
Iron Dyshomeostasis in Neurodegeneration with Brain Iron Accumulation (NBIA): Is It the Cause or the Effect?



Abstract
1. Introduction
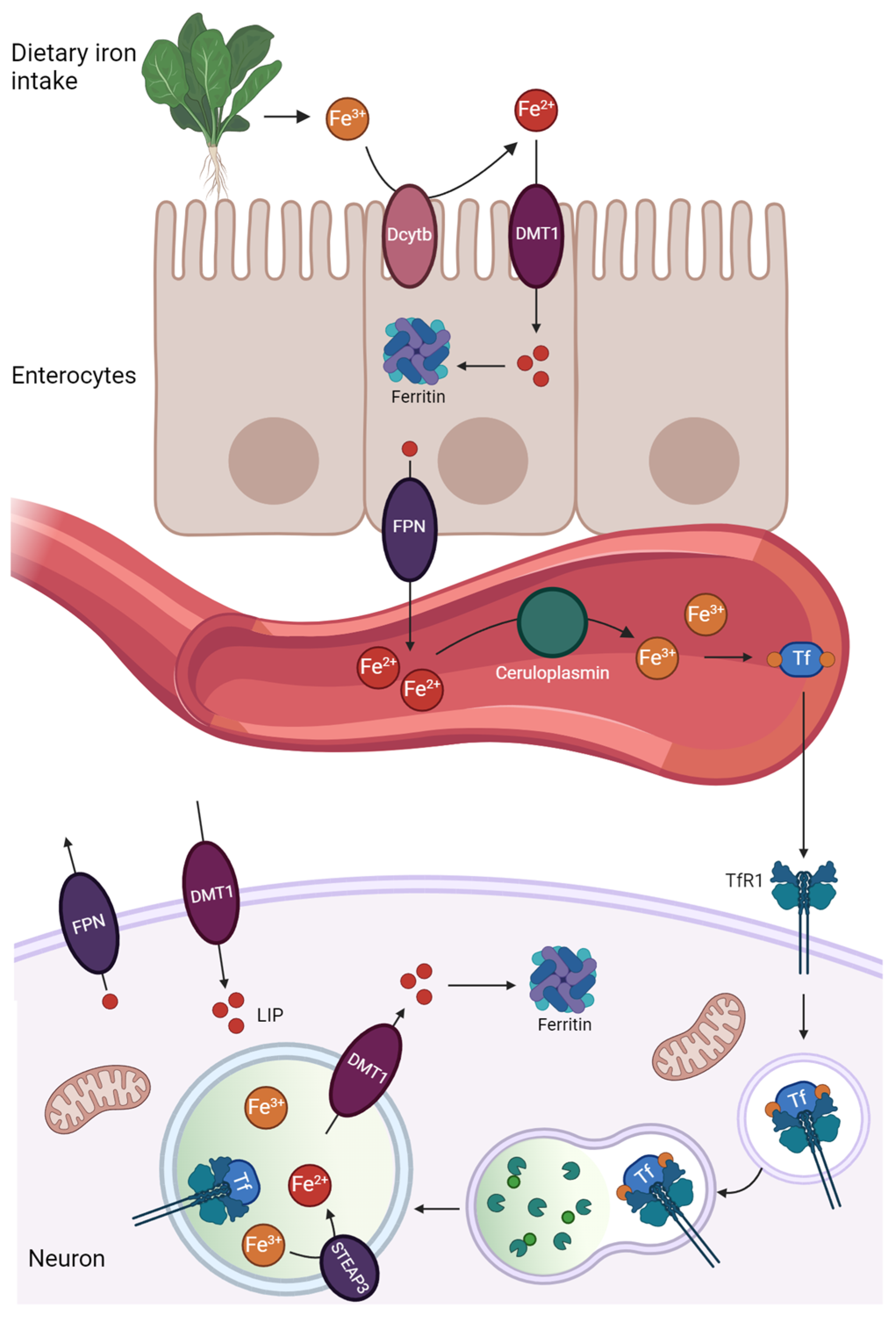
2. Iron Homeostasis
Mitochondria and Lysosomes Participation in Iron Homeostasis
3. Iron Dyshomeostasis and Ferroptosis
4. NBIA, Clinical and Histopathological Aspects
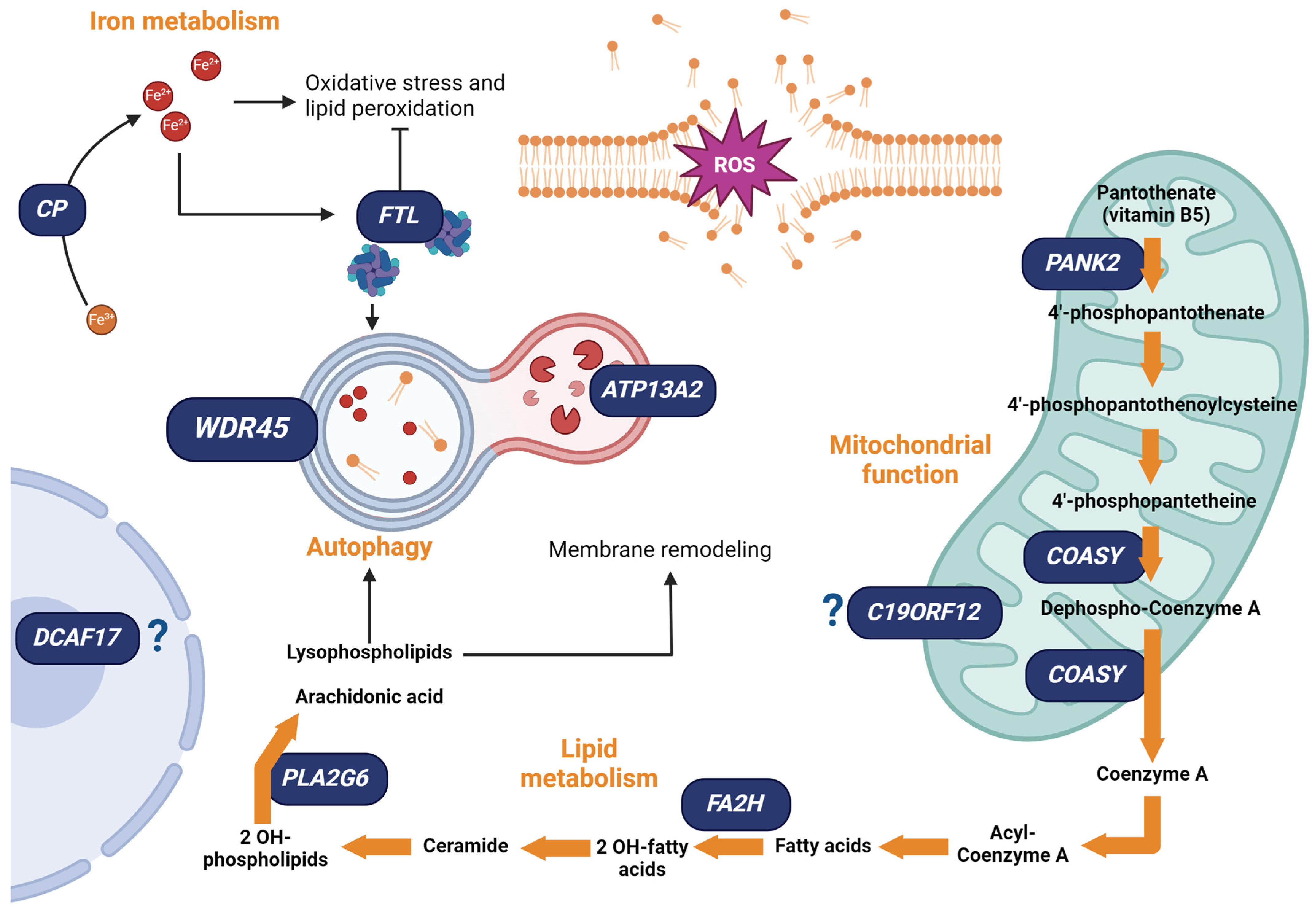
5. NBIA: Genetic and Physiopathological Aspects
5.1. NBIA Associated with Iron Homeostasis
5.2. NBIA Associated with Lipid Metabolism
Lipid and Iron Crosstalk in NBIA
5.3. NBIA Associated with Mitochondrial Function
Mitochondria and Iron Homeostasis in NBIA
5.4. NBIA Associated with Autophagy
5.4.1. Autophagy and Iron Homeostasis in NBIA
5.4.2. Mitochondria–Lysosomes Interplay in Iron Metabolism
5.5. NBIA Associated with DCAF17
6. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anderson, E.R.; Shah, Y.M. Iron homeostasis in the liver. Compr. Physiol. 2013, 3, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Hare, D.; Ayton, S.; Bush, A.; Lei, P. A delicate balance: Iron metabolism and diseases of the brain. Front. Aging Neurosci. 2013, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Illés, E.; Patra, S.G.; Marks, V.; Mizrahi, A.; Meyerstein, D. The FeII(citrate) Fenton reaction under physiological conditions. J. Inorg. Biochem. 2020, 206, 111018. [Google Scholar] [CrossRef] [PubMed]
- Kehrer, J.P. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology 2000, 149, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, R.S.; Han, S.M.; Leeuwenburgh, C.; Xiao, R. Iron homeostasis and organismal aging. Ageing Res. Rev. 2021, 72, 101510. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Dhorajia, V.V.; Kim, J.; Kim, Y. Mitochondrial iron metabolism and neurodegenerative diseases. Neurotoxicology 2022, 88, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yu, X.; Xie, J.; Xu, H. New Insights into the Role of Ferritin in Iron Homeostasis and Neurodegenerative Diseases. Mol. Neurobiol. 2021, 58, 2812–2823. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.Y.; Oliva, C.R.; Flor, S.; Griguer, C.E. Mitoferrin, Cellular and Mitochondrial Iron Homeostasis. Cells 2022, 11, 3464. [Google Scholar] [CrossRef] [PubMed]
- Rizzollo, F.; More, S.; Vangheluwe, P.; Agostinis, P. The lysosome as a master regulator of iron metabolism. Trends Biochem. Sci. 2021, 46, 960–975. [Google Scholar] [CrossRef]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef]
- Yambire, K.F.; Rostosky, C.; Watanabe, T.; Pacheu-Grau, D.; Torres-Odio, S.; Sanchez-Guerrero, A.; Senderovich, O.; Meyron-Holtz, E.G.; Milosevic, I.; Frahm, J.; et al. Impaired lysosomal acidification triggers iron deficiency and inflammation in vivo. Elife 2019, 8, e51031. [Google Scholar] [CrossRef]
- Wolff, N.A.; Garrick, M.D.; Zhao, L.; Garrick, L.M.; Ghio, A.J.; Thévenod, F. A role for divalent metal transporter (dmt1) in mitochondrial uptake of iron and manganese. Sci. Rep. 2018, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Kim, S.; Peng, W.; Krainc, D. Regulation and Function of Mitochondria–Lysosome Membrane Contact Sites in Cellular Homeostasis. Trends Cell Biol. 2019, 29, 500–513. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Hirose, W.; Ikematsu, K.; Tsuda, R. Age-associated increases in heme oxygenase-1 and ferritin immunoreactivity in the autopsied brain. Leg. Med. 2003, 5, S360–S366. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.R.; Snyder, B.S.; Arosio, P.; Loeffler, D.A.; LeWitt, P. A Quantitative Analysis of Isoferritins in Select Regions of Aged, Parkinsonian, and Alzheimer’s Diseased Brains. J. Neurochem. 1995, 65, 717–724. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef]
