
Population Genetics of California Gray Foxes Clarify Origins of the Island Fox


Abstract
:1. Introduction
2. Materials and Methods
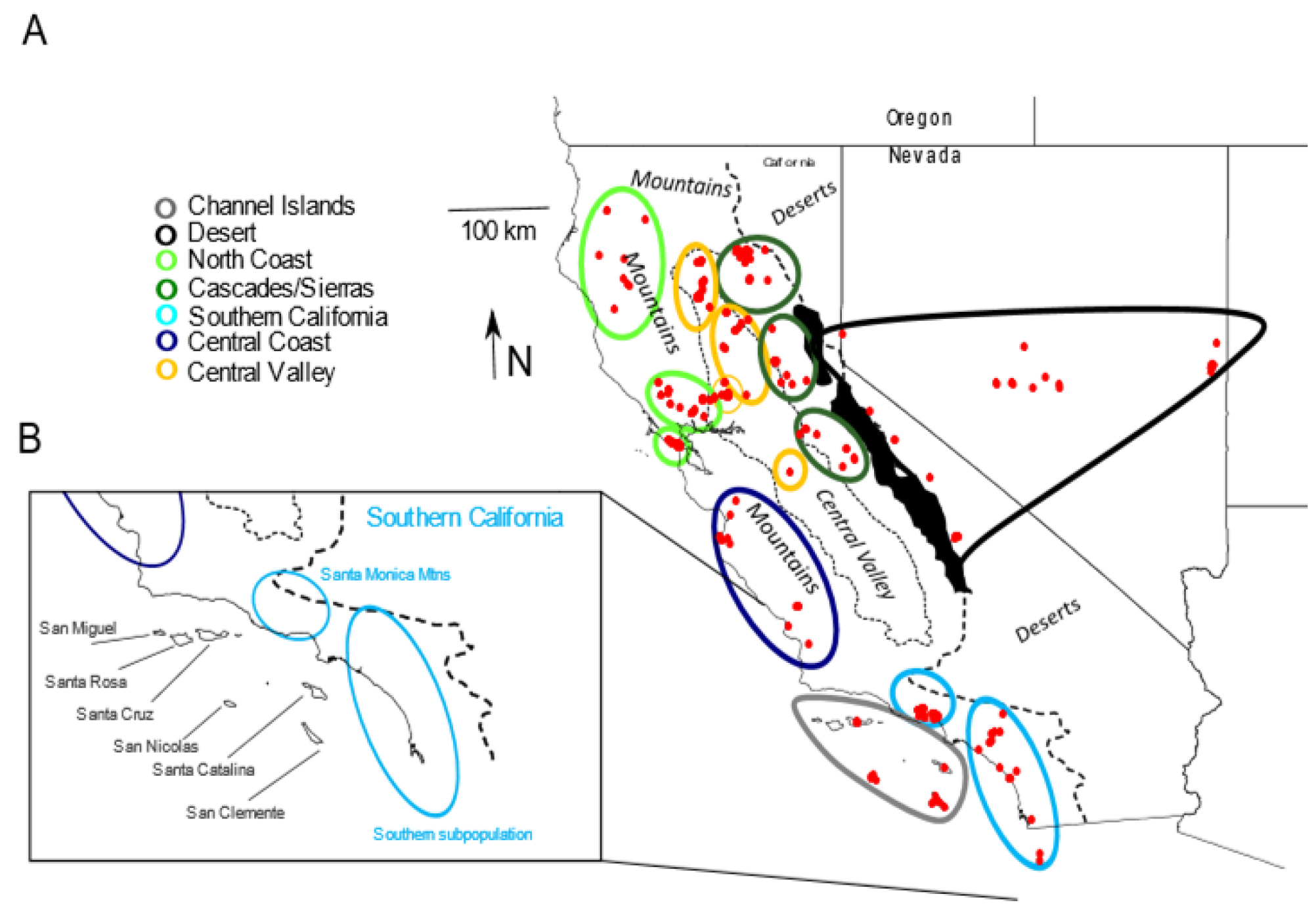
2.1. Samples
2.2. Laboratory Procedures
