Abstract
Transient receptor potential (TRP) channels are broadly implicated in the developmental programs of most tissues. Amongst these tissues, skeletal muscle and adipose are noteworthy for being essential in establishing systemic metabolic balance. TRP channels respond to environmental stimuli by supplying intracellular calcium that instigates enzymatic cascades of developmental consequence and often impinge on mitochondrial function and biogenesis. Critically, aminoglycoside antibiotics (AGAs) have been shown to block the capacity of TRP channels to conduct calcium entry into the cell in response to a wide range of developmental stimuli of a biophysical nature, including mechanical, electromagnetic, thermal, and chemical. Paradoxically, in vitro paradigms commonly used to understand organismal muscle and adipose development may have been led astray by the conventional use of streptomycin, an AGA, to help prevent bacterial contamination. Accordingly, streptomycin has been shown to disrupt both in vitro and in vivo myogenesis, as well as the phenotypic switch of white adipose into beige thermogenic status. In vivo, streptomycin has been shown to disrupt TRP-mediated calcium-dependent exercise adaptations of importance to systemic metabolism. Alternatively, streptomycin has also been used to curb detrimental levels of calcium leakage into dystrophic skeletal muscle through aberrantly gated TRPC1 channels that have been shown to be involved in the etiology of X-linked muscular dystrophies. TRP channels susceptible to AGA antagonism are critically involved in modulating the development of muscle and adipose tissues that, if administered to behaving animals, may translate to systemwide metabolic disruption. Regenerative medicine and clinical communities need to be made aware of this caveat of AGA usage and seek viable alternatives, to prevent contamination or infection in in vitro and in vivo paradigms, respectively.
1. Background
Muscle and adipose are the body’s two most critical tissues for establishing systemic metabolic balance. These two tissues synchronize each other’s metabolic status via their mutual exchange of paracrine factors—myokines and adipokines, respectively—that have both trophic and metabolism-modulating activities [1,2,3]. Ultimately, this system of paracrine crosstalk expands beyond the realm of muscle and fat to encompass the other tissues of the body [4], including the liver [5,6], heart [6,7], bone [8,9], and various inflammation-regulating systems [10,11].
Skeletal muscle is the largest tissue mass in healthy individuals and, by design as a weight-supporting tissue, is constantly mechanically loaded by the unyielding force of gravity. Weight-bearing provides a basal level of muscle mitochondrial activation (to sustain posture) that sets the enzymatic backdrop for the muscle secretome response [12]. Consequently, the experiences of prolonged periods of exposure to microgravity and bed rest share many of the same physiological disruptions [13]. Unlike skeletal muscle, adipose tissue is not organized within the body to resist the force of gravity, whereas skeletal muscle is organized for the maintenance of a functional posture for the execution of daily activities. Skeletal muscle is hence the initial and principal point for modulation by gravitational/mechanical loading. Consequently, in non-obese individuals, the influence of skeletal muscle presides in establishing the balance between myokine–adipokine exchange.
If muscular interaction with gravity is further augmented by the addition of movement, otherwise known as exercise [4,14], the muscle secretome response may become sufficiently augmented to attenuate the inflammatory status of white adipose. A reduction in white adipose inflammation, in turn, would then be reflected as a reduction in the inflammatory status of the adipose secretome [15]. Specifically, white adipose phenotypically “beiges” when subjected to anti-inflammatory myokines (exerkines), characterized by adipose mitochondrial uncoupled respiration and non-shivering thermogenesis [15,16]. Adequately beiged adipose tissue, in turn, reciprocates with the release of brown adipose-associated adipokines, or batokines, that reinforce the anti-inflammatory status of muscle [12,17,18,19]. Activating muscle via “exercise” hence improves the systemic metabolism and inflammatory status by first targeting adipose tissue in a way that later transcends to the entire system [10,11].
On the other hand, excessive caloric intake in combination with a sedentary lifestyle creates a physiological scenario via which the inflammatory status of adipose tissue is left relatively unchecked under the reduced influence of the muscle secretome. This results in an accumulation of inflamed white adipose tissue in the thoracic subcutaneous and visceral deposits, as well as ectopic adipose deposition in the liver, heart, and skeletal muscle, that in turn, assumes detrimental metabolic dominance by augmenting the release of pro-inflammatory adipokines into the systemic circulation [15], instilling a state of systemic inflammation [20]. Obesity-associated systemic inflammation, in turn, compromises muscle development [21] as well as attenuating muscle’s ability to metabolically adapt to exercise [22] that, in turn, undermines muscle’s secretome modulation of adipose [2,3,4,10,15]. Key in tipping this delicate tissue balance in favor of a healthful metabolic status is the attenuation of white adipose inflammation via the activation of muscle with exercise.
Fortunately, key aspects of the exercise muscular metabolic cascade remain intact during systemic inflammation. Despite the existence of systemic insulin resistance, acute exercise must still stimulate mitochondrial respiration, increasing the demand for oxygen and metabolic substrates for energy production [23]. The exhaustion of molecular oxygen by respiring mitochondria during exercise is a key factor in this innate muscular tolerance to systemic inflammation that has been shown to promote cell survival [24]. If the execution of exercise results in an insufficiency of oxygen to respiring mitochondria [25], then mitochondriogenesis and mitohormetic adaptations may be stimulated [26,27]. Inherent to this mitohormetic response is an oxidative shift in skeletal muscle mitochondrial substrate utilization towards more energy-enriched fatty acids [28,29] that ultimately assists in reducing adipose reserves. Hence, the vicious cycle set in motion by white adipose inflammation can be interrupted, or potentially reversed, by exercise. Exercise re-establishes an anti-inflammatory muscle secretome response that reinstates systemic metabolic balance [4,14].
Critically, aminoglycoside antibiotics (AGAs) have been shown to block the ability of muscles to adapt to exercise [30,31,32], particularly towards an oxidative phenotype [33,34] (also see Table 1). This detrimental aspect of AGAs can have critical implications for human health status. Oxidative muscle is associated with the greatest metabolic benefits [32,33,35,36], as well as possessing stronger metabolism-stabilizing secretomes [37,38]. Evidence also exists that the AGAs interfere with the transition of white adipose tissue into the beiged state [39] or preferentially stabilize the white adipose phenotype [40]. These combined antagonisms of the AGAs over both muscle and adipose determination could severely disrupt human metabolic adaptations to exercise, as depicted in Figure 1.
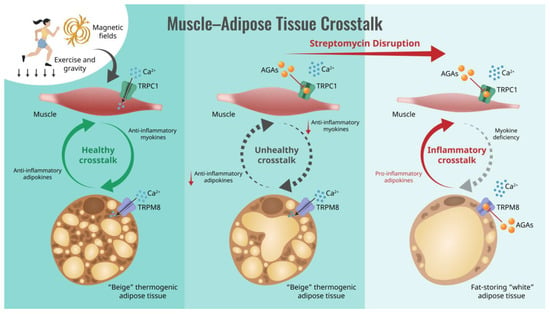
Figure 1.
Schematic of how the aminoglycoside antibiotics (AGAs) interrupt muscle–adipose crosstalk by inhibiting Ca2+ entry via transient receptor potential (TRP) cation channel classes, causing systemic metabolic disruption. Semicircular arrows indicate the direction of muscle-adipose paracrine crosstalk ranging between anti-inflammatory (green; left) to pro-inflammatory (red; right) character.
Streptomycin (100 μg/mL), but not penicillin (150 U/mL) or amphotericin B (5 μg/mL), was shown to preferentially block protein accretion into primary myogenic cultures under identical conditions [41], indicating that the aminoglycosides exert a distinct form of developmental disruption. Indeed, subsequent studies revealed that the AGAs exhibit a form of developmental antagonism that is unique amongst the other antibiotic classes, since it entails the immediate interference of ion channel-mediated signal transduction arising from diverse biophysical stimuli, such as mechanical force, magnetism, light, or temperature, into developmental responses (see Section 3: TRP Channels Transduce Biophysical Stimuli into Mitohormetic Responses), rather than the more protracted inhibition of protein translation commonly attributed to a wide scope of antibiotics [42]. As a point of distinction, AGAs need only to be present during the presentation of the biophysical stimuli to effectively inhibit developmental signal transduction, and therefore, in this respect, their requisite time course of action can be shown to be remarkably brief [34]. Speed of action is particularly evident when the experiment is specifically designed with this intent in mind. The duration of exposure to the AGAs has varied at the discretion of the researcher and ranged from days [30], to 10 s of minutes [43], to instantaneous, as when examined cation channel activity is measured via the patch electrode [44]. For instance, Iversen et al. recently demonstrated that streptomycin applied during a brief exposure to light, magnetic fields, or combined light and magnetic fields precluded the induction of cell proliferation normally induced by analogous exposure, whereas streptomycin applied immediately after exposure did not [43] (Figure 2). As the duration of the electromagnetic exposure was only 15 min and cell analyses were conducted 24 h following exposure, the difference in time spent in streptomycin (30 min) was relatively negligible and unable to account for the antagonism observed when applied during exposure. Although commonly overlooked in clinical literature, this aspect of AGAs could have profound developmental consequences.
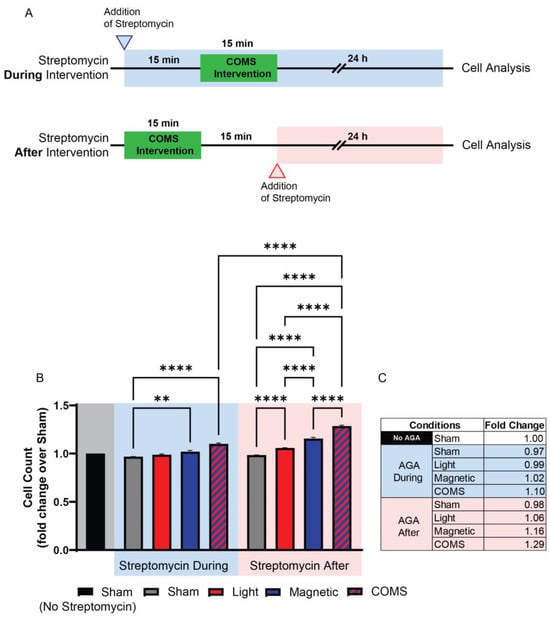
Figure 2.
Streptomycin present during exposure suffices to preclude developmental responses to biophysical stimuli. Streptomycin (100 μg/mL) added to the bathing media 15 min before, but not 15 min after, exposure to light (optical), magnetic fields, or concurrent optical and magnetic field stimulation (COMS), inhibited the typical stimulation of myoblast proliferation. (A) Experimental timeline depicting when streptomycin was administered in the “During” (top/blue) or “After” (below/red) scenarios with reference to electromagnetic exposure (green shaded region). (B) Murine myoblast proliferation as aforementioned, with streptomycin added to the bathing media (100 μg/mL) either 15 min before (blue-shaded region) or after (red-shaded region) exposure to the different exposure paradigms. The shaded areas indicate either the absence of streptomycin (gray) or its application before and during (blue) or after exposure (red) to the indicated conditions. (C) Tabulated fold changes of live cell number for the different exposure conditions relative to sham (no biophysical exposure, no streptomycin). In summary, a criterion for streptomycin antagonism of electromagnetic regenerative responses is that it be present at the time of exposure. AGA: aminoglycoside antibiotic. Data were analysed using one-way ANOVA followed by multiple comparison tests. Significance levels are indicated as follows: ** p < 0.01, and **** p < 0.0001. The error bars represent the standard error of the mean. Adapted from Iversen et al. 2024 [43].

Table 1.
AGA antagonism of exercise effects.
Table 1.
AGA antagonism of exercise effects.
| AGA Antagonism of Exercise Effects | ||||||
|---|---|---|---|---|---|---|
| Year | Tissue/Cell Type | Animal | Context | AGA (dose) | Relevant Findings | Ref. |
| 2003 | Muscle (Tibialis Anterior) | Rat (female Sprague Dawley rats, 3 months of age) | Electrical Eccentric Contractions (Stretch-induced depolarizations in Tibialis Anterior muscles) | Streptomycin (4 g/L: drink water) Streptomycin given in drinking water 6 days prior to Electrical Eccentric Exercise and terminated immediately before analysis. Other SAC Blockers: GdCl3 (10 μM) | Streptomycin blocked accommodation of eccentric contraction-induced depolarization and c-fos increase. Streptomycin blocked adaptive muscle hypertrophy after eccentric contraction muscle training. In vitro GdCl3 augmented streptomycin effects. Implicated SACs in response. | [30] |
| 2006 | Muscle (Tibialis Anterior) | Rat (female Sprague Dawley rats, 3 months of age) | Electric Eccentric Contractions (stretch-induced activation of Akt-mammalian target of rapamycin-p70 S6 kinase (p70S6K) signaling pathway) | Streptomycin (4 g/L; drinking water) Streptomycin given in drinking water 6 days prior to Electrical Eccentric Exercise and terminated immediately before analysis. Other SAC Blockers: GdCl3 (80 μmol/kg IV). | Streptomycin blocked rise in Akt and phosphorylation of p70S6K associated with eccentric contraction. Streptomycin also blocked the phosphorylation of GSK-3b and S6 in response to muscle contraction. Both streptomycin and GdCl3 blocked contraction-induced hyperpolarization of the muscle resting membrane potential without altering force production. Implicated SACs in response. | [35] |
| 2009 | Muscle (Dorsiflexors) | Rabbit (New Zealand White) | 12 bouts Electric Eccentric Exercise over four weeks | Streptomycin (800 mg/mL injected daily for 40 days intramuscularly at a dose of 300 mg/kg body weight). | Streptomycin eliminates functional adaptations of muscle after eccentric exercise. Implicated SACs in response. | [32] |
| 2015 | Muscle (cell line) | Mouse muscle cell line (C2C12) | Electrical chronic low-frequency stimulation of engineered muscle | Streptomycin (added to culture media at a concentration of 100 μg/mL). | Streptomycin inhibited fast-to-slow phenotype shift of C2C12-engineered muscle as indicated by depressions of contraction-induced fatigue resistance, GLUT4 levels, and the mitochondrial proteins succinate dehydrogenase and ATP synthase. Implicated SACs and voltage-gated Ca2+ and K+ channels in response. | [33] |
| 2015 | Bone (Tibialis Anterior) | Rat (F344; seven-week-old males) | Electrical muscle stimulation for 30 min/day for 6 days/week for one week. | Streptomycin (4 g/L in drinking water administered for 6 days prior to surgery). | Streptomycin did not induce bone loss, but attenuated protection offered by electrically stimulated muscle contraction against disuse-induced trabecular bone loss in muscle-denervated rats. | [36] |
| 2018 | Muscle (Tibialis Anterior) | Rat (Fischer 344; seven-week-old males) | Electric Eccentric Contractions | Streptomycin (4 g/L in drinking water administered for 6 days prior to Electrical Eccentric Exercise). | Streptomycin reduced eccentric contraction-induced Evans blue dye uptake by muscle fibers, but did not increase in cross-sectional area or decrease in roundness. | [45] |
Abbreviations: SAC: stretch-activated channel; aminoglycoside antibiotic (AGA).
