligiuri, M.; Chiocca, E.A. Modeling tumor immunity of mouse glioblastoma by exhausted CD8(+) T cells. Sci. Rep. 2018, 8, 208. [Google Scholar] [CrossRef]
- Filley, A.C.; Henriquez, M.; Dey, M. Recurrent glioma clinical trial, CheckMate-143: The game is Not over yet. Oncotarget 2017, 8, 91779–91794. [Google Scholar] [CrossRef]
- Wainwright, D.A.; Chang, A.L.; Dey, M.; Balyasnikova, I.V.; Kim, C.K.; Tobias, A.; Cheng, Y.; Kim, J.W.; Qiao, J.; Zhang, L.; et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 5290–5301. [Google Scholar] [CrossRef]
- Genoud, V.; Marinari, E.; Nikolaev, S.I.; Castle, J.C.; Bukur, V.; Dietrich, P.Y.; Okada, H.; Walker, P.R. Responsiveness to anti-PD-1 and anti-CTLA-4 immune checkpoint blockade in SB28 and GL261 mouse glioma models. Oncoimmunology 2018, 7, e1501137. [Google Scholar] [CrossRef]
- Reynolds, B.A.; Weiss, S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 1992, 255, 1707–1710. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Pellegatta, S.; Poliani, P.L.; Corno, D.; Menghi, F.; Ghielmetti, F.; Suarez-Merino, B.; Caldera, V.; Nava, S.; Ravanini, M.; Facchetti, F.; et al. Neurospheres enriched in cancer stem-like cells are highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas. Cancer Res. 2006, 66, 10247–10252. [Google Scholar] [CrossRef]
- Telentschak, S.; Soliwoda, M.; Nohroudi, K.; Addicks, K.; Klinz, F.J. Cytokinesis failure and successful multipolar mitoses drive aneuploidy in glioblastoma cells. Oncol. Rep. 2015, 33, 2001–2008. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakraborty, A.; Yang, C.; Kresak, J.L.; Silver, A.J.; Feier, D.; Tian, G.; Andrews, M.; Sobanjo, O.O.; Hodge, E.D.; Engelbart, M.K.; et al. KR158 Spheres Harboring Slow-Cycling Cells Recapitulate High-Grade Glioma Features in an Immunocompetent System. Cells 2024, 13, 938. https://doi.org/10.3390/cells13110938
Chakraborty A, Yang C, Kresak JL, Silver AJ, Feier D, Tian G, Andrews M, Sobanjo OO, Hodge ED, Engelbart MK, et al. KR158 Spheres Harboring Slow-Cycling Cells Recapitulate High-Grade Glioma Features in an Immunocompetent System. Cells. 2024; 13(11):938. https://doi.org/10.3390/cells13110938
Chicago/Turabian StyleChakraborty, Avirup, Changlin Yang, Jesse L. Kresak, Aryeh J. Silver, Diana Feier, Guimei Tian, Michael Andrews, Olusegun O. Sobanjo, Ethan D. Hodge, Mia K. Engelbart, and et al. 2024. "KR158 Spheres Harboring Slow-Cycling Cells Recapitulate High-Grade Glioma Features in an Immunocompetent System" Cells 13, no. 11: 938. https://doi.org/10.3390/cells13110938
APA StyleChakraborty, A., Yang, C., Kresak, J. L., Silver, A. J., Feier, D., Tian, G., Andrews, M., Sobanjo, O. O., Hodge, E. D., Engelbart, M. K., Huang, J., Harrison, J. K., Sarkisian, M. R., Mitchell, D. A., & Deleyrolle, L. P. (2024). KR158 Spheres Harboring Slow-Cycling Cells Recapitulate High-Grade Glioma Features in an Immunocompetent System. Cells, 13(11), 938. https://doi.org/10.3390/cells13110938