- Stoyanovsky, D.A.; Tyurina, Y.Y.; Shrivastava, I.; Bahar, I.; Tyurin, V.A.; Protchenko, O.; Jadhav, S.; Bolevich, S.B.; Kozlov, A.V.; Vladimirov, Y.A.; et al. Iron catalysis of lipid peroxidation in ferroptosis: Regulated enzymatic or random free radical reaction? Free Radic. Biol. Med. 2019, 133, 153–161. [Google Scholar] [CrossRef]
- Bouchaoui, H.; Mahoney-Sanchez, L.; Garçon, G.; Berdeaux, O.; Alleman, L.Y.; Devos, D.; Duce, J.A.; Devedjian, J.C. ACSL4 and the lipoxygenases 15/15B are pivotal for ferroptosis induced by iron and PUFA dyshomeostasis in dopaminergic neurons. Free Radic. Biol. Med. 2023, 195, 145–157. [Google Scholar] [CrossRef]
- Pratt, D.A. Targeting lipoxygenases to suppress ferroptotic cell death. Proc. Natl. Acad. Sci. USA 2023, 120, 10–12. [Google Scholar] [CrossRef] [PubMed]
- DeHart, D.N.; Fang, D.; Heslop, K.; Li, L.; Lemasters, J.J.; Maldonado, E.N. Opening of voltage dependent anion channels promotes reactive oxygen species generation, mitochondrial dysfunction and cell death in cancer cells. Biochem. Pharmacol. 2018, 148, 155–162. [Google Scholar] [CrossRef]
- Vianello, C.; Dal Bello, F.; Shin, S.H.; Schiavon, S.; Bean, C.; Magalhães Rebelo, A.P.; Knedlík, T.; Esfahani, E.N.; Costiniti, V.; Lacruz, R.S.; et al. High-Throughput Microscopy Analysis of Mitochondrial Membrane Potential in 2D and 3D Models. Cells 2023, 12, 1089. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, C.; Zhao, Y.; Gao, G. Mitochondria regulation in ferroptosis. Eur. J. Cell Biol. 2020, 99, 151058. [Google Scholar] [CrossRef]
- Jelinek, A.; Heyder, L.; Daude, M.; Plessner, M.; Krippner, S.; Grosse, R.; Diederich, W.E.; Culmsee, C. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic. Biol. Med. 2018, 117, 45–57. [Google Scholar] [CrossRef]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef]
- Ganguly, U.; Singh, S.; Bir, A.; Ghosh, A.; Chakrabarti, S.S.; Saini, R.V.; Saso, L.; Bisaglia, M.; Chakrabarti, S. Alpha-synuclein interaction with mitochondria is the final mechanism of ferroptotic death induced by erastin in SH-SY5Y cells. Free Radic. Res. 2024, 58, 217–228. [Google Scholar] [CrossRef]
- Krenn, M.A.; Schürz, M.; Teufl, B.; Uchida, K.; Eckl, P.M.; Bresgen, N. Ferritin-stimulated lipid peroxidation, lysosomal leak, and macroautophagy promote lysosomal “metastability” in primary hepatocytes determining in vitro cell survival. Free Radic. Biol. Med. 2015, 80, 48–58. [Google Scholar] [CrossRef]
- Mai, T.T.; Hamaï, A.; Hienzsch, A.; Cañeque, T.; Wicinski, J.; Cabaud, O.; Leroy, C.; David, A.; Ryo, A.; Ginestier, C.; et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat. Chem. 2018, 9, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Abarientos, A.; Hong, J.; Hadi Hashemi, S.; Yan, R.; Drager, N.; Leng, K.; Nalls, M.A.; Singleton, A.B.; Xu, K.; et al. Genome-wide CRISPRi/a screens in human neurons link lysosomal failure to ferroptosis. Nat. Neurosci. 2021, 24, 1020–1034. [Google Scholar] [CrossRef] [PubMed]
- Torii, S.; Shintoku, R.; Kubota, C.; Yaegashi, M.; Torii, R.; Sasaki, M.; Suzuki, T.; Mori, M.; Yoshimoto, Y.; Takeuchi, T.; et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem. J. 2016, 473, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef]
- Hayflick, S.J. A Brief History of NBIA Gene Discovery. J. Mov. Disord. 2023, 16, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, S.J.; Westaway, S.K.; Levinson, B.; Zhou, B.; Johnson, M.A.; Ching, K.H.L.; Gitschier, J. Genetic, Clinical, and Radiographic Delineation of Hallervorden–Spatz Syndrome. N. Engl. J. Med. 2003, 348, 33–40. [Google Scholar] [CrossRef]
- Hayflick, S.J.; Kurian, M.A.; Hogarth, P. Neurodegeneration with brain iron accumulation. Handb. Clin. Neurol. 2018, 147, 293–305. [Google Scholar] [CrossRef]