2. Exercise Mitohormetic Adaptations Are Antagonized by AGAs
The increase in mitochondrial respiration that, by necessity, accompanies exercise serves as the basis for the metabolic adaptations that are initially instilled in skeletal muscle and later spread to the rest of the organism. The ensuing energy deficit and oxidative stress instigates a compensatory transcriptional response within muscle that is designed to improve metabolic efficiency, stimulates mitochondriogenesis, enhances the antioxidant defenses of mitochondria, and promotes cell survival in the face of oxidative stressors [27,38,46]. This adaptive process is known as mitohormesis [47,48]. Via mitohormetic principles, low levels of oxidative stress promote cell survival, whereas excessive oxidative stress results in cell demise. Secretome activation is one of the ensuing response limbs of the mitohormetic cascades invoked by exercise [47,49], which is then translated to the rest of the organism [14,50,51]. Because of the mitohormetic adaptations of muscle, exercised muscles increase their oxidative capacities and become better “secreters” in response to exercise as well as during moments of rest [52]. Muscle mitohormetic adaptations hence translate into improved basal systemic metabolism and overall health. Nonetheless, they are susceptible to environmental confounders.
AGAs may be one such class of mitohormetic disruptor. AGAs have been shown to block myogenic secretome release [53] as well as secretome action [52]. Importantly, any circumstance, or agent, that would interfere with muscle’s ability to respond to exercise would leave adipose tissue unchecked and facilitate systemic inflammation to preside. AGAs are hence valid candidates for culpable agents that may lead to metabolic disruption by interfering with the mitohormetic effects of exercise. Evidence exists that AGAs exert their antagonism of mitohormetic adaptations by interfering with biophysical signal transduction by transient receptor potential (TRP) channels [34].
3. TRP Channels Transduce Biophysical Stimuli into Mitohormetic Responses
Underappreciated is the fact that biophysical stimuli are fundamental developmental determinants [54,55]. At the cellular level, TRP channels are responsible for transducing biophysical stimuli into developmental responses [56]. Additionally, for a biophysical stimulus to be transduced into an enzymatic cascade by a TRPC channel complex (composed of tetramers of TRPC channel subunits), it need not occur in isolation but may be combined with other biophysical stimuli of distinct modalities simultaneously impinging upon the cell and transduced by other TRPC channel complexes or subunits. The manner in which TRPC channel complexes are capacitated for transducing diverse biophysical stimuli is via a process of heterologous multimerization (heterotetramers) of different “C” subunits possessing sensitivities to distinct forms of biophysical stimuli [57,58]. TRPC subunit heteromultimerization effectively unifies diverse gating sensitivities into a single-channel complex [59] to produce a unified cell response. In this manner, TRPC channel complexes subserve the role of integrators of diverse biophysical stimuli of developmental consequence. In summary, heteromultimerization is the modus operandi of TRP subunits, and cellular signal integration is the benefit it confers.
The TRPC1 subunit appears to be pivotal for the integration capacity of a TRP channel complex to be realized. The TRPC1 subunit regulates the activity of the other TRPC family members by forming heteromultimers with them [57,58]. The TRPC1 subunit also heteromultimerizes with the subunits of other TRP families to further extend the range of sensitivities of the channel complex to biophysical stimuli [60,61]. Accordingly, activation modes associated with the TRPC1 subunit include mechanotransduction [62,63,64,65], magnetotransduction [34,66,67,68], phototransduction [69,70,71], thermosensation [72], mitogen activation [57,58], phospholipid activation [62,73], calcium-sensing/SOCE (store-operated calcium entry) [74], redox sensing [75,76,77,78], and mitogen sensing [57,58]. TRP channel-mediated stimuli signal transduction must ultimately be transformed into mitohormetic adaptations.
Mitochondria provide the molecular energy (ATP) that underlies any cellular response to an impinging stimulus, noxious or beneficial. Mitochondria are thus fundamental to the adaptive mitohormetic adaptations that govern our health and lifespan [48,79]. Capacitating mitohormetic adaptations, and in accordance with their role as biophysical stimuli receptors, the activation of TRPC channels is relayed to the mitochondria. Mitochondrial responses are hence a common response limb of TRPC channel activation [34,80]. Mechanistically, the TRPC family members are regulated by, and responsible for, sustained calcium entry [81,82,83]. TRPC channel-mediated calcium entry also modulates mitochondrial function [84,85,86]. Conversely, mitochondrial-derived reactive oxygen species (ROS) regulate TRPC channel activity [75,76,77]. The communication between TRPC channels and the mitochondria is thus two-way [87] and serves to mutually consolidate cellular responses to developmental stimuli.
4. TRP-Mediated Mitohormetic Responses Are Antagonized by AGAs
TRP channels are widely distributed amongst the different tissues of the body [57,88] and have been implicated in diverse developmental programs [89,90]. Noteworthily, certain TRP channel families have been implicated in the mitohormetic adaptations of muscle and adipose tissues. Critically, AGAs possess the ability to block calcium entry via these TRP channels (Table 2) to preclude downstream mitochondrial response cascades in these tissues [34,39]. The antagonism of these TRP channels by the AGAs will thus limit the capacity of muscle and adipose tissues to adapt to exercise and thus positively modulate systemic metabolism. The mechanism of acute antagonism of these TRP channels by the AGAs goes beyond the more commonly ascribed inhibition of the protein synthetic cellular machinery shared by most antibiotics [42]. Specifically, the AGAs disrupt cellular signal transduction by physically impeding Ca2+ permeation through TRP channels. This is achieved by virtue of their high affinity to bind within, and thereby sterically obstruct, the pore region of TRP channels [44,61,91,92,93]. Given the structural similarity of the pore regions between certain TRP channel classes [60,94], AGA antagonism may represent a general feature of multiple TRP channel classes (also see Section 10: AGA antagonism of other TRP channels). To be capable of responding to biophysical stimuli, TRPC channels are likely required to interact with other subcellular components, rather than to directly perceive and respond to biophysical stimuli per se [95]. The integrative nature of TRPC1 hence holds broad developmental importance that can be undermined by AGAs. The AGAs would hence interfere with the establishment of muscle and fat TRP channel-mediated metabolic adaptations arising from exercise or other forms of biophysical stimulation.
5. Gravity and the Geomagnetic Field Are Unyielding Biophysical Developmental Forces
The Earth’s gravitational and geomagnetic fields are the two biophysical forces to which we are always exposed on Earth. Ample evidence exists demonstrating that both gravity and the geomagnetic field are authentic and fundamental developmental forces [96]. Most importantly, gravitational and geomagnetic fields modulate oxidative muscle development, also known as “antigravity” muscle [38,96,97,98]. By contrast, other forms of biophysical stimuli such as temperature and light change diurnally in the natural environment, as well as being intervened with at the hands of man. Notably, gravitational force, manifested as weight bearing [60,62,65], and magnetic fields [38,96,99] are amongst the biophysical forces transduced by TRPC1-containing channels. TRPC1 is ubiquitously expressed [39,57,59,88] and, alone or in combination with other TRP channel subunits, is involved in the transduction of both mechanical forces [63,64,92,100,101] and magnetic fields [34,53,66,67,68,102]. The mechanistic interplay between the mechano- and magneto-sensitive modes of TRPC1 has been previously postulated and the implications discussed [96,103]. Most critically for the context of the current discussion, TRPC1 function is essential for oxidative/antigravity muscle development (see Section 6: Myogenesis), and since oxidative muscle is constantly activated by the force of Earth’s gravity [97,98], their blockage by AGAs would have profound physiological ramifications.
The Earth’s standing gravitational field is a constant developmental imperative. Yet, due to its ever-present nature, its relevance to human health is commonly misunderstood by the layperson. In practice, however, interaction with the Earth’s gravitational field is under our own control, predominantly implicates muscle, and is modulated by physical movement that is otherwise referred to as exercise. Mechanical force is transmitted at the organismal level, from cells to tissues, by gravitational mechanical loading of the musculoskeletal frame. Exercise can hence be classified as movement under the force of gravity and/or muscle contraction against the inertia of objects weighted by gravity. In hominoids, the assumption of an upright posture fostered the development of the antigravity, or oxidative, musculature [97]. Conversely, oxidative muscle is lost under conditions of reduced gravitational force, such as during manned space flight [104,105]. Oxidative muscle exhibits the greatest resistance to fatiguing and is associated with systemic metabolic balance and longevity [97,106]. Oxidative muscle is exemplified by the soleus muscle that runs underneath and along either side of the calve musculature of the lower leg. The soleus allows us to maintain a standing posture under the constant force of gravity and therefore, must be capable of sustaining contractions for long periods of time without fatiguing. In particular, the activation of the soleus muscle has been shown to be intimately associated with systemwide metabolic health [107]. Therefore, the basic act of maintaining a standing posture, or walking, under the constant force of gravity is important in establishing basal metabolism. Therefore, exercise, in its most fundamental form, is a state of gravitational loading of metabolic consequence.
On the other hand, our physiological interaction with magnetism is a bit more enigmatic. Outside of the Earth’s standing geomagnetic field, which varies in inclination and amplitude at different regions of the planet, and intentional magnetic therapies, exposure to exogenous magnetism, both man-made (power lines, etc.) and natural (lodestones made of magnetite and geomagnetic storms, etc.) forms, is largely haphazard [108,109]. Nonetheless, evidence is accumulating indicating that appropriately conceived magnetic therapies can have beneficial consequences with regards to human health [38,110,111]. Indeed, unequivocal evidence exists that electromagnetic fields are an authentic developmental force [96,112], as shielding cells from all magnetic fields halts their development [34]. Notably, the development of the soleus muscle is preferentially promoted by sustained mechanical loading [113] as well as certain forms of magnetic field exposure [114]. In terms of stimulus transduction, TRPC1-mediated calcium entry is stimulated by mechanical loading [115,116] as well as magnetic field exposure [34,114] as a prerequisite for soleus development. Yet, neither magnetism nor mechanical forces vicariously act in the form of the other. That is, the magnetic exposure paradigms that have been implicated in TRPC1 activation do not inadvertently produce mechanical stress [34], and likewise, the mechanical stimuli commonly used in in vitro studies should not induce ionic fluxes of a sufficient magnitude to produce appreciable magnetic fields in the micro-milliTesla range [109,117,118,119]. Notably, AGAs have been shown to antagonize both the mechanical and electromagnetic cellular responses commonly attributed to TRPC1 and, when explicitly tested, can be emulated by established blockers of mechanosensitive or TRPC1 channels (Table 2). These responses can be further extrapolated to the AGA-mediated inhibition of exercise-induced adaptations (Table 1). For historical reasons, streptomycin is the most thoroughly tested of the AGAs, although neomycin may demonstrate a higher efficacy of blocking for both the voltage-gated [120] and stretch-activated/TRP (Table 2) Ca2+-permeable channels. The inhibition of TRPC1-mediated Ca2+ entry by AGAs will hence simultaneously counteract the actions of our two most fundamental developmental forces, gravitational mechanical loading and magnetism [54,121,122,123], for oxidative skeletal muscle that is intimately linked to systemic metabolic balance.
Table 2.
AGA antagonism of stretch-activated/mechanosensitive channel.
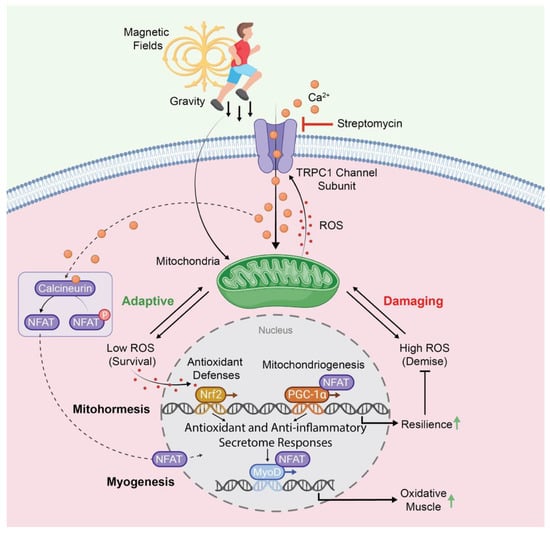
Figure 3.
TRPC1-dependent transcriptional and enzymatic pathways co-activated by gravity-mediated (mechanical loading) or electromagnetic stimuli impinging on muscle. The orange circles represent Ca2+ and the red dots represent ROS. TRPC1 activation enhances mitochondrial biogenesis and myogenesis towards the oxidative (slow) phenotype by contributing to the mitohormetic adaptations associated with resistance to oxidative stress. The TRPC1-mediated activation of these same pathways should forestall the development of sarcopenia and be reversed by aminoglycoside antibiotics, such as streptomycin. Abbreviations: ROS: reactive oxygen species; NFAT: Nuclear Factor of Activated T-Cells; PGC-1α: Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-alpha; Nrf2: Nuclear Factor Erythroid 2-Related Factor 2; MyoD: Myoblast Determination Protein 1. For further details, kindly refer to references [34,96].
Table 2.