2.3. Data Analysis
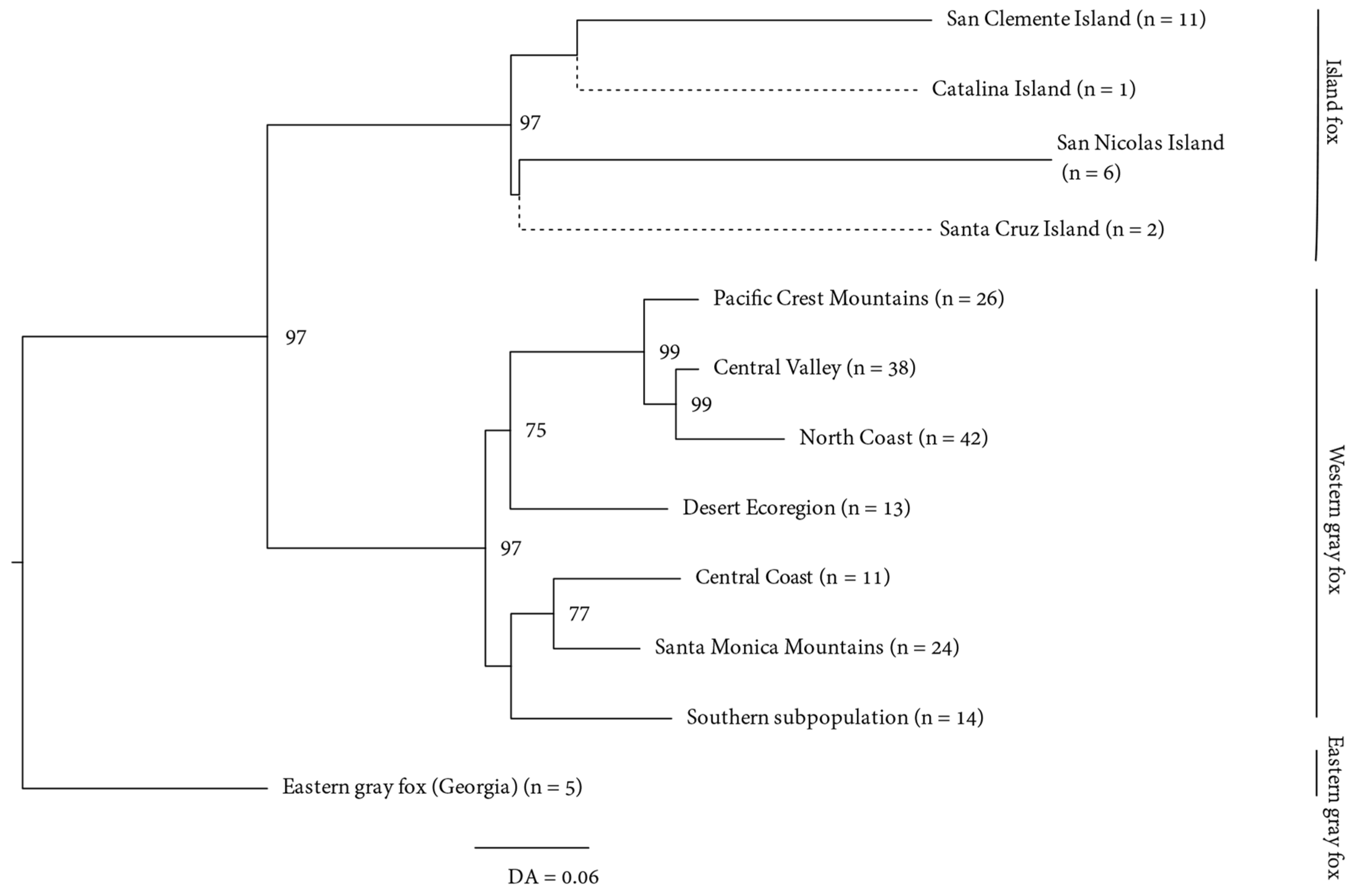
3. Results
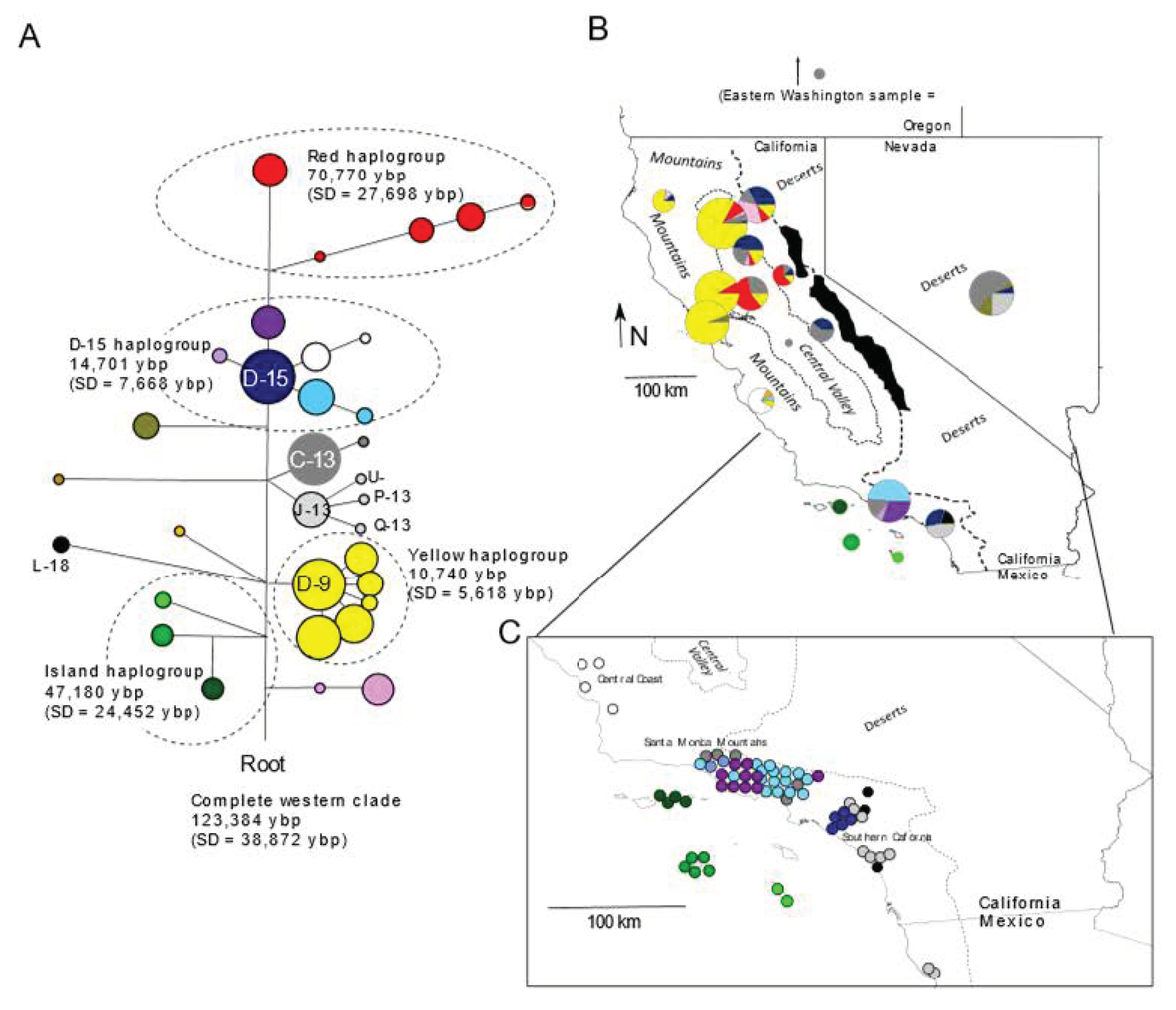