- Gregory, A.; Polster, B.J.; Hayflick, S.J. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J. Med. Genet. 2009, 46, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Pellecchia, M.T.; Valente, E.M.; Cif, L.; Salvi, S.; Albanese, A.; Scarano, V.; Bonuccelli, U.; Bentivoglio, A.R.; D’Amico, A.; Marelli, C.; et al. The diverse phenotype and genotype of pantothenate kinase-associated neurodegeneration. Neurology 2005, 64, 1810–1812. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.A.; Bhatia, K.P. Syndromes of Neurodegeneration with Brain Iron Accumulation. Semin. Pediatr. Neurol. 2012, 19, 57–66. [Google Scholar] [CrossRef]
- Lehericy, S.; Roze, E.; Goizet, C.; Mochel, F. MRI of neurodegeneration with brain iron accumulation. Curr. Opin. Neurol. 2020, 3, 462–473. [Google Scholar] [CrossRef]
- Koeppen, A.H.; Dickson, A.C. Iron in the Hallervorden-Spatz Syndrome. Pediatr. Neurol. 2001, 25, 148–155. [Google Scholar] [CrossRef]
- Javed, N.; Cascella, M. Neuroanatomy, Globus Pallidus; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Hayflick, S.J.; Hartman, M.; Coryell, J.; Gitschier, J.; Rowley, H. Brain MRI in neurodegeneration with brain iron accumulation with and without PANK2 mutations. Am. J. Neuroradiol. 2006, 27, 1230–1233. [Google Scholar]
- Sadeh, M. Neurodegeneration associated with genetic defects in phospholipase A2. Neurology 2009, 73, 819. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Kawai, M.; Inoue, K.; Sasaki, R.; Arai, H.; Nanba, E.; Kuzuhara, S.; Ihara, Y.; Kanazawa, I.; Murayama, S. Widespread expression of α-synuclein and τ immunoreactivity in Hallervorden-Spatz syndrome with protracted clinical course. J. Neurol. Sci. 2000, 177, 48–59. [Google Scholar] [CrossRef]
- Arber, C.E.; Li, A.; Houlden, H.; Wray, S. Review: Insights into molecular mechanisms of disease in neurodegeneration with brain iron accumulation: Unifying theories. Neuropathol. Appl. Neurobiol. 2016, 42, 220–241. [Google Scholar] [CrossRef]
- Hinarejos, I.; Machuca-Arellano, C.; Sancho, P.; Espinós, C. Mitochondrial dysfunction, oxidative stress and neuroinflammation in neurodegeneration with brain iron accumulation (Nbia). Antioxidants 2020, 9, 1020. [Google Scholar] [CrossRef] [PubMed]
- Dusek, P.; Hofer, T.; Alexander, J.; Roos, P.M.; Aaseth, J.O. Cerebral Iron Deposition in Neurodegeneration. Biomolecules 2022, 12, 714. [Google Scholar] [CrossRef]
- White, K.N.; Conesa, C.; Sánchez, L.; Amini, M.; Farnaud, S.; Lorvoralak, C.; Evans, R.W. The transfer of iron between ceruloplasmin and transferrins. Biochim. Biophys. Acta-Gen. Subj. 2012, 1820, 411–416. [Google Scholar] [CrossRef]
- Marchi, G.; Busti, F.; Zidanes, A.L.; Castagna, A.; Girelli, D. Aceruloplasminemia: A severe neurodegenerative disorder deserving an early diagnosis. Front. Neurosci. 2019, 13, 325. [Google Scholar] [CrossRef]
- Holzman, G.; Gernon, C. Aceruloplasminemia. Appl. Radiol. 2010, 39, 28–29. [Google Scholar] [CrossRef]
- Niu, L.; Zhou, Y.; Lu, L.; Su, A.; Guo, X. Ceruloplasmin Deficiency Impaired Brain Iron Metabolism and Behavior in Mice. Cell Biochem. Biophys. 2022, 80, 385–393. [Google Scholar] [CrossRef]
- Yamamoto, K.; Yoshida, K.; Miyagoe, Y.; Ishikawa, A.; Hanaoka, K.; Nomoto, S.; Kaneko, K.; Ikeda, S.I.; Takeda, S. Quantitative evaluation of expression of iron-metabolism genes in ceruloplasmin-deficient mice. Biochim. Biophys. Acta-Mol. Basis Dis. 2002, 1588, 195–202. [Google Scholar] [CrossRef]
- Patel, B.N.; Dunn, R.J.; Jeong, S.Y.; Zhu, Q.; Julien, J.P.; David, S. Ceruloplasmin regulates iron levels in the CNS and prevents free radical injury. J. Neurosci. 2002, 22, 6578–6586. [Google Scholar] [CrossRef]