AGA antagonism of stretch-activated/mechanosensitive channel.
| AGA Antagonism of Stretch-Activated/Mechanosensitive Cation Channels | |||||||
|---|---|---|---|---|---|---|---|
| Year | Tissue | Animal | Context | Modulator | Channel | Findings | Ref. |
| 1982 | Sacculus Hair Cells | Bullfrog (Rana catesbeiana) | Hair Bundle Deflection | Dihydrostreptomycin (~10 μM near bundle calculated from 400 mM within iontophoretic electrode) | SACs | Dihydrostreptomycin blocked sinusoidal deflection-activated currents within the hair bundle/stereocilia. | [124] |
| 1985 | Vestibular Hair Cells | Chicken | Hair Bundle Deflection | Streptomycin (500 μM) Neomycin (750 μM) | SACs | Streptomycin and meomycin blocked mechanosensitive/deflection-activated Ca2+ currents. Streptomycin was a more potent blocker than neomycin. | [125] |
| 1989 | Sacculus Hair Cells | Bullfrog (Rana catesbeiana) | Otolithic Membrane Displacement | Dihydrostreptomycin (500 μM) Gentamicin (500 μM) | SACs | AGAs reversibly blocked displacement-activated transduction channels in the otolithic membrane. | [126] |
| 1993 | Muscle | Chicken | General Review of SAC Modulators (Patch Clamp Recordings) | 0 Vm KDs: Neomycin (2 pM) Streptomycin (21 pM) Ribostamycin (32 pM) Dibekacin (48 pM) Kanamycin (55 pM) | SACs | Aminoglycoside antibiotics block SAC single-channel currents in a dose- and voltage-dependent manner in the sequence neomycin > streptomycin > ribostamycin > dibekacin > kanamycin. GdCl3 is also identified as an SAC blocker. The dissociation constants of the aminoglycoside antibiotics for SACs are lower than for other Ca2+ or K+ channel classes. | [44] |
| 1994 | Heart Ventricular Myocytes | Guinea pig (Cavia porcellus) | Stretch Ca2+ Transients | Streptomycin (40 μM) Penicillin (40 μM) | SACs | Streptomycin blocked stretch-induced Ca2+ transients but had no effect on L-type voltage-gated Ca2+ currents. Streptomycin exhibited faster blocking action than penicillin and was inferred from published literature to be more specific for SACs than GdCl3. | [127] |
| 1996 | General Review | Vertebrates and C. elegans | General Review of SAC Pharmacology | AGAs (2–60 μM) Table 2: IC50 values of different AGAs on different channel classes | SACs | General review of SAC modulators including AGAs and GdCl3. IC50 values are generally lowest for SACs compared to other cation channel classes. | [128] |
| 1996 | Muscle (Flexor Digitorum Brevis) | Mouse (C57BL/6) | Suction via Patch Electrode During Single-Channel Recordings | Dihydrostreptomycin (50–100 μM) Neomycin (50–100 μM) Gentamicin (1000–2000 μM) Amikacin (100–400 μM) Streptomycin (500–800 μM) Kanamycin (800–100 μM) | SACs | AGAs reversibly blocked stretch-activated channel currents at discrete subconductance levels. Aminoglycoside antibiotic blocking potency: dihydrostreptomycin, neomycin > gentamicin > amikacin, streptomycin > kanamycin. Table 1: KDs of AGAs. | [91] |
| 2000 | Muscle (Tibialis Anterior) | Rat (Sprague Dawley) | Electrical Eccentric Contraction | Streptomycin (4 g/L in drinking water provided six days prior to contractures) Streptomycin (1 mM in vitro) GdCl3 (10 μM) | SACs | Streptomycin blocked stretch-induced Na+ depolarizations. The effects of GdCl3 and streptomycin were qualitatively similar. | [129] |
| 2000 | Muscle (Tibialis Anterior) | Rat (Sprague Dawley; 3 months old) (Fisher 344:BN F1 hybrids; 32 months old) | Electrical Eccentric Contraction | Streptomycin (4 g/L in drinking water provided six days prior to contractures) GdCl3 (10 μM) | SACs | Stretch-induced NaCl depolarizations in aged skeletal muscle are less sensitive to streptomycin due to the presence of non-specific leakage. The effects of GdCl3 and streptomycin were qualitatively similar. | [130] |
| 2003 | Muscle (Tibialis Anterior) | Rat (Sprague-Dawley) | Electrical Eccentric Contraction | Streptomycin (4 g/L in drinking water provided six days prior to contractures) GdCl3 (10 μM) | SACs | Streptomycin blocked contraction-induced hypertrophy but not increases in contraction force generation. The effects of GdCl3 and streptomycin were qualitatively similar. | [30] |
| 2003 | Cancer SiHa Cervical Cancer Cells (Epithelial) | Human (Homo sapiens) | Hypotonic Swelling | Streptomycin (10–1000 μM) Gentamicin (10–1000 μM) Netilmicin (10–1000 μM) | SACs | Streptomycin disrupted hypotonicity-induced Ca2+ influx but did not influence basal Ca2+ level. Hypotonic cell swelling-induced Ca2+ influx, in turn, activated a compensatory osmoregulatory Cl- conductance. IC50 inhibit initial peak [Ca2+]i (μM): Streptomycin (25) Gentamicin (90) Netilmicin (250) | [131] |
| 2003 | Muscle (Flexor Digitorum Brevis) | Mouse (mdx and C57 BL/10 ScSn (wt)) | Electrical Eccentric Contraction | Streptomycin (100 μM) GdCl3 (20 μM) | SACs TRPCs | Streptomycin abolished Na+ depolarization, reduced force and muscle damage after electrical contracture. Smaller effect size by streptomycin observed in wild-type muscle. TRPCs were cited to represent the aberrantly regulated SACs in dystrophic muscle leading to elevated resting Ca2+ entry. The effects of GdCl3 and streptomycin were qualitatively similar, suggesting that the spontaneous SAC activity apparent in mdx muscle is silent in wild-type muscle. | [132] |
| 2003 | Muscle (Flexor Digitorum Brevis (Mouse), Extensor Digitorum Longus (Rat)) | Rat and Mouse (Sprague Dawley) | Electrical Eccentric Contraction | Streptomycin (100 μM) GdCl3 (20 μM) | SACs | Streptomycin abolished Na+1 depolarization following electrical contracture. Similar to streptomycin, GdCl3 did not change resting [Na+]i but prevented its rise in response to eccentric contractions. | [133] |
| 2005 | Muscle (Tibialis Anterior) | Rat (Sprague Dawley) | Mechanical Stretch (40 stretches by ankle rotation) | Streptomycin (300 mg/kg/day; IP 3X/day) EDTA (150 mg/kg; IP 20 min before and 24 h after injury protocol) | SACs | Streptomycin significantly reduced stretch-induced muscle damage. Streptomycin produced greater protection than EDTA administration. | [134] |
| 2005 | Heart (Ventricular Myocytes) | Swine (Sus Scrofa Domesticus) | Baseball Impact (40 mph) (Commotio cordis) | Streptomycin (2 g intramuscularly; ~115 μM serum levels) | SACs | Streptomycin did not alter the frequency of ventricular fibrillations, but reduced the amplitude of the ventricular ST segment upon impact. | [135] |
| 2005 | Muscle (Flexor Digitorum Brevis, Extensor Digitorum Longus) | Mouse (mdx and C57 BL/10 ScSn (wt)) | Electrical Eccentric Contraction | Streptomycin Patch electrode: 100–200 μM Drinking water: 4 g/L water (3 mM) GsMTx4 (10 μM) GdCl3 (20 μM) | SACs | Streptomycin blocked stretch-induced Ca2+ increases measured with Patch Clamp recordings and reduced decline in tetanic force following stretch. Patch Clamp recordings revealed SAC blocked by GsMTx4 in both wild-type and mdx myotubes. Streptomycin added to drinking water reduced muscle pathology and incidence of central nuclei, in mdx muscle. GsMTx4, GdCl3, and extracellular Ca2+ removal recapitulated the streptomycin-associated reductions in stretch-induced Ca2+ increase and muscle force production following tetanic stretch. | [136] |
| 2005 | Muscle | Mouse cell line (C2C12) | Myogenic Differentiation | Streptomycin (500–5000 μM) GsMTx4 (0.5 μM) GdCl3 (10–100 μM) | SACs | Streptomycin blocks myogenic differentiation. The effect of streptomycin was recapitulated by GsMTx4 and GdCl3. GsMTx4 was the least effective and GdCl3 was the most effective. | [31] |
| 2009 | Fibroblasts (Embryonic Lung) | Human (Homo sapiens) | Cell Migration | Streptomycin (200 μM) EGTA (5 mM) MgCl2 (10 mM) Growth Factor: PDGF | TRPM7 SACs | Streptomycin reduced lamella Ca2+ flickers and the turning of cells. Ca2+ flickering is greatest on leading lamella of migrating cells. EGTA and Mg2+ also abolished flickers, implicating channel-mediated Ca2+ entry. Lamellar turning requires TRP channel-mediated Ca2+ entry and can be activated by platelet-derived growth factor (PDGF). TRPM7 knock-out abolished flickers. | [137] |
| 2005 | Muscle | Mouse (mdx and C57 BL/10 ScSn (wt)) Human (Homo sapiens) | General Review with Emphasis on Muscular Dystrophy | Streptomycin (100–200 μM) GdCl3 GsMTx4 | TRPC1 | Stretch-induced damage and Ca2+ overload are more common in X-linked muscular dystrophies. Streptomycin inhibits stretch-induced Na+/Ca2+ rise, dye uptake, and muscle damage. The other most common stretch-activated channel blockers (GdCl3 and GsMTx4) were highlighted to produce a similar protection as streptomycin. | [138] |
| 2006 | Neurons (Spinal Cord Neurons) | Amphibian (Xenopus laevis) | Neurite Outgrowth | Streptomycin, Gentamicin (100–200 μM) GsMTx4 (5 μM) GdCl3 (100 μM) SKF-96365 (3 μM) 2-APB (100 μM) Ruthenium Red (1 μM) Ca2+ Chelators: BAPTA (5 μm) EGTA (5–25 μM) | TRPCs TRPVs | Blocking stretch-activated channel activity accelerates neurite outgrowth. Gentamicin and SAC/TRP channel blockers (GdCl3, GsMTx4, Ruthenium Red, 2APB) slowed neurite outgrowth and reduced growth Ca2+ transients (2APB). Removing streptomycin and gentamicin from bathing increased spontaneous Ca2+ transient activity in growth cones. Specific Ca2+ chelation (BAPTA) slowed neurite outgrowth. A mixture of blockers for voltage-gated Ca2+ channels (w-conotoxin (1 μM), nifedipine (100 nM), NiCl (50 μM), w-agatoxin (60 nM), CdCl2 (50 μM), SKF-96365 (3 μM)) did not slow neurite outgrowth and did not inhibit growth cone Ca2+ transients in response to hypoosmotic swelling. | [139] |
| 2006 | Muscle (Flexor Digitorum Brevis) | Mouse (mdx and C57 BL/10 ScSn (wt)) | Exercise of Intact Mice Single-Channel Patch Clamp Recordings from Isolated Muscle Fibers | Streptomycin (200 μM) Ruthenium Red (50 μM) GdCl3 (50 μM) Growth Factor: IGF-1 | TRPV2 TRPC1 | Streptomycin inhibited single-channel currents on the surface of muscle fibers. Myofibers from dystrophic or exercised mice exhibited higher single-channel activity than unexercised wt fibers. Ruthenium Red and GdCl3 similarly inhibited single-channel currents. Insulin-like Growth Factor 1 (IGF-1) increased stretch-activated channel activity in wild-type, but not mdx muscles, reflecting channel deregulation. | [140] |
| 2006 | Muscle (Extensor Digitorum Longus) | Mouse (mdx and C57 BL/10 ScSn (wt)) | Downhill Treadmill Running of Intact Mice Eccentric Contractions of Isolated EDL Muscles | Streptomycin: Mice: IP injection (182 mg/kg; 0.5% volume to body mass; ~500 μM in blood) Isolated Muscles: 200 μM in bathing solution. | TRPC1 | Streptomycin reduced the increase in stretch-induced (run/electrode) membrane permeability (dye uptake) and preserved force generation in mdx fibers. These effects were less apparent and less sensitive to streptomycin in wt muscle fibers. The effects of streptomycin were effectively recapitulated by GsMTx4, a specific SAC blocker. Authors implicated SACs in stretch-induced membrane disruption in muscular dystrophy. | [141] |
| 2006 | Muscle | Mouse | General Review with Emphasis on Muscular Dystrophy | Streptomycin Gentamicin GdCl3 GsMTx4 | TRPC1 | Myoplasmic Ca2+ overload is strongly implicated in the pathology of X-linked muscular dystrophies. Streptomycin blocked stretch-induced Na+/Ca2+ rise, dye uptake, and muscle damage. Other stretch-activated channel blockers (GdCl3 and GsMTx4) were highlighted to produce similar protection as streptomycin. | [142] |
| 2007 | Neurons | Vertebrates | General Review with Emphasis on Neurite Outgrowth | Gentamicin (200 μM) | TRPCs | Neurite outgrowth is modulated by mechanical input. Blocking SACs accelerates neurite outgrowth. Gentamicin increases neurite outgrowth. | [143] |
| 2007 | Muscle | Mouse (mdx mouse) General Review | Stretch-Activated Single-Channel Recordings via Patch Electrode | General AGAs Neomycin (~200 μM) | TRPCs TRPVs | Neomycin blocked the high spontaneous activity of SACs in dystrophic muscle. The AGAs and lanthanide (trivalent cations such as GdCl3) block SACs by directly plugging the channel pore via a process known as open channel block, rather than interacting with the lipid bilayer or via enzymatic means, such as with the phorbol esters or 2-APB. Open channel block exhibits sensitivity to transmembrane voltage. | [93] |
| 2007 | Muscle | General Review | SAC Inhibitors | Streptomycin GsMTx-4 | TRPC1 TRPC6 TRPA1 | Streptomycin, GsMTx-4, and GdCl3 all block SAC activity. | [144] |
| 2008 | Muscle (Flexor Digitorum Brevis and Extensor Digitorum Longus Muscles) Muscle Cell Line (C2C12) | Mouse (mdx and C57 BL/10 ScSn (wt)) | Reactive Oxygen Species (ROS)-Induced Ca2+ Increments | Streptomycin (200 μM) | TRPC1 | Streptomycin blocked ROS-induced Ca2+ increase in mdx, but not in wild-type muscle fibers. ROS scavengers reduced stretch-induced Ca2+ increase. ROS increased Ca2+ influx only in muscle cells expressing TRPC1. TRPC1 expression is higher in mdx muscle. | [75] |
| 2011 | Muscle (leg musculature) | Mouse (mdx and C57 C57BL/10 (wt)) | Muscle Degeneration | Streptomycin (4 g/L in drinking water) In utero: upon vaginal plug observed up to 6 months post-birth. | TRPC1 | In utero treatment with streptomycin delayed the onset of dystrophic symptoms in the limbs of young mdx mice but not dystrophic muscular progression in later life. Long-term treatment improved muscle pathology and improved muscle regeneration in older mice. | [145] |
| 2011 | Muscle (Diaphragm, Sternomastoid, biceps brachii, Tibialis Anterior) | Mouse (mdx and C57BL/10ScCr) | Muscle Degeneration in Response to Exercise | Streptomycin (182 mg/kg bodyweight; IP injections for 18 days) | TRPC1 | Streptomycin reduced serum creatine kinase, exercise-induced Ca2+ entry & Evans blue dye uptake into the diaphragm and sternomastoid muscles of mdx mice. | [146] |
| 2012 | Muscle (Extensor Digitorum Longus) | Mouse (Balb/c C57BL/6J (wt) and C57BL/6J TRPC1 knock out) | Electrical Eccentric Contraction | Streptomycin (200 μM) Administered to muscle 30 min before tetanic stimulation. | TRPC1 | Streptomycin prevented loss of force after an electrical eccentric contraction in wild-type, but not TRPC1 knock-out muscle. Eccentric contraction-induced loss of desmin, titin, and dystrophin were prevented by streptomycin and TRPC1 knock down. | [147] |
| 2013 | Neurons (Spinal Cord Neurons) | Amphibian (Xenopus laevis) | Neurite Outgrowth | Gentamicin (100–200 μg/mL) GdCl3 (100 μM) GsMTx (10 μM) | TRPC1 | Blocking SACs accelerates neurite outgrowth. TRPC1 knock down precluded effect of gentamicin on axonal outgrowth and Ca2+ transients. Effects of gentamicin were observed within 10 s of seconds of addition to the bath. GdCl3 and GsMTx recapitulated some effects of gentamicin on axonal outgrowth. | [148] |
| 2019 | Muscle Muscle Cell Line (C2C12) | Mouse | Brief (10 min) Pulsing Magnetic Field Exposure (1 mT) (Myogenic Differentiation) | Streptomycin (100 μg/mL) Neomycin (50 μg/mL) Gentamycin (50 μg/mL) Penicillin/Streptomycin (100 U/mL & 100 μg/mL, respectively). SKF-96365 (50 μM) 2-APB (10 μM) Ruthenium Red (10 μM) | TRPC1 | Streptomycin, neomycin, gentamycin, and penicillin/streptomycin blocked magnetic induction of proliferation when added shortly before exposure, whereas removal of AGAs a few minutes before exposure did not. Penicillin/streptomycin blocked magnetic induction of Ca2+ entry, ROS production, and TRPC1 gene expression. Penicillin/streptomycin blocked magnetic induction of myogenic differentiation. The TRP channel blockers, SKF-96365, 2-APB, and Ruthenium Red and magnetic shielding emulated the effects of AGA treatment across several outcome measures. Magnetic sensitivity most closely followed TRPC1 expression and could be abolished with TRPC1 silencing, but not TRPM8 silencing. Authors deduced that AGAs interfere with mitohormetic adaptations within skeletal muscle cells (Figure 3). | [34] |
| 2021 | Stem Cells (Dental Pulp) | Human (Primary Dental Pulp Stem Cells) | Brief (10 min) Pulsing Magnetic Field Exposure (2 mT) Neurogenic Differentiation | Penicillin and Streptomycin (1%) | TRPC1 | Penicillin and streptomycin suppressed human dental pulp stem cell proliferation. Magnetic sensitivity followed TRPC1 expression. | [68] |
| 2022 | Muscle | Mouse Muscle Cell Line (C2C12) | Brief (10 min) Pulsing Magnetic Field Exposure (1.5 mT) | Streptomycin (100 μg/mL) | TRPC1 | Streptomycin blocked magnetic activation of the myogenic secretome response. Streptomycin inhibited TRPC1 protein expression. | [53] |
| 2024 | Muscle | Mouse Muscle Cell Line (C2C12) | Brief (10 min) Pulsing Magnetic Field Exposure (1.5 mT) | Gentamicin (50 μg/mL) | TRPC1 | Gentamicin prevented magnetically induced myogenic secretome from slowing breast cancer cell migration. | [52,53] |
| 2024 | Muscle | Mouse Muscle Cell Line (C2C12) | Brief (10 min) Pulsing Magnetic Field Exposure (1.5 mT) | Streptomycin (100 μg/mL) | TRPC1 | Streptomycin prevented combined magnetic field- and light-induced myogenic proliferation when applied during, but not after, exposure. Streptomycin was either applied 15 min prior to or after photo–electromagnetic exposure. | [43] |
Abbreviations: 2-Aminoethyl diphenylborinate (2-APB); aminoglycoside antibiotic (AGA); IP: intraperitoneal; SACs: stretch-activated channels; wt: wild type.