- Cadenas, B.; Fita-Torró, J.; Bermúdez-Cortés, M.; Hernandez-Rodriguez, I.; Fuster, J.L.; Llinares, M.E.; Galera, A.M.; Romero, J.L.; Pérez-Montero, S.; Tornador, C.; et al. L-ferritin: One gene, five diseases; from hereditary hyperferritinemia to hypoferritinemia—Report of new cases. Pharmaceuticals 2019, 12, 17. [Google Scholar] [CrossRef]
- Carmona, U.; Li, L.; Zhang, L.; Knez, M. Ferritin light-chain subunits: Key elements for the electron transfer across the protein cage. Chem. Commun. 2014, 50, 15358–15361. [Google Scholar] [CrossRef] [PubMed]
- Muhoberac, B.B.; Vidal, R. Abnormal iron homeostasis and neurodegeneration. Front. Aging Neurosci. 2013, 5, 32. [Google Scholar] [CrossRef]
- Muhoberac, B.B.; Vidal, R. Iron, Ferritin, Hereditary Ferritinopathy, and Neurodegeneration. Front. Neurosci. 2019, 13, 1195. [Google Scholar] [CrossRef]
- Barbeito, A.G.; Garringer, H.J.; Baraibar, M.A.; Gao, X.; Arredondo, M.; Núñez, M.T.; Smith, M.A.; Ghetti, B.; Vidal, R. Abnormal iron metabolism and oxidative stress in mice expressing a mutant form of the ferritin light polypeptide gene. J. Neurochem. 2009, 109, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.A.; Stolzing, A. The role of lipid metabolism in aging, lifespan regulation, and age-related disease. Aging Cell 2019, 18, e13048. [Google Scholar] [CrossRef]
- Galper, J.; Dean, N.J.; Pickford, R.; Lewis, S.J.G.; Halliday, G.M.; Kim, W.S.; Dzamko, N. Lipid pathway dysfunction is prevalent in patients with Parkinson’s disease. Brain 2022, 145, 3472–3487. [Google Scholar] [CrossRef]
- Vieira, S.R.L.; Schapira, A.H.V. Glucocerebrosidase mutations and Parkinson disease. J. Neural Transm. 2022, 129, 1105–1117. [Google Scholar] [CrossRef]
- Deng, X.; Yuan, L.; Jankovic, J.; Deng, H. The role of the PLA2G6 gene in neurodegenerative diseases. Ageing Res. Rev. 2023, 89, 101957. [Google Scholar] [CrossRef] [PubMed]
- Kiel, M.; Kozaric, A.; Singer-Berk, M.; Wood, J.; Evangelista, E.; Hope, A.; Panwala, L.; Heinrich, S.; Baxter, S. P170: An estimation of global genetic prevalence of PLA2G6-associated neurodegeneration. Genet. Med. Open 2024, 2, 101067. [Google Scholar] [CrossRef]
- Malley, K.R.; Koroleva, O.; Miller, I.; Sanishvili, R.; Jenkins, C.M.; Gross, R.W.; Korolev, S. The structure of iPLA2β reveals dimeric active sites and suggests mechanisms of regulation and localization. Nat. Commun. 2018, 9, 765. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tan, J.; Tang, B.; Guo, J. Unveiling the role of iPLA2β in neurodegeneration: From molecular mechanisms to advanced therapies. Pharmacol. Res. 2024, 202, 107114. [Google Scholar] [CrossRef]
- Malik, I.; Turk, J.; Mancuso, D.J.; Montier, L.; Wohltmann, M.; Wozniak, D.F.; Schmidt, R.E.; Gross, R.W.; Kotzbauer, P.T. Disrupted membrane homeostasis and accumulation of ubiquitinated proteins in a mouse model of infantile neuroaxonal dystrophy caused by PLA2G6 mutations. Am. J. Pathol. 2008, 172, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Kinghorn, K.J.; Castillo-Quan, J.I.; Bartolome, F.; Angelova, P.R.; Li, L.; Pope, S.; Cochemé, H.M.; Khan, S.; Asghari, S.; Bhatia, K.P.; et al. Loss of PLA2G6 leads to elevated mitochondrial lipid peroxidation and mitochondrial dysfunction. Brain 2015, 138, 1801–1816. [Google Scholar] [CrossRef]
- Gregory, A.; Venkateswaran, S.; Hayflick, S.J. Fatty Acid Hydroxylase-Associated Neurodegeneration Summary Genetic Counseling Suggestive Findings. In GeneReviews; Adam, M.P., Feldman, J., Mirzaa, G.M., Eds.; University of Washington: Seattle, WA, USA, 2020; pp. 1–16. [Google Scholar]
- Eckhardt, M. Fatty acid 2-Hydroxylase and 2-Hydroxylated Sphingolipids: Metabolism and Function in Health and Diseases. Int. J. Mol. Sci. 2023, 24, 4908. [Google Scholar] [CrossRef]
- Eckhardt, M.; Yaghootfam, A.; Fewou, S.N.; Zöller, I.; Gieselmann, V. A mammalian fatty acid hydroxylase responsible for the formation of α-hydroxylated galactosylceramide in myelin. Biochem. J. 2005, 388, 245–254. [Google Scholar] [CrossRef]
- Potter, K.A.; Kern, M.J.; Fullbright, G.; Bielawski, J.; Scherer, S.S.; Yum, S.W.; Li, J.J.; Cheng, H.; Han, X.; Venkata, J.K.; et al. Central nervous system dysfunction in a mouse model of Fa2H deficiency. Glia 2011, 59, 1009–1021. [Google Scholar] [CrossRef]
- Mandik, F.; Kanana, Y.; Rody, J.; Misera, S.; Wilken, B.; Laabs von Holt, B.-H.; Klein, C.; Vos, M. A new model for fatty acid hydroxylase-associated neurodegeneration reveals mitochondrial and autophagy abnormalities. Front. Cell Dev. Biol. 2022, 10, 1000553. [Google Scholar] [CrossRef] [PubMed]
- Rockfield, S.; Chhabra, R.; Robertson, M.; Rehman, N.; Bisht, R.; Nanjundan, M. Links between iron and lipids: Implications in some major human diseases. Pharmaceuticals 2018, 11, 113. [Google Scholar] [CrossRef]
- Cheli, V.T.; Correale, J.; Paez, P.M.; Pasquini, J.M. Iron Metabolism in Oligodendrocytes and Astrocytes, Implications for Myelination and Remyelination. ASN Neuro 2020, 12, 1759091420962681. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Dogon, G.; Rigal, E.; Zeller, M.; Cottin, Y.; Vergely, C. Lipid Peroxidation and Iron Metabolism: Two Corner stones in the Homeostasis Control of Ferroptosis. Int. J. Mol. Sci. 2023, 24, 449. [Google Scholar] [CrossRef]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer disease brain. Free Radic. Biol. Med. 2013, 62, 157–169. [Google Scholar] [CrossRef]
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More Than Just a Powerhouse. Curr. Biol. 2006, 16, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.N.; Fiskum, G.; Flint Beal, M. Mitochondria in neurodegeneration: Bioenergetic function in cell life and death. J. Cereb. Blood Flow Metab. 1999, 19, 231–245. [Google Scholar] [CrossRef]
- Tatsuta, T.; Langer, T. Quality control of mitochondria: Protection against neurodegeneration and ageing. EMBO J. 2008, 27, 306–314. [Google Scholar] [CrossRef]
- Pan, S.; Zhu, C. Atypical pantothenate kinase-associated neurodegeneration with PANK2 mutations: Clinical description and a review of the literature. Neurocase 2020, 26, 175–182. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Rock, C.O.; Jackowski, S. Biochemical properties of human pantothenate kinase 2 isoforms and mutations linked to pantothenate kinase-associated neurodegeneration. J. Biol. Chem. 2006, 281, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Bum, S.H.; Senisterra, G.; Rabeh, W.M.; Vedadi, M.; Leonardi, R.; Zhang, Y.M.; Rock, C.O.; Jackowski, S.; Park, H.W. Crystal structures of human pantothenate kinases: Insights into allosteric regulation and mutations linked to a neurodegeneration disorder. J. Biol. Chem. 2007, 282, 27984–27993. [Google Scholar] [CrossRef]
- Johnson, M.A.; Kuo, Y.M.; Westaway, S.K.; Parker, S.M.; Ching, K.H.L.; Gitschier, J.; Hayflick, S.J. Mitochondrial localization of human PANK2 and hypotheses of secondary iron accumulation in pantothenate kinase-associated neurodegeneration. Ann. N. Y. Acad. Sci. 2004, 1012, 282–298. [Google Scholar] [CrossRef]
- Brunetti, D.; Dusi, S.; Morbin, M.; Uggetti, A.; Moda, F.; D’Amato, I.; Giordano, C.; d’Amati, G.; Cozzi, A.; Levi, S.; et al. Pantothenate kinase-associated neurodegeneration: Altered mitochondria membrane potential and defective respiration in Pank2 Knock-out mouse model. Hum. Mol. Genet. 2012, 21, 5294–5305. [Google Scholar] [CrossRef]