The same calcium-sensitive (calcineurin/NFAT), mitochondrial-regulating, transcriptional pathway (PGC-1α) that is activated by TRPC1 [34,114,149] (Figure 3) is downregulated in diabetes [150]. Accordingly, TRPC1/PGC-1 α-dependent oxidative muscle development [34,114,149] is depressed in diabetes [150]. Inhibiting TRPC1 activity hence holds the potential to disrupt metabolism by attenuating oxidative muscle development and activity. On the transcriptional level, the brief activation of TRPC1 by low-energy magnetic fields [34] was capable of provoking the nuclear translocation of the NFATC1 isoform responsible for oxidative muscle determination [151]. On the systemic level, muscular modulation of the whole-body metabolism is conveyed via its secretome. Of relevance is the fact that AGAs have been shown to inhibit TRPC1-mediated growth factor release [53], as well as blocking the actions of growth factors released by TRPC1 activation [52], which could have profound metabolic implications. AGAs may thus potentially disrupt muscle–adipose paracrine crosstalk and systemic metabolism.
6. Myogenesis
TRPC1 function is critical for both in vitro [34,101,152,153,154,155] and in vivo [115,116] myogeneses. Muscle phenotype determination is exquisitely responsive to the form of mechanical loading. Prolonged mechanical loading, such as that produced by supporting one’s weight under the force of gravity or during endurance exercise, favors the oxidative phenotype of skeletal muscle [113]. Notably, TRPC1 is essential for muscle’s adaptation to sustained mechanical loading and accordingly, for oxidative muscle development [116,156]. Oxidative muscle is distinguished from glycolytic muscle by its predilection for fatty acids as substrates for mitochondrial energy production, instead of carbohydrates [157,158,159,160,161]. By virtue of its elevated mitochondrial content and associated mitohormetic adaptations [29], the secretome of oxidative muscle is particularly anti-inflammatory [37,38,162]. Oxidative muscle is hence imperative for systemic metabolic balance and resilience to disease. It was previously shown that streptomycin selectively blocked oxidative muscle determination in vitro [33,34,163] and in vivo [146], in alignment with the requirement for TRPC1-mediated calcium entry for oxidative muscle development that reinforces TRPC1 expression [115,116,164]. Streptomycin was also shown to impede TRPC1-mediated muscle secretome release [53] and action [52] in vitro. This same AGA antagonism of skeletal muscle mitohormetic responses would likely be manifested in vivo, undermining the ability of exercise, or other relevant biophysical stimuli, to positively modulate systemic metabolism and health.
Sarcopenia describes the condition of age-related muscle loss that is linked to mitochondrial dysfunction downstream of accruing oxidative stress and chronic inflammation [165,166]. Sarcopenia is characterized by a slow-to-fast muscle fiber shift [167] and attenuated myokine response [168]. By contrast, TRPC1-mediated oxidative enhancement of skeletal muscle is characterized by a fast-to-slow muscle fiber shift (Figure 3) and an anti-inflammatory secretome response (Figure 2). As streptomycin was shown to inhibit oxidative muscle development [33,34], AGAs, in principle, may aggravate the progression of sarcopenia and frailty. Finally, progressive alterations in the cellular mechanical environment have also been proposed to underlie the slowing in muscle development with advancing age. Specifically, a connection between age-dependent changes in the extracellular matrix and mitochondrial dysfunction is being explored in association with sarcopenia [169]. Given that TRPC1 subserves some forms of mechanotransduction, the systemic presence of AGAs may aggravate the regenerative deficits associated with unfavorable alterations in the mechanical environment that may contribute to the development of sarcopenia.
7. Adipogenesis
The excessive storage of triglycerides by white adipose tissue is associated with systemic metabolic dysfunction [170] that can be corrected via adipose’s interaction with the muscle secretome [1,2,3]. Critically, AGAs hold the potential to interrupt this beneficial muscle–adipose crosstalk. Non-shivering thermogenesis is the cornerstone of white adipose adaptations (browning or beiging), contributing to systemic metabolic improvement. The induction of non-shivering thermogenesis in white adipocytes relies on the enhanced expression of the mitochondrial protein, uncoupled protein 1 (UCP-1) [171], which is upregulated upon exposure to the muscle secretome [172]. Accordingly, it was previously shown that muscle mitochondrial activation using brief magnetic field exposure conferred a thermogenic effect over white, but not brown, adipose tissue, in association with a significant upregulation of UCP-1 in white adipose [114].
Adipocytes also exhibit AGA-dependent metabolic disruption. However, the antagonism was attributed to a TRP family member other than TRPC1. A comprehensive expression profile analysis of all known 27 human TRP genes was undertaken during white or brown adipogenesis derived from human progenitor cells of either bone marrow or a stromal vascular fraction of subcutaneous adipose origins [39]. The induction of adipose browning (UCP1 expression) was most strongly correlated with two TRP channels, TRPM8 (cold temperature-responsive) and TRPP3 (pH-responsive), in adipose tissue differentiated from either progenitor cell class. Adenylate cyclase modulated the activity of either channel, linking their activity back to mitochondrial respiratory efficiency [39]. The induction of thermogenesis in these mesenchymal cell-derived adipocytes was found to be exquisitely sensitive to the presence of streptomycin. In this context, AGA antagonism of TRPM8 was shown to interrupt adipocyte thermogenesis. Menthol is an agent that chemically recreates the sensation of low temperatures and is a known activator of TRPM8 [173] but not of TRPC1. Streptomycin was capable of competitively inhibiting menthol-activated TRPM8-mediated calcium influx and interfering with the browning of white adipocytes. As TRPC1 expression was equally elevated throughout all stages of adipogenesis (progenitor cell, white and brown adipocytes), it was presumably not required for the induction of uncoupled respiration per se, and its antagonism by streptomycin was inconsequential to adipose browning. Other collateral consequences of AGA-TRPC1 antagonism were not pursued.
AGA antagonism of adipose browning could hence play out on two fronts, systemically (muscle) and locally (adipose). First, the AGAs would attenuate systemic muscle–adipose paracrine crosstalk via the antagonism of TRPC1 on muscle and second, prevent white adipose browning via the antagonism of TRPM8 on white adipocytes. The metabolic disruption associated with AGAs would hence be double-edged with reference to our most inflammatory tissue, adipose.
8. In Vivo Antagonism of Mechanotransduction by AGAs
Exercise on the organismal level is fundamentally mechanotransduction on the cellular level. Indeed, mechanotransduction may be a unifying feature of most TRP channels [174,175]. Both stretch-activated channels [93,176,177] and TRPC1 [34] are expressed at their highest levels in muscles during early development. AGAs can hence interfere with muscle development in response to mechanical stimulation. Accordingly, muscle adaptations to exercise have been shown to be interrupted by AGAs (Table 1). Although mechanosensitive channel-mediated calcium entry is necessary for skeletal muscle development and exercise adaptations, deregulated channel activity leading to calcium overload may result in muscle degeneration. Such a deregulation of stretch-activated calcium channels has been implicated in Duchenne and Becker X-linked muscular dystrophies [178,179]. In these conditions, a disruption in normal channel gating is observed, leading to abnormal calcium entry, elevated sarcoplasmic calcium levels, and consequent calcium-dependent necrosis. Evidence exists that the deregulated stretch-activated channel detected in mdx muscle has TRP channel origins [174,175], with emphasis on the TRPC family [65], particularly the TRPC1 subunit of the channel [92,138,180].
Streptomycin use is compatible with in vivo studies examining mechanosensitive processes due to its ability to be administered to animals in their drinking water without severe toxicity. Given its noted antagonism over TRPC1, streptomycin has been employed in numerous studies to attenuate either elevated basal sarcoplasmic calcium entry into dystrophic muscle or that stimulated in response to exaggerated muscle contractures (Table 1). Streptomycin was shown to directly block a TRP channel species in skeletal muscle fibers isolated from mdx mice, a mouse model for human X-linked muscular dystrophy [140], as well as from myotubes grown from mdx muscle biopsies [136]. Acutely applied streptomycin reduced the eccentric contraction-induced calcium entry and associated muscle damage of isolated mdx extensor digitorum longus (EDL) muscles [136]. Streptomycin also reduced force loss after the onset of eccentric contractures in wild-type muscle fibers, but not in the EDLs of TRPC1 knock-out mice [147], supporting the hypothesis that the demonstrated stretch-activated channels are synonymous with TRPC1. An analogous effect was observed in normal rat tibialis anterior muscles, whereby acutely applied streptomycin reduced eccentric contracture-induced muscle damage [45]. Previous animal trials have also shown that systemically administered AGAs protected against muscle damage in dystrophic mice by specifically targeting TRPC1 [141,145,146] or generic stretch-activated channel activity [136], which evidence indicates represents the same channel complex [92,138]. Streptomycin in these instances may have protective physiological consequences with reference to skeletal muscle calcium overload and resultant myonecrosis.
An additional protective mechanism for gentamicin has also been described in mdx mice that is attributed to its ability to suppress the premature dystrophin stop codon, restoring dystrophin expression [181]. Consequently, gentamicin was employed in a human clinical trial of Duchenne muscular dystrophy (DMD) with modest success [182]. While no change in dystrophin expression was observed following the gentamicin treatment in DMD patients, reductions in creatine kinase leakage into the serum from muscle were observed, indicating improved muscle membrane integrity, resembling preclinical results from mdx mice administered streptomycin [136] (also see Table 2). Given that gentamicin, like streptomycin, has been shown to block stretch-activated/TRPC1 channels in diverse cell types [34,44,91,126,131,139], it would be important to determine the relative contribution of each of these protective mechanisms in dystrophic muscle [141,142].
Developmentally troubling is the widespread use of antibiotics during pregnancy [183]. In infants and the young, the antagonism of signal transduction by AGAs would not be entirely determinant of developmental alterations but would contribute to biosynthetic and microbiomal disruptions of developmental consequence common to most antibiotic classes. Of special concern in the context of the present discussion is the common use of gentamicin in neonatal and pediatric clinical scenarios where it is routinely administered intravenously or intramuscularly [184]. Interestingly, the heart physiology appears to be less sensitive to streptomycin application (Table 3), which may be related to its relatively dampened hypertrophic response to physiological levels of stretch (mechanical stimulation).
Table 3.
AGA antagonism of heart stretch-activated channels reveals lower efficacy.
9. Non-TRP Channel Classes Shown to Be Sensitive to AGAs
Promiscuity is a feature that plagues most ion channel antagonists and, in this respect, AGAs are no exception. AGAs also have been shown to block other calcium channel classes but with a lower affinity than for TRPC1 [128,141]. For instance, voltage-gated calcium channels (L-type) are also blocked by AGAs in diverse species and cell classes [120,191,192]. Notably, the affinity of AGAs for the L-type voltage-gated calcium channel pore [120,192,193] is lower than that for the pore region of TRPC/SAC channels [91], binding in the mM range rather than the μM range, respectively [128,141]. Streptomycin also has been shown to block the mechanosensitive Ca2+-permeable channel, Piezo1, in a use-dependent manner [194]. Streptomycin was shown to bind to an open conformation of the Piezo1 channel in the mM range. Importantly, Piezo1 has been recently shown to contribute to muscle regeneration [195] and maintenance [196]. Finally, AGAs have also been shown to “plug” the Maxi-K+ and Ca2+-activated K+ channels in the low mM range [197,198]. These other (non-TRP) channel classes, due to their lower sensitivity to AGAs, would contribute comparatively less to systemic AGA-induced metabolic disruption [128].
10. AGA Antagonism of Other TRP Channels
Certain TRP channels have also been implicated in the transduction of sound and body position relative to gravity originating in the saccular and vestibular organs, respectively [199,200], in their capacity as acoustic vibration and shear stress sensors, respectively [124,126]. These TRP channels include TRPV4, TRPN1, TRPA1, and TRPML3 [199,200,201] and are essentially mechanosensory modalities. Accordingly, AGAs are also implicated in ototoxicity [202], as well as having been directly shown to block vibrational mechanosensitive gating via an open channel-dependent binding of AGAs to the channel’s cation conduction pathway [124,126].
TRPV channels in a variety of tissues are also susceptible to AGA antagonism. In sensory neurons, Ca2+ entry via TRPV1 channels was blocked by AGAs [203], exhibiting the highest sensitivity to neomycin and streptomycin (IC50 ~400 nM) and the least sensitivity to gentamicin. In the nervous system, TRPV1 is necessary for the perception of heat and pain. The TRPV1 and TRPV4 channels have also been shown to play a decisive role in vascular muscular remodeling [204], wherein AGA-mediated TRPV1 antagonism may be ultimately found to have critical ramifications. In skeletal muscle cells, however, magnetotransduction was unaffected by TRPV4 antagonism with Ruthenium Red (10 μM), and TRPV1 developmental expression was induced after myoblast expansion when magnetosensitivity was greatest [34], whereas TRPC1 expression was found to be necessary and sufficient to endow magnetoreception in myoblasts [67]. On the other hand, gentamicin has been shown to permeate TRPV1 [205,206,207] and TRPV4 [207,208] channels in various cell types. The TRPM7 and TRPM8 channels are blocked by streptomycin in human embryonic lung fibroblasts [137] and human adipocytes [39], respectively.
This review purposefully focused on the acute blockage of the TRPC1 and TRPM8 channels’ conduction pores by AGAs, particularly with regards to the muscle–adipose crosstalk (Figure 1). The more generalized and protracted roles of AGAs over cellular protein synthesis [42], antimicrobial resistance [209] and the microbiome [210] were not explored, despite their importance to human physiology. Of note, the knock out of the TRPV1 and TRPA1 channels, independently and combined, previously implicated in cellular responses antagonized by AGAs (see above), led to a form of systemic dysbiosis in mice that was associated with higher lipid biosynthesis [211]. Sensory neurons innervating the gut were proposed to be the source of the implicated TRPA1 and TRPV1 channels, presumably modulating the microbiome by influencing the permeability of the gut epithelial lining. It may be thus inferred that systemic AGA administration may similarly contribute to lipid accretion and a predisposition to systemic inflammation. This provocative scenario merits its own dedicated review, as it implicates both antimicrobial resistance and antagonism towards TRP channel function as a result of the presence of AGAs.
11. Conclusions
TRP channels are broadly implicated in tissue determination and development. TRPC1 is the founding member of the TRPC subfamily and, for that matter, the entire TRP superfamily. TRPC1 is the most ubiquitously expressed of all TRP channel subunits, underscoring its importance to human physiology. Ample evidence implicates TRPC1 expression in the transduction of two unyielding biophysical stimuli of developmental importance, gravitational force (mechanical loading) and magnetism. An underappreciated feature of AGAs is their ability to impede Ca2+ permeation through various TRP channel complexes, including TRPC1- and TRPM8-containing channels. The use of AGAs during in vitro or in vivo studies will hence alter the natural course of tissue development and may lead to erroneous interpretations of the data generated therein. The disruption by AGAs of TRPC1-mediated oxidative muscle development could have profound repercussions for systemic inflammation, metabolic balance, frailty, and aging. Streptomycin has also been shown to cause another form of metabolic disruption by blocking Ca2+ permeation via the thermosensitive TRPM8 channel. AGA antagonism of TRPM8 in white adipocytes prevented the induction of uncoupled respiration and non-shivering thermogenesis, which effectively entrenched the white phenotype. This effect would facilitate the establishment of systemic inflammation, as well as interfering with the metabolically beneficial muscle–adipose crosstalk that is dependent on the conversion of white to beige adipocytes. The combined antagonism of TRPC1 and TRPM8 channels by systemic AGA administration could thus have profound consequences on the whole-body metabolic balance by first, compromising mitochondrial (mitohormetic) adaptations at the cellular level and second, by attenuating muscle–fat paracrine crosstalk at the systemic level. The implications of AGA antagonism on other TRP channel complexes predominantly expressed in other tissues remain to be elucidated but may be equally critical. Given the broad developmental implications of the TRP channel family and the described antagonism of several TRP channel species by AGAs, prudence must be exercised when employing these antibiotics during in vitro or in vivo developmental studies.
Author Contributions
Conceptualization, A.F.-O.; writing—original draft preparation, A.F.-O.; writing—review and editing, Y.K.T.; visualization, Y.K.T.; funding acquisition, A.F.-O. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Pulsing Magnetic Field Therapy Research, Singapore (E-551-00-0004-01).
Acknowledgments
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Li, F.; Li, Y.; Duan, Y.; Hu, C.A.; Tang, Y.; Yin, Y. Myokines and adipokines: Involvement in the crosstalk between skeletal muscle and adipose tissue. Cytokine Growth Factor Rev. 2017, 33, 73–82. [Google Scholar] [CrossRef]
- Leal, L.G.; Lopes, M.A.; Batista, M.L., Jr. Physical Exercise-Induced Myokines and Muscle-Adipose Tissue Crosstalk: A Review of Current Knowledge and the Implications for Health and Metabolic Diseases. Front. Physiol. 2018, 9, 1307. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Catalan, V.; Ramirez, B.; Unamuno, X.; Portincasa, P.; Gomez-Ambrosi, J.; Fruhbeck, G.; Becerril, S. Impact of adipokines and myokines on fat browning. J. Physiol. Biochem. 2020, 76, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gil, A.M.; Elizondo-Montemayor, L. The Role of Exercise in the Interplay between Myokines, Hepatokines, Osteokines, Adipokines, and Modulation of Inflammation for Energy Substrate Redistribution and Fat Mass Loss: A Review. Nutrients 2020, 12, 1899. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.J.; Lee, D.S.; Kim, W.K.; Han, B.S.; Lee, S.C.; Bae, K.H. Metabolic Adaptation in Obesity and Type II Diabetes: Myokines, Adipokines and Hepatokines. Int. J. Mol. Sci. 2016, 18, 8. [Google Scholar] [CrossRef]
- Mounesan, A.; Samadian, Z. The effect of exercise on metabolic crosstalk between heart and liver. J. Exerc. Organ Cross Talk 2023, 3, 216–224. [Google Scholar] [CrossRef]
- Romero-Becera, R.; Santamans, A.M.; Arcones, A.C.; Sabio, G. From Beats to Metabolism: The Heart at the Core of Interorgan Metabolic Cross Talk. Physiology 2024, 39, 98–125. [Google Scholar] [CrossRef]
- Leal, D.V.; Ferreira, A.; Watson, E.L.; Wilund, K.R.; Viana, J.L. Muscle-Bone Crosstalk in Chronic Kidney Disease: The Potential Modulatory Effects of Exercise. Calcif. Tissue Int. 2021, 108, 461–475. [Google Scholar] [CrossRef]
- Walowski, C.O.; Herpich, C.; Enderle, J.; Braun, W.; Both, M.; Hasler, M.; Muller, M.J.; Norman, K.; Bosy-Westphal, A. Determinants of bone mass in older adults with normal- and overweight derived from the crosstalk with muscle and adipose tissue. Sci. Rep. 2023, 13, 5030. [Google Scholar] [CrossRef]
- Laurens, C.; Bergouignan, A.; Moro, C. Exercise-Released Myokines in the Control of Energy Metabolism. Front. Physiol. 2020, 11, 91. [Google Scholar] [CrossRef]
- Severinsen, M.C.K.; Pedersen, B.K. Muscle-Organ Crosstalk: The Emerging Roles of Myokines. Endocr. Rev. 2020, 41, 594–609. [Google Scholar] [CrossRef] [PubMed]
- Scheele, C.; Wolfrum, C. Brown Adipose Crosstalk in Tissue Plasticity and Human Metabolism. Endocr. Rev. 2020, 41, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Lau, P.; Vico, L.; Rittweger, J. Dissociation of Bone Resorption and Formation in Spaceflight and Simulated Microgravity: Potential Role of Myokines and Osteokines? Biomedicines 2022, 10, 342. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.S.; Gerszten, R.E.; Taylor, J.M.; Pedersen, B.K.; van Praag, H.; Trappe, S.; Febbraio, M.A.; Galis, Z.S.; Gao, Y.; Haus, J.M.; et al. Exerkines in health, resilience and disease. Nat. Rev. Endocrinol. 2022, 18, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Febbraio, M.A. Muscles, exercise and obesity: Skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 2012, 8, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Bargut, T.C.L.; Souza-Mello, V.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Browning of white adipose tissue: Lessons from experimental models. Horm. Mol. Biol. Clin. Investig. 2017, 31. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Kajimura, S. The cellular and functional complexity of thermogenic fat. Nat. Rev. Mol. Cell Biol. 2021, 22, 393–409. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.T.; Stanford, K.I. Batokines: Mediators of Inter-Tissue Communication (a Mini-Review). Curr. Obes. Rep. 2022, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, Q.; Wang, C.; Wang, T. The crosstalk between BAT thermogenesis and skeletal muscle dysfunction. Front. Physiol. 2023, 14, 1132830. [Google Scholar] [CrossRef]
- Piche, M.E.; Tchernof, A.; Despres, J.P. Obesity Phenotypes, Diabetes, and Cardiovascular Diseases. Circ. Res. 2020, 126, 1477–1500. [Google Scholar] [CrossRef]
- Wu, H.; Ballantyne, C.M. Skeletal muscle inflammation and insulin resistance in obesity. J. Clin. Investig. 2017, 127, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Bohm, A.; Hoffmann, C.; Irmler, M.; Schneeweiss, P.; Schnauder, G.; Sailer, C.; Schmid, V.; Hudemann, J.; Machann, J.; Schick, F.; et al. TGF-beta Contributes to Impaired Exercise Response by Suppression of Mitochondrial Key Regulators in Skeletal Muscle. Diabetes 2016, 65, 2849–2861. [Google Scholar] [CrossRef] [PubMed]
- Soo, J.; Raman, A.; Lawler, N.G.; Goods, P.S.R.; Deldicque, L.; Girard, O.; Fairchild, T.J. The role of exercise and hypoxia on glucose transport and regulation. Eur. J. Appl. Physiol. 2023, 123, 1147–1165. [Google Scholar] [CrossRef] [PubMed]
- Snyder, C.M.; Chandel, N.S. Mitochondrial regulation of cell survival and death during low-oxygen conditions. Antioxid. Redox Signal. 2009, 11, 2673–2683. [Google Scholar] [CrossRef] [PubMed]
- Durand, F.; Raberin, A. Exercise-Induced Hypoxemia in Endurance Athletes: Consequences for Altitude Exposure. Front. Sports Act. Living 2021, 3, 663674. [Google Scholar] [CrossRef]
- Beyfuss, K.; Hood, D.A. A systematic review of p53 regulation of oxidative stress in skeletal muscle. Redox Rep. 2018, 23, 100–117. [Google Scholar] [CrossRef]
- Bennett, C.F.; Latorre-Muro, P.; Puigserver, P. Mechanisms of mitochondrial respiratory adaptation. Nat. Rev. Mol. Cell Biol. 2022, 23, 817–835. [Google Scholar] [CrossRef]
- Melanson, E.L.; MacLean, P.S.; Hill, J.O. Exercise improves fat metabolism in muscle but does not increase 24-h fat oxidation. Exerc. Sport. Sci. Rev. 2009, 37, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef]
- McBride, T.A. Stretch-activated ion channels and c-fos expression remain active after repeated eccentric bouts. J. Appl. Physiol. 2003, 94, 2296–2302. [Google Scholar] [CrossRef]
- Wedhas, N.; Klamut, H.J.; Dogra, C.; Srivastava, A.K.; Mohan, S.; Kumar, A. Inhibition of mechanosensitive cation channels inhibits myogenic differentiation by suppressing the expression of myogenic regulatory factors and caspase-3 activity. FASEB J. 2005, 19, 1986–1997. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, T.A.; Best, T.M. Stretch-activated ion channel blockade attenuates adaptations to eccentric exercise. Med. Sci. Sports Exerc. 2009, 41, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Khodabukus, A.; Baar, K. Streptomycin decreases the functional shift to a slow phenotype induced by electrical stimulation in engineered muscle. Tissue Eng. Part A 2015, 21, 1003–1012. [Google Scholar] [CrossRef]
- Yap, J.L.Y.; Tai, Y.K.; Fröhlich, J.; Fong, C.H.H.; Yin, J.N.; Foo, Z.L.; Ramanan, S.; Beyer, C.; Toh, S.J.; Casarosa, M.; et al. Ambient and supplemental magnetic fields promote myogenesis via a TRPC1-mitochondrial axis: Evidence of a magnetic mitohormetic mechanism. FASEB J. 2019, 33, 12853–12872. [Google Scholar] [CrossRef] [PubMed]
- Spangenburg, E.E.; McBride, T.A. Inhibition of stretch-activated channels during eccentric muscle contraction attenuates p70S6K activation. J. Appl. Physiol. 2006, 100, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, H.; Yotani, K.; Ogita, F.; Sugawara, K.; Kirimto, H.; Onishi, H.; Kasuga, N.; Yamamoto, N. Effect of electrical stimulation-induced muscle force and streptomycin treatment on muscle and trabecular bone mass in early-stage disuse musculoskeletal atrophy. J. Musculoskelet. Neuronal Interact. 2015, 15, 270–278. [Google Scholar]
- Piccirillo, R. Exercise-Induced Myokines With Therapeutic Potential for Muscle Wasting. Front. Physiol. 2019, 10, 287. [Google Scholar] [CrossRef] [PubMed]
- Franco-Obregón, A.; Tai, Y.K.; Wu, K.Y.; Iversen, J.N.; Wong, C.J.K. The Developmental Implications of Muscle-Targeted Magnetic Mitohormesis: A Human Health and Longevity Perspective. Bioengineering 2023, 10, 956. [Google Scholar] [CrossRef]