- Orellana, D.I.; Santambrogio, P.; Rubio, A.; Yekhlef, L.; Cancellieri, C.; Dusi, S.; Giannelli, S.G.; Venco, P.; Mazzara, P.G.; Cozzi, A.; et al. Coenzyme a corrects pathological defects in human neurons of PANK 2-associated neurodegeneration. EMBO Mol. Med. 2016, 8, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Arber, C.; Angelova, P.R.; Wiethoff, S.; Tsuchiya, Y.; Mazzacuva, F.; Preza, E.; Bhatia, K.P.; Mills, K.; Gout, I.; Abramov, A.Y.; et al. iPSC-derived neuronal models of PANK2-associated neurodegeneration reveal mitochondrial dysfunction contributing to early disease. PLoS ONE 2017, 12, e0184104. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, R.; Zhang, Y.M.; Rock, C.O.; Jackowski, S. Coenzyme A: Back in action. Prog. Lipid Res. 2005, 44, 125–153. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Liu, C.; Li, L.; Yang, M.; Jiang, N.; Luo, S.; Sun, L. Acyl-CoA synthase ACSL4: An essential target in ferroptosis and fatty acid metabolism. Chin. Med. J. 2023, 136, 2521–2537. [Google Scholar] [CrossRef] [PubMed]
- Cavestro, C.; Morra, F.; Legati, A.; D’Amato, M.; Nasca, A.; Iuso, A.; Lubarr, N.; Morrison, J.L.; Wheeler, P.G.; Serra-Juhé, C.; et al. Emerging variants, unique phenotypes, and transcriptomic signatures: An integrated study of COASY-Associated diseases. Ann. Clin. Transl. Neurol. 2024, 11, 1615–1629. [Google Scholar] [CrossRef]
- Berti, C.C.; Dallabona, C.; Lazzaretti, M.; Dusi, S.; Tosi, E.; Tiranti, V.; Goffrini, P. Modeling human Coenzyme a synthase mutation in yeast reveals altered mitochondrial function, lipid content and iron metabolism. Microb. Cell 2015, 2, 126–135. [Google Scholar] [CrossRef]
- Ramesh, R.; Deenadayalu, A.; Bhattacharjee, S.; Paramanandam, V. C19orf12 mutation causing mitochondrial membrane-protein Associated Neurodegeneration masquerading as spastic paraplegia. Park. Relat. Disord. 2021, 89, 146–147. [Google Scholar] [CrossRef] [PubMed]
- Fraser, S.; Koenig, M.; Farach, L.; Mancias, P.; Mowrey, K. A De Novo case of autosomal dominant mitochondrial membrane protein-associated neurodegeneration. Mol. Genet. Genom. Med. 2021, 9, e1706. [Google Scholar] [CrossRef]
- Balicza, P.; Bencsik, R.; Lengyel, A.; Gal, A.; Grosz, Z.; Csaban, D.; Rudas, G.; Danics, K.; Kovacs, G.G.; Molnar, M.J. Novel dominant MPAN family with a complex genetic architecture as a basis for phenotypic variability. Neurol. Genet. 2020, 6, e515. [Google Scholar] [CrossRef]
- Shao, C.; Zhu, J.; Ma, X.; Siedlak, S.L.; Cohen, M.L.; Lerner, A.; Wang, W. C19orf12 ablation causes ferroptosis in mitochondrial membrane protein-associated with neurodegeneration. Free Radic. Biol. Med. 2022, 182, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Venco, P.; Bonora, M.; Giorgi, C.; Papaleo, E.; Iuso, A.; Prokisch, H.; Pinton, P.; Tiranti, V. Mutations of C19orf12, Coding for a transmembrane glycine zipper containing mitochondrial protein, cause mis-localization of the protein, inability to respond to oxidative stress and increased mitochondrial Ca2+. Front. Genet. 2015, 6, 185. [Google Scholar] [CrossRef]
- Zanuttigh, E.; Derderian, K.; Güra, M.A.; Geerlof, A.; Di Meo, I.; Cavestro, C.; Hempfling, S.; Ortiz-Collazos, S.; Mauthe, M.; Kmieć, T.; et al. Identification of Autophagy as a Functional Target Suitable for the Pharmacological Treatment of Mitochondrial Membrane Protein-Associated Neurodegeneration (MPAN) In Vitro. Pharmaceutics 2023, 15, 267. [Google Scholar] [CrossRef]
- Sreejith, P.; Lolo, S.; Patten, K.R.; Gunasinghe, M.; More, N.; Pallanck, L.J.; Bharadwaj, R. Nazo, the Drosophila homolog of the NBIAmutated protein c19orf12, is required for triglyceride homeostasis. PLoS Genet. 2024, 20, e1011137. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef]
- Martini-Stoica, H.; Xu, Y.; Ballabio, A.; Zheng, H. The Autophagy–Lysosomal Pathway in Neurodegeneration: A TFEB Perspective. Trends Neurosci. 2016, 39, 221–234. [Google Scholar] [CrossRef]
- Cong, Y.; So, V.; Tijssen, M.A.J.; Verbeek, D.S.; Reggiori, F.; Mauthe, M. WDR45, one gene associated with multiple neurodevelopmental disorders. Autophagy 2021, 17, 3908–3923. [Google Scholar] [CrossRef]