- Goralczyk, A.; van Vijven, M.; Koch, M.; Badowski, C.; Yassin, M.S.; Toh, S.A.; Shabbir, A.; Franco-Obregón, A.; Raghunath, M. TRP channels in brown and white adipogenesis from human progenitors: New therapeutic targets and the caveats associated with the common antibiotic, streptomycin. FASEB J. 2017, 31, 3251–3266. [Google Scholar] [CrossRef]
- Skubis, A.; Gola, J.; Sikora, B.; Hybiak, J.; Paul-Samojedny, M.; Mazurek, U.; Los, M.J. Impact of Antibiotics on the Proliferation and Differentiation of Human Adipose-Derived Mesenchymal Stem Cells. Int. J. Mol. Sci. 2017, 18, 2522. [Google Scholar] [CrossRef]
- Moss, P.S.; Spector, D.H.; Glass, C.A.; Strohman, R.C. Streptomycin retards the phenotypic maturation of chick myogenic cells. In Vitro 1984, 20, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Khondker, A.; Bider, R.C.; Passos-Gastaldo, I.; Wright, G.D.; Rheinstadter, M.C. Membrane interactions of non-membrane targeting antibiotics: The case of aminoglycosides, macrolides, and fluoroquinolones. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183448. [Google Scholar] [CrossRef] [PubMed]
- Iversen, J.N.; Fröhlich, J.; Tai, Y.K.; Franco-Obregón, A. Synergistic Cellular Responses Conferred by Concurrent Optical and Magnetic Stimulation Are Attenuated by Simultaneous Exposure to Streptomycin: An Antibiotic Dilemma. Bioengineering 2024, 11, 637. [Google Scholar] [CrossRef] [PubMed]
- Sokabe, M.; Hasegawa, N.; Yamamori, K. Blockers and activators for stretch-activated ion channels of chick skeletal muscle. Ann. N. Y Acad. Sci. 1993, 707, 417–420. [Google Scholar] [CrossRef]
- Hayao, K.; Tamaki, H.; Nakagawa, K.; Tamakoshi, K.; Takahashi, H.; Yotani, K.; Ogita, F.; Yamamoto, N.; Onishi, H. Effects of Streptomycin Administration on Increases in Skeletal Muscle Fiber Permeability and Size Following Eccentric Muscle Contractions. Anat. Rec. 2018, 301, 1096–1102. [Google Scholar] [CrossRef]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef]
- Cheng, Y.W.; Liu, J.; Finkel, T. Mitohormesis. Cell Metab. 2023, 35, 1872–1886. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose Response 2014, 12, 288–341. [Google Scholar] [CrossRef] [PubMed]
- Scheele, C.; Nielsen, S.; Pedersen, B.K. ROS and myokines promote muscle adaptation to exercise. Trends Endocrinol. Metab. 2009, 20, 95–99. [Google Scholar] [CrossRef]
- Louzada, R.A.; Bouviere, J.; Matta, L.P.; Werneck-de-Castro, J.P.; Dupuy, C.; Carvalho, D.P.; Fortunato, R.S. Redox Signaling in Widespread Health Benefits of Exercise. Antioxid. Redox Signal. 2020, 33, 745–760. [Google Scholar] [CrossRef]
- Thyfault, J.P.; Bergouignan, A. Exercise and metabolic health: Beyond skeletal muscle. Diabetologia 2020, 63, 1464–1474. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.K.; Iversen, J.N.; Chan, K.K.W.; Fong, C.H.H.; Abdul Razar, R.B.; Ramanan, S.; Yap, L.Y.J.; Yin, J.N.; Toh, S.J.; Wong, C.J.K.; et al. Secretome from Magnetically Stimulated Muscle Exhibits Anticancer Potency: Novel Preconditioning Methodology Highlighting HTRA1 Action. Cells 2024, 13, 460. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.J.K.; Tai, Y.K.; Yap, J.L.Y.; Fong, C.H.H.; Loo, L.S.W.; Kukumberg, M.; Frohlich, J.; Zhang, S.; Li, J.Z.; Wang, J.W.; et al. Brief exposure to directionally-specific pulsed electromagnetic fields stimulates extracellular vesicle release and is antagonized by streptomycin: A potential regenerative medicine and food industry paradigm. Biomaterials 2022, 287, 121658. [Google Scholar] [CrossRef] [PubMed]
- Bielfeldt, M.; Rebl, H.; Peters, K.; Sridharan, K.; Staehlke, S.; Nebe, J.B. Sensing of Physical Factors by Cells: Electric Field, Mechanical Forces, Physical Plasma and Light—Importance for Tissue Regeneration. Biomed. Mater. Devices 2023, 1, 146–161. [Google Scholar] [CrossRef]
- Murugan, N.J.; Cariba, S.; Abeygunawardena, S.; Rouleau, N.; Payne, S.L. Biophysical control of plasticity and patterning in regeneration and cancer. Cell Mol. Life Sci. 2023, 81, 9. [Google Scholar] [CrossRef] [PubMed]
- Marini, M.; Titiz, M.; Souza Monteiro de Araújo, D.; Geppetti, P.; Nassini, R.; De Logu, F. TRP Channels in Cancer: Signaling Mechanisms and Translational Approaches. Biomolecules 2023, 13, 1557. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Lee, Y.; Kim, S.M.; Yang, Y.D.; Jung, J.; Oh, U. Quantitative analysis of TRP channel genes in mouse organs. Arch. Pharm. Res. 2012, 35, 1823–1830. [Google Scholar] [CrossRef] [PubMed]
- Van den Eynde, C.; De Clercq, K.; Vriens, J. Transient Receptor Potential Channels in the Epithelial-to-Mesenchymal Transition. Int. J. Mol. Sci. 2021, 22, 8188. [Google Scholar] [CrossRef]
- Vazquez, E.; Valverde, M.A. A review of TRP channels splicing. Semin. Cell Dev. Biol. 2006, 17, 607–617. [Google Scholar] [CrossRef]
- Eijkelkamp, N.; Quick, K.; Wood, J.N. Transient receptor potential channels and mechanosensation. Annu. Rev. Neurosci. 2013, 36, 519–546. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Faria, R.X. TRPing on the pore phenomenon: What do we know about transient receptor potential ion channel-related pore dilation up to now? J. Bioenerg. Biomembr. 2016, 48, 1–12. [Google Scholar] [CrossRef]
- Kiselyov, K.; Patterson, R.L. The integrative function of TRPC channels. Front. Biosci. 2009, 14, 45–58. [Google Scholar] [CrossRef]
- Benavides Damm, T.; Franco-Obregón, A.; Egli, M. Gravitational force modulates G2/M phase exit in mechanically unloaded myoblasts. Cell Cycle 2013, 12, 3001–3012. [Google Scholar] [CrossRef] [PubMed]
- Benavides Damm, T.; Richard, S.; Tanner, S.; Wyss, F.; Egli, M.; Franco-Obregón, A. Calcium-dependent deceleration of the cell cycle in muscle cells by simulated microgravity. FASEB J. 2013, 27, 2045–2054. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Sharif-Naeini, R.; Folgering, J.R.; Bichet, D.; Duprat, F.; Honore, E. Canonical TRP channels and mechanotransduction: From physiology to disease states. Pflugers Arch. 2010, 460, 571–581. [Google Scholar] [CrossRef]
- Parate, D.; Franco-Obregón, A.; Fröhlich, J.; Beyer, C.; Abbas, A.A.; Kamarul, T.; Hui, J.H.P.; Yang, Z. Enhancement of mesenchymal stem cell chondrogenesis with short-term low intensity pulsed electromagnetic fields. Sci. Rep. 2017, 7, 9421. [Google Scholar] [CrossRef]
- Kurth, F.; Tai, Y.K.; Parate, D.; van Oostrum, M.; Schmid, Y.R.F.; Toh, S.J.; Yap, J.L.Y.; Wollscheid, B.; Othman, A.; Dittrich, P.S.; et al. Cell-Derived Vesicles as TRPC1 Channel Delivery Systems for the Recovery of Cellular Respiratory and Proliferative Capacities. Adv. Biosyst. 2020, 4, e2000146. [Google Scholar] [CrossRef] [PubMed]
- Madanagopal, T.T.; Tai, Y.K.; Lim, S.H.; Fong, C.H.; Cao, T.; Rosa, V.; Franco-Obregón, A. Pulsed electromagnetic fields synergize with graphene to enhance dental pulp stem cell-derived neurogenesis by selectively targeting TRPC1 channels. Eur. Cell Mater. 2021, 41, 216–232. [Google Scholar] [CrossRef]
- Detwiler, P.B. Phototransduction in Retinal Ganglion Cells. Yale J. Biol. Med. 2018, 91, 49–52. [Google Scholar]
- Contreras, E.; Nobleman, A.P.; Robinson, P.R.; Schmidt, T.M. Melanopsin phototransduction: Beyond canonical cascades. J. Exp. Biol. 2021, 224, jeb226522. [Google Scholar] [CrossRef]
- Moraes, M.N.; de Assis, L.V.M.; Provencio, I.; Castrucci, A.M.L. Opsins outside the eye and the skin: A more complex scenario than originally thought for a classical light sensor. Cell Tissue Res. 2021, 385, 519–538. [Google Scholar] [CrossRef] [PubMed]
- Bellemer, A. Thermotaxis, circadian rhythms, and TRP channels in Drosophila. Temperature 2015, 2, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cheng, X.; Tian, J.; Xiao, Y.; Tian, T.; Xu, F.; Hong, X.; Zhu, M.X. TRPC channels: Structure, function, regulation and recent advances in small molecular probes. Pharmacol. Ther. 2020, 209, 107497. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, I.S.; de Souza, L.B.; Ong, H.L. TRPC1, Orai1, and STIM1 in SOCE: Friends in tight spaces. Cell Calcium 2017, 63, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Gervasio, O.L.; Whitehead, N.P.; Yeung, E.W.; Phillips, W.D.; Allen, D.G. TRPC1 binds to caveolin-3 and is regulated by Src kinase—Role in Duchenne muscular dystrophy. J. Cell Sci. 2008, 121 Pt 13, 2246–2255. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, D.L. Redox regulation of endothelial canonical transient receptor potential channels. Antioxid. Redox Signal. 2011, 15, 1567–1582. [Google Scholar] [CrossRef] [PubMed]
- Song, M.Y.; Makino, A.; Yuan, J.X. Role of reactive oxygen species and redox in regulating the function of transient receptor potential channels. Antioxid. Redox Signal. 2011, 15, 1549–1565. [Google Scholar] [CrossRef] [PubMed]
- Piciu, F.; Balas, M.; Badea, M.A.; Cucu, D. TRP Channels in Tumoral Processes Mediated by Oxidative Stress and Inflammation. Antioxidants 2023, 12, 1327. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef]
- Takahashi, N.; Mori, Y. TRP Channels as Sensors and Signal Integrators of Redox Status Changes. Front. Pharmacol. 2011, 2, 58. [Google Scholar] [CrossRef]
- Birnbaumer, L. The TRPC class of ion channels: A critical review of their roles in slow, sustained increases in intracellular Ca(2+) concentrations. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 395–426. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Fu, M.; Zhang, Y.; You, Q.; Wang, L. Small molecules targeting canonical transient receptor potential channels: An update. Drug Discov. Today 2024, 29, 103951. [Google Scholar] [CrossRef] [PubMed]
- Baron, J.; Groschner, K.; Tiapko, O. Calcium transport and sensing in TRPC channels—New insights into a complex feedback regulation. Cell Calcium 2023, 116, 102816. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Li, H.; Tai, Y.; Huang, J.; Su, Y.; Abramowitz, J.; Zhu, M.X.; Birnbaumer, L.; Wang, Y. Canonical transient receptor potential 3 channels regulate mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 11011–11016. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Farfariello, V.; Gordienko, D.V.; Mesilmany, L.; Touil, Y.; Germain, E.; Fliniaux, I.; Desruelles, E.; Gkika, D.; Roudbaraki, M.; Shapovalov, G.; et al. TRPC3 shapes the ER-mitochondria Ca(2+) transfer characterizing tumour-promoting senescence. Nat. Commun. 2022, 13, 956. [Google Scholar] [CrossRef] [PubMed]
- Trebak, M.; Ginnan, R.; Singer, H.A.; Jourd’heuil, D. Interplay between calcium and reactive oxygen/nitrogen species: An essential paradigm for vascular smooth muscle signaling. Antioxid. Redox Signal. 2010, 12, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Kunert-Keil, C.; Bisping, F.; Kruger, J.; Brinkmeier, H. Tissue-specific expression of TRP channel genes in the mouse and its variation in three different mouse strains. BMC Genom. 2006, 7, 159. [Google Scholar] [CrossRef]
- Dadon, D.; Minke, B. Cellular functions of transient receptor potential channels. Int. J. Biochem. Cell Biol. 2010, 42, 1430–1445. [Google Scholar] [CrossRef][Green Version]
- Zhang, M.; Ma, Y.; Ye, X.; Zhang, N.; Pan, L.; Wang, B. TRP (transient receptor potential) ion channel family: Structures, biological functions and therapeutic interventions for diseases. Signal Transduct. Target. Ther. 2023, 8, 261. [Google Scholar] [CrossRef]
- Winegar, B.D.; Haws, C.M.; Lansman, J.B. Subconductance block of single mechanosensitive ion channels in skeletal muscle fibers by aminoglycoside antibiotics. J. Gen. Physiol. 1996, 107, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Maroto, R.; Raso, A.; Wood, T.G.; Kurosky, A.; Martinac, B.; Hamill, O.P. TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nat. Cell Biol. 2005, 7, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Lansman, J.B. Mechanosensitive ion channels in dystrophic muscle. Curr. Top. Membr. 2007, 59, 467–484. [Google Scholar] [CrossRef]
- Zhao, Y.; McVeigh, B.M.; Moiseenkova-Bell, V.Y. Structural Pharmacology of TRP Channels. J. Mol. Biol. 2021, 433, 166914. [Google Scholar] [CrossRef]
- Goretzki, B.; Guhl, C.; Tebbe, F.; Harder, J.M.; Hellmich, U.A. Unstructural Biology of TRP Ion Channels: The Role of Intrinsically Disordered Regions in Channel Function and Regulation. J. Mol. Biol. 2021, 433, 166931. [Google Scholar] [CrossRef] [PubMed]
- Franco-Obregón, A. Harmonizing Magnetic Mitohormetic Regenerative Strategies: Developmental Implications of a Calcium-Mitochondrial Axis Invoked by Magnetic Field Exposure. Bioengineering 2023, 10, 1176. [Google Scholar] [CrossRef] [PubMed]
- Atomi, Y. Gravitational Effects on Human Physiology. Subcell. Biochem. 2015, 72, 627–659. [Google Scholar] [CrossRef]
- Najrana, T.; Sanchez-Esteban, J. Mechanotransduction as an Adaptation to Gravity. Front. Pediatr. 2016, 4, 140. [Google Scholar] [CrossRef] [PubMed]
- Varani, K.; Vincenzi, F.; Pasquini, S.; Blo, I.; Salati, S.; Cadossi, M.; De Mattei, M. Pulsed Electromagnetic Field Stimulation in Osteogenesis and Chondrogenesis: Signaling Pathways and Therapeutic Implications. Int. J. Mol. Sci. 2021, 22, 809. [Google Scholar] [CrossRef]
- Formigli, L.; Sassoli, C.; Squecco, R.; Bini, F.; Martinesi, M.; Chellini, F.; Luciani, G.; Sbrana, F.; Zecchi-Orlandini, S.; Francini, F.; et al. Regulation of transient receptor potential canonical channel 1 (TRPC1) by sphingosine 1-phosphate in C2C12 myoblasts and its relevance for a role of mechanotransduction in skeletal muscle differentiation. J. Cell Sci. 2009, 122 Pt 9, 1322–1333. [Google Scholar] [CrossRef]
- Crocetti, S.; Beyer, C.; Unternahrer, S.; Benavides Damm, T.; Schade-Kampmann, G.; Hebeisen, M.; Di Berardino, M.; Frohlich, J.; Franco-Obregón, A. Impedance flow cytometry gauges proliferative capacity by detecting TRPC1 expression. Cytom. A 2014, 85, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Chen, C.; Deng, P.; Zhu, G.; Lin, M.; Zhang, L.; Xu, S.; He, M.; Lu, Y.; Duan, W.; et al. Extremely Low-Frequency Electromagnetic Fields Promote In Vitro Neuronal Differentiation and Neurite Outgrowth of Embryonic Neural Stem Cells via Up-Regulating TRPC1. PLoS ONE 2016, 11, e0150923. [Google Scholar] [CrossRef] [PubMed]
- Mocanu-Dobranici, A.E.; Costache, M.; Dinescu, S. Insights into the Molecular Mechanisms Regulating Cell Behavior in Response to Magnetic Materials and Magnetic Stimulation in Stem Cell (Neurogenic) Differentiation. Int. J. Mol. Sci. 2023, 24, 2028. [Google Scholar] [CrossRef] [PubMed]
- Fitts, R.H.; Riley, D.R.; Widrick, J.J. Functional and structural adaptations of skeletal muscle to microgravity. J. Exp. Biol. 2001, 204 Pt 18, 3201–3208. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.R.; Caiozzo, V.J.; Baldwin, K.M. Skeletal muscle unweighting: Spaceflight and ground-based models. J. Appl. Physiol. 2003, 95, 2185–2201. [Google Scholar] [CrossRef] [PubMed]
- Zampino, M.; Semba, R.D.; Adelnia, F.; Spencer, R.G.; Fishbein, K.W.; Schrack, J.A.; Simonsick, E.M.; Ferrucci, L. Greater Skeletal Muscle Oxidative Capacity Is Associated With Higher Resting Metabolic Rate: Results From the Baltimore Longitudinal Study of Aging. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 2262–2268. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.T.; Hamilton, D.G.; Zderic, T.W. A potent physiological method to magnify and sustain soleus oxidative metabolism improves glucose and lipid regulation. iScience 2022, 25, 104869. [Google Scholar] [CrossRef] [PubMed]
- Sarimov, R.M.; Serov, D.A.; Gudkov, S.V. Biological Effects of Magnetic Storms and ELF Magnetic Fields. Biology 2023, 12, 1506. [Google Scholar] [CrossRef] [PubMed]
- Hart, D.A. The Influence of Magnetic Fields, Including the Planetary Magnetic Field, on Complex Life Forms: How Do Biological Systems Function in This Field and in Electromagnetic Fields? Biophysica 2024, 4, 1–21. [Google Scholar] [CrossRef]
- Hu, H.; Yang, W.; Zeng, Q.; Chen, W.; Zhu, Y.; Liu, W.; Wang, S.; Wang, B.; Shao, Z.; Zhang, Y. Promising application of Pulsed Electromagnetic Fields (PEMFs) in musculoskeletal disorders. Biomed. Pharmacother. 2020, 131, 110767. [Google Scholar] [CrossRef]
- Lai, H.; Levitt, B.B. Cellular and molecular effects of non-ionizing electromagnetic fields. Rev. Environ. Health 2023. [Google Scholar] [CrossRef] [PubMed]
- Zadeh-Haghighi, H.; Rishabh, R.; Simon, C. Hypomagnetic field effects as a potential avenue for testing the radical pair mechanism in biology. Front. Phys. 2023, 11, 1026460. [Google Scholar] [CrossRef]
- Fraysse, B.; Desaphy, J.F.; Pierno, S.; De Luca, A.; Liantonio, A.; Mitolo, C.I.; Camerino, D.C. Decrease in resting calcium and calcium entry associated with slow-to-fast transition in unloaded rat soleus muscle. FASEB J. 2003, 17, 1916–1918. [Google Scholar] [CrossRef]
- Tai, Y.K.; Ng, C.; Purnamawati, K.; Yap, J.L.Y.; Yin, J.N.; Wong, C.; Patel, B.K.; Soong, P.L.; Pelczar, P.; Frohlich, J.; et al. Magnetic fields modulate metabolism and gut microbiome in correlation with Pgc-1alpha expression: Follow-up to an in vitro magnetic mitohormetic study. FASEB J. 2020, 34, 11143–11167. [Google Scholar] [CrossRef] [PubMed]
- Zanou, N.; Shapovalov, G.; Louis, M.; Tajeddine, N.; Gallo, C.; Van Schoor, M.; Anguish, I.; Cao, M.L.; Schakman, O.; Dietrich, A.; et al. Role of TRPC1 channel in skeletal muscle function. Am. J. Physiol. Cell Physiol. 2010, 298, C149–C162. [Google Scholar] [CrossRef]
- Xia, L.; Cheung, K.K.; Yeung, S.S.; Yeung, E.W. The involvement of transient receptor potential canonical type 1 in skeletal muscle regrowth after unloading-induced atrophy. J. Physiol. 2016, 594, 3111–3126. [Google Scholar] [CrossRef] [PubMed]
- Di, X.; Gao, X.; Peng, L.; Ai, J.; Jin, X.; Qi, S.; Li, H.; Wang, K.; Luo, D. Cellular mechanotransduction in health and diseases: From molecular mechanism to therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 282. [Google Scholar] [CrossRef] [PubMed]
- LaGuardia, J.S.; Shariati, K.; Bedar, M.; Ren, X.; Moghadam, S.; Huang, K.X.; Chen, W.; Kang, Y.; Yamaguchi, D.T.; Lee, J.C. Convergence of Calcium Channel Regulation and Mechanotransduction in Skeletal Regenerative Biomaterial Design. Adv. Healthc. Mater. 2023, 12, e2301081. [Google Scholar] [CrossRef]
- Saraswathibhatla, A.; Indana, D.; Chaudhuri, O. Cell-extracellular matrix mechanotransduction in 3D. Nat. Rev. Mol. Cell Biol. 2023, 24, 495–516. [Google Scholar] [CrossRef]
- Haws, C.M.; Winegar, B.D.; Lansman, J.B. Block of single L-type Ca2+ channels in skeletal muscle fibers by aminoglycoside antibiotics. J. Gen. Physiol. 1996, 107, 421–432. [Google Scholar] [CrossRef]
- Uzieliene, I.; Bernotas, P.; Mobasheri, A.; Bernotiene, E. The Role of Physical Stimuli on Calcium Channels in Chondrogenic Differentiation of Mesenchymal Stem Cells. Int. J. Mol. Sci. 2018, 19, 2998. [Google Scholar] [CrossRef]
- Chen, X.; Sooch, G.; Demaree, I.S.; White, F.A.; Obukhov, A.G. Transient Receptor Potential Canonical (TRPC) Channels: Then and Now. Cells 2020, 9, 1983. [Google Scholar] [CrossRef] [PubMed]
- Franco-Obregón, A. Magnetic mitohormesis: A non-invasive therapy for inflammatory disorders? Biocell 2022, 47, 239–244. [Google Scholar] [CrossRef]
- Hudspeth, A.J. Extracellular current flow and the site of transduction by vertebrate hair cells. J. Neurosci. 1982, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, H. Mechano-electrical transduction currents in isolated vestibular hair cells of the chick. J. Physiol. 1985, 359, 189–217. [Google Scholar] [CrossRef] [PubMed]
- Kroese, A.B.; Das, A.; Hudspeth, A.J. Blockage of the transduction channels of hair cells in the bullfrog’s sacculus by aminoglycoside antibiotics. Hear. Res. 1989, 37, 203–217. [Google Scholar] [CrossRef]
- Gannier, F.; White, E.; Lacampagne, A.; Garnier, D.; Le Guennec, J.Y. Streptomycin reverses a large stretch induced increases in [Ca2+]i in isolated guinea pig ventricular myocytes. Cardiovasc. Res. 1994, 28, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Hamill, O.P.; McBride, D.W., Jr. The pharmacology of mechanogated membrane ion channels. Pharmacol. Rev. 1996, 48, 231–252. [Google Scholar]
- McBride, T.A.; Stockert, B.W.; Gorin, F.A.; Carlsen, R.C. Stretch-activated ion channels contribute to membrane depolarization after eccentric contractions. J. Appl. Physiol. 2000, 88, 91–101. [Google Scholar] [CrossRef]
- McBride, T. Increased depolarization, prolonged recovery and reduced adaptation of the resting membrane potential in aged rat skeletal muscles following eccentric contractions. Mech. Ageing Dev. 2000, 115, 127–138. [Google Scholar] [CrossRef]
- Shen, M.R.; Chou, C.Y.; Chiu, W.T. Streptomycin and its analogues are potent inhibitors of the hypotonicity-induced Ca2+ entry and Cl- channel activity. FEBS Lett. 2003, 554, 494–500. [Google Scholar] [CrossRef]
- Yeung, E.W.; Head, S.I.; Allen, D.G. Gadolinium reduces short-term stretch-induced muscle damage in isolated mdx mouse muscle fibres. J. Physiol. 2003, 552 Pt 2, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Yeung, E.W.; Ballard, H.J.; Bourreau, J.P.; Allen, D.G. Intracellular sodium in mammalian muscle fibers after eccentric contractions. J. Appl. Physiol. 2003, 94, 2475–2482. [Google Scholar] [CrossRef] [PubMed]
- Willems, M.E.; Stauber, W.T. Streptomycin and EDTA decrease the number of desmin-negative fibers following stretch injury. Muscle Nerve 2005, 32, 310–315. [Google Scholar] [CrossRef]
- Garan, A.R.; Maron, B.J.; Wang, P.J.; Estes, N.A., 3rd; Link, M.S. Role of streptomycin-sensitive stretch-activated channel in chest wall impact induced sudden death (commotio cordis). J. Cardiovasc. Electrophysiol. 2005, 16, 433–438. [Google Scholar] [CrossRef]
- Yeung, E.W.; Whitehead, N.P.; Suchyna, T.M.; Gottlieb, P.A.; Sachs, F.; Allen, D.G. Effects of stretch-activated channel blockers on [Ca2+]i and muscle damage in the mdx mouse. J. Physiol. 2005, 562 Pt 2, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Wang, X.; Chen, M.; Ouyang, K.; Song, L.S.; Cheng, H. Calcium flickers steer cell migration. Nature 2009, 457, 901–905. [Google Scholar] [CrossRef]
- Allen, D.G.; Whitehead, N.P.; Yeung, E.W. Mechanisms of stretch-induced muscle damage in normal and dystrophic muscle: Role of ionic changes. J. Physiol. 2005, 567 Pt 3, 723–735. [Google Scholar] [CrossRef]
- Jacques-Fricke, B.T.; Seow, Y.; Gottlieb, P.A.; Sachs, F.; Gomez, T.M. Ca2+ influx through mechanosensitive channels inhibits neurite outgrowth in opposition to other influx pathways and release from intracellular stores. J. Neurosci. 2006, 26, 5656–5664. [Google Scholar] [CrossRef]
- Rolland, J.F.; De Luca, A.; Burdi, R.; Andreetta, F.; Confalonieri, P.; Conte Camerino, D. Overactivity of exercise-sensitive cation channels and their impaired modulation by IGF-1 in mdx native muscle fibers: Beneficial effect of pentoxifylline. Neurobiol. Dis. 2006, 24, 466–474. [Google Scholar] [CrossRef]
- Whitehead, N.P.; Streamer, M.; Lusambili, L.I.; Sachs, F.; Allen, D.G. Streptomycin reduces stretch-induced membrane permeability in muscles from mdx mice. Neuromuscul. Disord. 2006, 16, 845–854. [Google Scholar] [CrossRef]
- Whitehead, N.P.; Yeung, E.W.; Allen, D.G. Muscle damage in mdx (dystrophic) mice: Role of calcium and reactive oxygen species. Clin. Exp. Pharmacol. Physiol. 2006, 33, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, M.; Pellegrini, M. Mechanosensitive channels in neurite outgrowth. Curr. Top. Membr. 2007, 59, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Bowman, C.L.; Gottlieb, P.A.; Suchyna, T.M.; Murphy, Y.K.; Sachs, F. Mechanosensitive ion channels and the peptide inhibitor GsMTx-4: History, properties, mechanisms and pharmacology. Toxicon 2007, 49, 249–270. [Google Scholar] [CrossRef]
- Jorgensen, L.H.; Blain, A.; Greally, E.; Laval, S.H.; Blamire, A.M.; Davison, B.J.; Brinkmeier, H.; MacGowan, G.A.; Schroder, H.D.; Bushby, K.; et al. Long-term blocking of calcium channels in mdx mice results in differential effects on heart and skeletal muscle. Am. J. Pathol. 2011, 178, 273–283. [Google Scholar] [CrossRef]
- Matsumura, C.Y.; Taniguti, A.P.; Pertille, A.; Santo Neto, H.; Marques, M.J. Stretch-activated calcium channel protein TRPC1 is correlated with the different degrees of the dystrophic phenotype in mdx mice. Am. J. Physiol. Cell Physiol. 2011, 301, C1344–C1350. [Google Scholar] [CrossRef]
- Zhang, B.T.; Whitehead, N.P.; Gervasio, O.L.; Reardon, T.F.; Vale, M.; Fatkin, D.; Dietrich, A.; Yeung, E.W.; Allen, D.G. Pathways of Ca(2)(+) entry and cytoskeletal damage following eccentric contractions in mouse skeletal muscle. J. Appl. Physiol. 2012, 112, 2077–2086. [Google Scholar] [CrossRef] [PubMed]
- Kerstein, P.C.; Jacques-Fricke, B.T.; Rengifo, J.; Mogen, B.J.; Williams, J.C.; Gottlieb, P.A.; Sachs, F.; Gomez, T.M. Mechanosensitive TRPC1 channels promote calpain proteolysis of talin to regulate spinal axon outgrowth. J. Neurosci. 2013, 33, 273–285. [Google Scholar] [CrossRef]
- Rocha, G.L.D.; Rupcic, I.F.; Mizobuti, D.S.; Hermes, T.A.; Covatti, C.; Silva, H.; Araujo, H.N.; Lourenco, C.C.; Silveira, L.D.R.; Pereira, E.C.L.; et al. Cross-talk between TRPC-1, mTOR, PGC-1alpha and PPARdelta in the dystrophic muscle cells treated with tempol. Free Radic. Res. 2022, 56, 245–257. [Google Scholar] [CrossRef]
- Roberts-Wilson, T.K.; Reddy, R.N.; Bailey, J.L.; Zheng, B.; Ordas, R.; Gooch, J.L.; Price, S.R. Calcineurin signaling and PGC-1alpha expression are suppressed during muscle atrophy due to diabetes. Biochim. Biophys. Acta 2010, 1803, 960–967. [Google Scholar] [CrossRef]
- Calabria, E.; Ciciliot, S.; Moretti, I.; Garcia, M.; Picard, A.; Dyar, K.A.; Pallafacchina, G.; Tothova, J.; Schiaffino, S.; Murgia, M. NFAT isoforms control activity-dependent muscle fiber type specification. Proc. Natl. Acad. Sci. USA 2009, 106, 13335–13340. [Google Scholar] [CrossRef] [PubMed]
- Louis, M.; Zanou, N.; Van Schoor, M.; Gailly, P. TRPC1 regulates skeletal myoblast migration and differentiation. J. Cell Sci. 2008, 121 Pt 23, 3951–3959. [Google Scholar] [CrossRef] [PubMed]