- Proikas-Cezanne, T.; Takacs, Z.; Dönnes, P.; Kohlbacher, O. WIPI proteins: Essential PtdIns3P effectors at the nascent autophagosome. J. Cell Sci. 2015, 128, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Zhao, H.; Chen, D.; Zhang, H.; Zhao, Y.G. β-propeller proteins WDR45 and WDR45B regulate autophagosome maturation into autolysosomes in neural cells. Curr. Biol. 2021, 31, 1666–1677.e6. [Google Scholar] [CrossRef]
- Diaw, S.H.; Ganos, C.; Zittel, S.; Plötze-Martin, K.; Kulikovskaja, L.; Vos, M.; Westenberger, A.; Rakovic, A.; Lohmann, K.; Dulovic-Mahlow, M. Mutant WDR45 Leads to Altered Ferritinophagy and Ferroptosis in β-Propeller Protein-Associated Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 9524. [Google Scholar] [CrossRef]
- Zhao, Y.G.; Sun, L.; Miao, G.; Ji, C.; Zhao, H.; Sun, H.; Miao, L.; Yoshii, S.R.; Mizushima, N.; Zhang, H. The autophagy gene Wdr45/Wipi4 regulates learning and memory function and axonal homeostasis. Autophagy 2015, 11, 881–890. [Google Scholar] [CrossRef] [PubMed]
- van Veen, S.; Martin, S.; Van den Haute, C.; Benoy, V.; Lyons, J.; Vanhoutte, R.; Kahler, J.P.; Decuypere, J.P.; Gelders, G.; Lambie, E.; et al. ATP13A2 deficiency disrupts lysosomal polyamine export. Nature 2020, 578, 419–424. [Google Scholar] [CrossRef]
- Croucher, K.M.; Fleming, S.M. ATP13A2 (PARK9) and basal ganglia function. Front. Neurol. 2023, 14, 1252400. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.; Heimbach, A.; Gründemann, J.; Stiller, B.; Hampshire, D.; Cid, L.P.; Goebel, I.; Mubaidin, A.F.; Wriekat, A.L.; Roeper, J.; et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006, 38, 1184–1191. [Google Scholar] [CrossRef]
- Gusdon, A.M.; Zhu, J.; Van Houten, B.; Chu, C.T. ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol. Dis. 2012, 45, 962–972. [Google Scholar] [CrossRef]
- Vrijsen, S.; Besora-Casals, L.; Van Veen, S.; Zielich, J.; Van Den Haute, C.; Hamouda, N.N.; Fischer, C.; Ghesquière, B.; Tournev, I.; Agostinis, P.; et al. ATP13A2-mediated endo-lysosomal polyamine export counters mitochondrial oxidative stress. Proc. Natl. Acad. Sci.USA 2020, 117, 31198–31207. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yu, C.; Kang, R.; Kroemer, G.; Tang, D. Cellular degradation systems in ferroptosis. Cell Death Differ. 2021, 28, 1135–1148. [Google Scholar] [CrossRef] [PubMed]
- Kurz, T.; Eaton, J.W.; Brunk, U.T. The role of lysosomes in iron metabolism and recycling. Int. J. Biochem. Cell Biol. 2011, 43, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Kurz, T.; Terman, A.; Gustafsson, B.; Brunk, U.T. Lysosomes in iron metabolism, ageing and apoptosis. Histochem. Cell Biol. 2008, 129, 389–406. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Hara, Y.; Yanatori, I.; Tanaka, A.; Kishi, F.; Lemasters, J.J.; Nishina, S.; Sasaki, K.; Hino, K. Iron loss triggers mitophagy through induction of mitochondrial ferritin. EMBO Rep. 2020, 21, e50202. [Google Scholar] [CrossRef] [PubMed]
- Deus, C.M.; Yambire, K.F.; Oliveira, P.J.; Raimundo, N. Mitochondria–Lysosome Crosstalk: From Physiology to Neurodegeneration. Trends Mol. Med. 2020, 26, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Mosquera, L.; Yambire, K.F.; Couto, R.; Pereyra, L.; Pabis, K.; Ponsford, A.H.; Diogo, C.V.; Stagi, M.; Milosevic, I.; Raimundo, N. Mitochondrial respiratory chain deficiency inhibits lysosomal hydrolysis. Autophagy 2019, 15, 1572–1591. [Google Scholar] [CrossRef]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef]
- Messina, C. Woodhouse-Sakati syndrome: A review. Rev. Neurol. 2024; in press. [Google Scholar] [CrossRef]