- Antigny, F.; Koenig, S.; Bernheim, L.; Frieden, M. During post-natal human myogenesis, normal myotube size requires TRPC1- and TRPC4-mediated Ca(2)(+) entry. J. Cell Sci. 2013, 126 Pt 11, 2525–2533. [Google Scholar] [CrossRef] [PubMed]
- Antigny, F.; Sabourin, J.; Sauc, S.; Bernheim, L.; Koenig, S.; Frieden, M. TRPC1 and TRPC4 channels functionally interact with STIM1L to promote myogenesis and maintain fast repetitive Ca(2+) release in human myotubes. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Shang, P.; Zhang, B.; Tian, X.; Nie, R.; Zhang, R.; Zhang, H. Function of the Porcine TRPC1 Gene in Myogenesis and Muscle Growth. Cells 2021, 10, 147. [Google Scholar] [CrossRef] [PubMed]
- Zanou, N.; Schakman, O.; Louis, P.; Ruegg, U.T.; Dietrich, A.; Birnbaumer, L.; Gailly, P. Trpc1 ion channel modulates phosphatidylinositol 3-kinase/Akt pathway during myoblast differentiation and muscle regeneration. J. Biol. Chem. 2012, 287, 14524–14534. [Google Scholar] [CrossRef] [PubMed]
- Supruniuk, E.; Miklosz, A.; Chabowski, A. The Implication of PGC-1alpha on Fatty Acid Transport across Plasma and Mitochondrial Membranes in the Insulin Sensitive Tissues. Front. Physiol. 2017, 8, 923. [Google Scholar] [CrossRef]
- Gemmink, A.; Schrauwen, P.; Hesselink, M.K.C. Exercising your fat (metabolism) into shape: A muscle-centred view. Diabetologia 2020, 63, 1453–1463. [Google Scholar] [CrossRef]
- Umek, N.; Horvat, S.; Cvetko, E. Skeletal muscle and fiber type-specific intramyocellular lipid accumulation in obese mice. Bosn. J. Basic. Med. Sci. 2021, 21, 730–738. [Google Scholar] [CrossRef]
- Pereyra, A.S.; Lin, C.T.; Sanchez, D.M.; Laskin, J.; Spangenburg, E.E.; Neufer, P.D.; Fisher-Wellman, K.; Ellis, J.M. Skeletal muscle undergoes fiber type metabolic switch without myosin heavy chain switch in response to defective fatty acid oxidation. Mol. Metab. 2022, 59, 101456. [Google Scholar] [CrossRef]
- Maunder, E.; Rothschild, J.A.; Fritzen, A.M.; Jordy, A.B.; Kiens, B.; Brick, M.J.; Leigh, W.B.; Chang, W.L.; Kilding, A.E. Skeletal muscle proteins involved in fatty acid transport influence fatty acid oxidation rates observed during exercise. Pflugers Arch. 2023, 475, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Leuchtmann, A.B.; Adak, V.; Dilbaz, S.; Handschin, C. The Role of the Skeletal Muscle Secretome in Mediating Endurance and Resistance Training Adaptations. Front. Physiol. 2021, 12, 709807. [Google Scholar] [CrossRef] [PubMed]
- Khodabukus, A.; Baar, K. Glucose concentration and streptomycin alter in vitro muscle function and metabolism. J. Cell Physiol. 2015, 230, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Morales, S.; Diez, A.; Puyet, A.; Camello, P.J.; Camello-Almaraz, C.; Bautista, J.M.; Pozo, M.J. Calcium controls smooth muscle TRPC gene transcription via the CaMK/calcineurin-dependent pathways. Am. J. Physiol. Cell Physiol. 2007, 292, C553–C563. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ji, Y.; Liu, R.; Zhu, X.; Wang, K.; Yang, X.; Liu, B.; Gao, Z.; Huang, Y.; Shen, Y.; et al. Mitochondrial dysfunction: Roles in skeletal muscle atrophy. J. Transl. Med. 2023, 21, 503. [Google Scholar] [CrossRef] [PubMed]
- Lo Buglio, A.; Bellanti, F.; Vendemiale, G. The aging muscle: Sarcopenia, mitochondrial function, and redox biology. J. Gerontol. Geriatr. 2024, 72, 1–10. [Google Scholar] [CrossRef]
- Dowling, P.; Gargan, S.; Swandulla, D.; Ohlendieck, K. Fiber-Type Shifting in Sarcopenia of Old Age: Proteomic Profiling of the Contractile Apparatus of Skeletal Muscles. Int. J. Mol. Sci. 2023, 24, 2415. [Google Scholar] [CrossRef] [PubMed]
- Bilski, J.; Pierzchalski, P.; Szczepanik, M.; Bonior, J.; Zoladz, J.A. Multifactorial Mechanism of Sarcopenia and Sarcopenic Obesity. Role of Physical Exercise, Microbiota and Myokines. Cells 2022, 11, 160. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Shi, L.; Peng, Z.; Sun, Y.; Chen, J. Ageing of skeletal muscle extracellular matrix and mitochondria: Finding a potential link. Ann. Med. 2023, 55, 2240707. [Google Scholar] [CrossRef]
- Klingelhuber, F.; Frendo-Cumbo, S.; Omar-Hmeadi, M.; Massier, L.; Kakimoto, P.; Taylor, A.J.; Couchet, M.; Ribicic, S.; Wabitsch, M.; Messias, A.C.; et al. A spatiotemporal proteomic map of human adipogenesis. Nat. Metab. 2024, 6, 861–879. [Google Scholar] [CrossRef]
- Tarantini, S.; Subramanian, M.; Butcher, J.T.; Yabluchanskiy, A.; Li, X.; Miller, R.A.; Balasubramanian, P. Revisiting adipose thermogenesis for delaying aging and age-related diseases: Opportunities and challenges. Ageing Res. Rev. 2023, 87, 101912. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Bae, J.H.; Wang, Y.; Song, W. The Potential Roles of Myokines in Adipose Tissue Metabolism with Exercise and Cold Exposure. Int. J. Mol. Sci. 2022, 23, 11523. [Google Scholar] [CrossRef] [PubMed]
- Hunter, A.M.; Grigson, C.; Wade, A. Influence of Topically Applied Menthol Cooling Gel on Soft Tissue Thermodynamics and Arterial and Cutaneous Blood Flow at Rest. Int. J. Sports Phys. Ther. 2018, 13, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Montell, C. Forcing open TRP channels: Mechanical gating as a unifying activation mechanism. Biochem. Biophys. Res. Commun. 2015, 460, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.D.; Poole, K.; Martinac, B. Re-evaluating TRP channel mechanosensitivity. Trends Biochem. Sci. 2024. [Google Scholar] [CrossRef] [PubMed]
- Franco, A., Jr.; Lansman, J.B. Stretch-sensitive channels in developing muscle cells from a mouse cell line. J. Physiol. 1990, 427, 361–380. [Google Scholar] [CrossRef] [PubMed]
- Haws, C.M.; Lansman, J.B. Developmental regulation of mechanosensitive calcium channels in skeletal muscle from normal and mdx mice. Proc. Biol. Sci. 1991, 245, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Franco, A., Jr.; Lansman, J.B. Calcium entry through stretch-inactivated ion channels in mdx myotubes. Nature 1990, 344, 670–673. [Google Scholar] [CrossRef] [PubMed]
- Franco-Obregón, A., Jr.; Lansman, J.B. Mechanosensitive ion channels in skeletal muscle from normal and dystrophic mice. J. Physiol. 1994, 481 Pt 2, 299–309. [Google Scholar] [CrossRef]
- Vandebrouck, C.; Martin, D.; Colson-Van Schoor, M.; Debaix, H.; Gailly, P. Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J. Cell Biol. 2002, 158, 1089–1096. [Google Scholar] [CrossRef]
- Barton-Davis, E.R.; Cordier, L.; Shoturma, D.I.; Leland, S.E.; Sweeney, H.L. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J. Clin. Investig. 1999, 104, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; Hamed, S.; Hadley, D.W.; Gropman, A.L.; Burstein, A.H.; Escolar, D.M.; Hoffman, E.P.; Fischbeck, K.H. Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann. Neurol. 2001, 49, 706–711. [Google Scholar] [CrossRef]
- Njotto, L.L.; Simin, J.; Fornes, R.; Odsbu, I.; Mussche, I.; Callens, S.; Engstrand, L.; Bruyndonckx, R.; Brusselaers, N. Maternal and Early-Life Exposure to Antibiotics and the Risk of Autism and Attention-Deficit Hyperactivity Disorder in Childhood: A Swedish Population-Based Cohort Study. Drug Saf. 2023, 46, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Germovsek, E.; Barker, C.I.; Sharland, M. What do I need to know about aminoglycoside antibiotics? Arch. Dis. Child. Educ. Pract. Ed. 2017, 102, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Komuro, I.; Kudoh, S.; Zou, Y.; Nagai, R.; Aikawa, R.; Uozumi, H.; Yazaki, Y. Role of ion channels and exchangers in mechanical stretch-induced cardiomyocyte hypertrophy. Circ. Res. 1998, 82, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Belus, A.; White, E. Streptomycin and intracellular calcium modulate the response of single guinea-pig ventricular myocytes to axial stretch. J. Physiol. 2003, 546 Pt 2, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Cooper, P.J.; Kohl, P. Species- and preparation-dependence of stretch effects on sino-atrial node pacemaking. Ann. N. Y Acad. Sci. 2005, 1047, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Cao, J.X.; Xie, R.S.; Li, J.; Han, Y.; Zhu, L.Q.; Dai, Y.N. The effect of streptomycin on stretch-induced electrophysiological changes of isolated acute myocardial infarcted hearts in rats. Europace 2007, 9, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Birla, R.K.; Huang, Y.C.; Dennis, R.G. Effect of streptomycin on the active force of bioengineered heart muscle in response to controlled stretch. In Vitro Cell Dev. Biol. Anim. 2008, 44, 253–260. [Google Scholar] [CrossRef]
- Ward, M.L.; Williams, I.A.; Chu, Y.; Cooper, P.J.; Ju, Y.K.; Allen, D.G. Stretch-activated channels in the heart: Contributions to length-dependence and to cardiomyopathy. Prog. Biophys. Mol. Biol. 2008, 97, 232–249. [Google Scholar] [CrossRef]
- Suarez-Kurtz, G. Inhibition of membrane calcium activation by neomycin and streptomycin in crab muscle fibers. Pflugers Arch. 1974, 349, 337–349. [Google Scholar] [CrossRef]
- Miller, A.L.; Langton, P.D. Streptomycin inhibition of myogenic tone, K+-induced force and block of L-type calcium current in rat cerebral arteries. J. Physiol. 1998, 508 Pt 3, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Audesirk, G.; Audesirk, T.; Ferguson, C.; Lomme, M.; Shugarts, D.; Rosack, J.; Caracciolo, P.; Gisi, T.; Nichols, P. L-type calcium channels may regulate neurite initiation in cultured chick embryo brain neurons and N1E-115 neuroblastoma cells. Brain Res. Dev. Brain Res. 1990, 55, 109–120. [Google Scholar] [CrossRef]
- Bae, C.; Sachs, F.; Gottlieb, P.A. The mechanosensitive ion channel Piezo1 is inhibited by the peptide GsMTx4. Biochemistry 2011, 50, 6295–6300. [Google Scholar] [CrossRef]
- Peng, Y.; Du, J.; Gunther, S.; Guo, X.; Wang, S.; Schneider, A.; Zhu, L.; Braun, T. Mechano-signaling via Piezo1 prevents activation and p53-mediated senescence of muscle stem cells. Redox Biol. 2022, 52, 102309. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Nomura, K.; Kato, D.; Tachibana, Y.; Niikura, T.; Uchiyama, K.; Hosooka, T.; Fukui, T.; Oe, K.; Kuroda, R.; et al. A Piezo1/KLF15/IL-6 axis mediates immobilization-induced muscle atrophy. J. Clin. Investig. 2022, 132, e154611. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, S.; Wangemann, P. Aminoglycoside antibiotics inhibit maxi-K+ channel in single isolated cochlear efferent nerve terminals. Hear. Res. 1993, 67, 13–19. [Google Scholar] [CrossRef]
- Dulon, D.; Sugasawa, M.; Blanchet, C.; Erostegui, C. Direct measurements of Ca(2+)-activated K+ currents in inner hair cells of the guinea-pig cochlea using photolabile Ca2+ chelators. Pflugers Arch. 1995, 430, 365–373. [Google Scholar] [CrossRef]
- Cuajungco, M.P.; Grimm, C.; Heller, S. TRP channels as candidates for hearing and balance abnormalities in vertebrates. Biochim. Biophys. Acta 2007, 1772, 1022–1027. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Montell, C. TRP channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef]
- Corey, D.P. New TRP channels in hearing and mechanosensation. Neuron 2003, 39, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Waguespack, J.R.; Ricci, A.J. Aminoglycoside ototoxicity: Permeant drugs cause permanent hair cell loss. J. Physiol. 2005, 567 Pt 2, 359–360. [Google Scholar] [CrossRef] [PubMed]
- Raisinghani, M.; Premkumar, L.S. Block of native and cloned vanilloid receptor 1 (TRPV1) by aminoglycoside antibiotics. Pain 2005, 113, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Dahan, D.; Cardouat, G.; Gillibert-Duplantier, J.; Marthan, R.; Savineau, J.P.; Ducret, T. Involvement of TRPV1 and TRPV4 channels in migration of rat pulmonary arterial smooth muscle cells. Pflugers Arch. 2012, 464, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Myrdal, S.E.; Steyger, P.S. TRPV1 regulators mediate gentamicin penetration of cultured kidney cells. Hear. Res. 2005, 204, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Vriens, J.; Appendino, G.; Nilius, B. Pharmacology of vanilloid transient receptor potential cation channels. Mol. Pharmacol. 2009, 75, 1262–1279. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Park, C.; Kim, S.J.; Kim, H.J.; Oh, G.S.; Shen, A.; So, H.S.; Park, R. Different uptake of gentamicin through TRPV1 and TRPV4 channels determines cochlear hair cell vulnerability. Exp. Mol. Med. 2013, 45, e12. [Google Scholar] [CrossRef] [PubMed]
- Karasawa, T.; Wang, Q.; Fu, Y.; Cohen, D.M.; Steyger, P.S. TRPV4 enhances the cellular uptake of aminoglycoside antibiotics. J. Cell Sci. 2008, 121 Pt 17, 2871–2879. [Google Scholar] [CrossRef]
- Krause, K.M.; Serio, A.W.; Kane, T.R.; Connolly, L.E. Aminoglycosides: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, a027029. [Google Scholar] [CrossRef]
- Reyman, M.; van Houten, M.A.; Watson, R.L.; Chu, M.; Arp, K.; de Waal, W.J.; Schiering, I.; Plotz, F.B.; Willems, R.J.L.; van Schaik, W.; et al. Effects of early-life antibiotics on the developing infant gut microbiome and resistome: A randomized trial. Nat. Commun. 2022, 13, 893. [Google Scholar] [CrossRef]
- Nagpal, R.; Mishra, S.K.; Deep, G.; Yadav, H. Role of TRP Channels in Shaping the Gut Microbiome. Pathogens 2020, 9, 753. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).