- Kohil, A.; Abdallah, A.M.; Hussain, K.; Al-Shafai, M. Genetic epidemiology of Woodhouse-Sakati Syndrome in the Greater Middle East region and beyond: A systematic review. Orphanet J. Rare Dis. 2023, 18, 22. [Google Scholar] [CrossRef]
- Levi, S.; Ripamonti, M.; Moro, A.S.; Cozzi, A. Iron imbalance in neurodegeneration. Mol. Psychiatry 2024, 29, 1139–1152. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Gene (Protein) | NBIA Form | Protein Function | Cellular Phenotype |
| Ceruloplasmin | Aceruloplasminemia | Oxidation of Fe2+ to Fe3+; trafficking and translocation of iron. | Oxidative stress; lipid peroxidation; cell death. |
| Ferritin light chain (ferritin subunit) | Neuroferritinopathy | Iron binding and storage. | Oxidative stress; lipid peroxidation; decrease of ferritin receptor level; ferritin aggregation. |
| Phospholipase A2 group IV (Ca2+-independent phospholipase A2β) | PLA2G6-Associated Neurodegeneration | Hydrolysis of membrane phospholipids. | Lipid peroxidation; alteration of phospholipids metabolism; impairment of membranes dynamics; accumulation of ubiquitinated proteins, α-synuclein and phosphorylated tau, mitochondrial dysfunctions. |
| Fatty acid 2-hydroxylase | Fatty Acid Hydroxylase-Associated Neurodegeneration | Generation of 2-hydroxylated phospholipids. | Demyelination and degeneration of neuronal membranes; impairments of autophagy; mitochondrial morphology alterations. |
| Pantothenate kinase 2 | Pantothenate Kinase-Associated Neurodegeneration | Phosphorylation of pantothenate (Coenzyme a biosynthesis). | Oxidative stress; lipid peroxidation; energy imbalance; increased apoptosis; alteration of mitochondrial membrane potential; decreased mitochondrial respiration; cell death. |
| Coenzyme A Synthase | COASY Protein-Associated Neurodegeneration | Coenzyme a biosynthesis. | Oxidative stress; lipid dyshomeostasis; decreased mitochondrial respiration. |
| C19orf12 | Mitochondrial Membrane Protein-Associated Neurodegeneration | Putative function in the response to oxidative stress at the mitochondrial level. | Mitochondrial fragmentation; reduced oxygen consumption; lipid alterations; autophagic defects; accumulation of α-synuclein and phosphorylated tau. |
| WD repeat domain 45 | β-propeller protein-associated neurodegeneration | Binding with phosphatidylinositol-3-phosphate. Autophagosomes formation, expansion and closure. | Mitochondrial morphology alterations; increased apoptosis; autophagic flux dysfunctions. |
| ATP13A | Kufor–Rakeb disease | Translocation of ions and polyamines across the lysosomal membrane. | Oxidative stress; mitochondrial fragmentation; autophagic defects; accumulation of insoluble proteins and organelles. |
| DCAF17 | Woodhouse–Sakati syndrome | Nucleolar protein of unknown function. | DNA methylation, alterations of cell cycle progression; increased apoptosis. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agostini, F.; Sgalletta, B.; Bisaglia, M. Iron Dyshomeostasis in Neurodegeneration with Brain Iron Accumulation (NBIA): Is It the Cause or the Effect? Cells 2024, 13, 1376. https://doi.org/10.3390/cells13161376
Agostini F, Sgalletta B, Bisaglia M. Iron Dyshomeostasis in Neurodegeneration with Brain Iron Accumulation (NBIA): Is It the Cause or the Effect? Cells. 2024; 13(16):1376. https://doi.org/10.3390/cells13161376
Chicago/Turabian StyleAgostini, Francesco, Bibiana Sgalletta, and Marco Bisaglia. 2024. "Iron Dyshomeostasis in Neurodegeneration with Brain Iron Accumulation (NBIA): Is It the Cause or the Effect?" Cells 13, no. 16: 1376. https://doi.org/10.3390/cells13161376
APA StyleAgostini, F., Sgalletta, B., & Bisaglia, M. (2024). Iron Dyshomeostasis in Neurodegeneration with Brain Iron Accumulation (NBIA): Is It the Cause or the Effect? Cells, 13(16), 1376. https://doi.org/10.3390/cells13161376