3.1. Mitochondrial
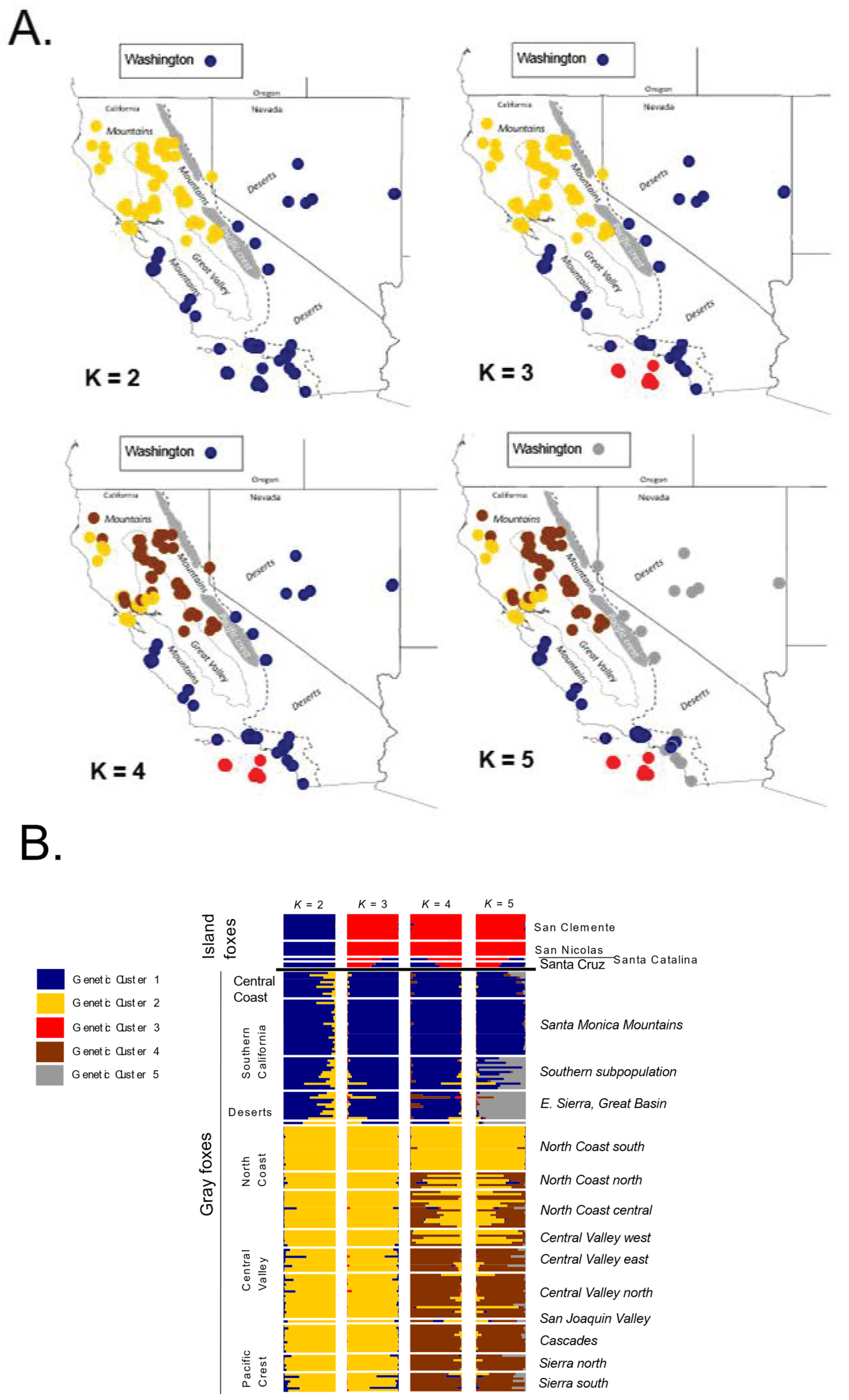
3.2. Microsatellites
3.2.1. Population Diversity
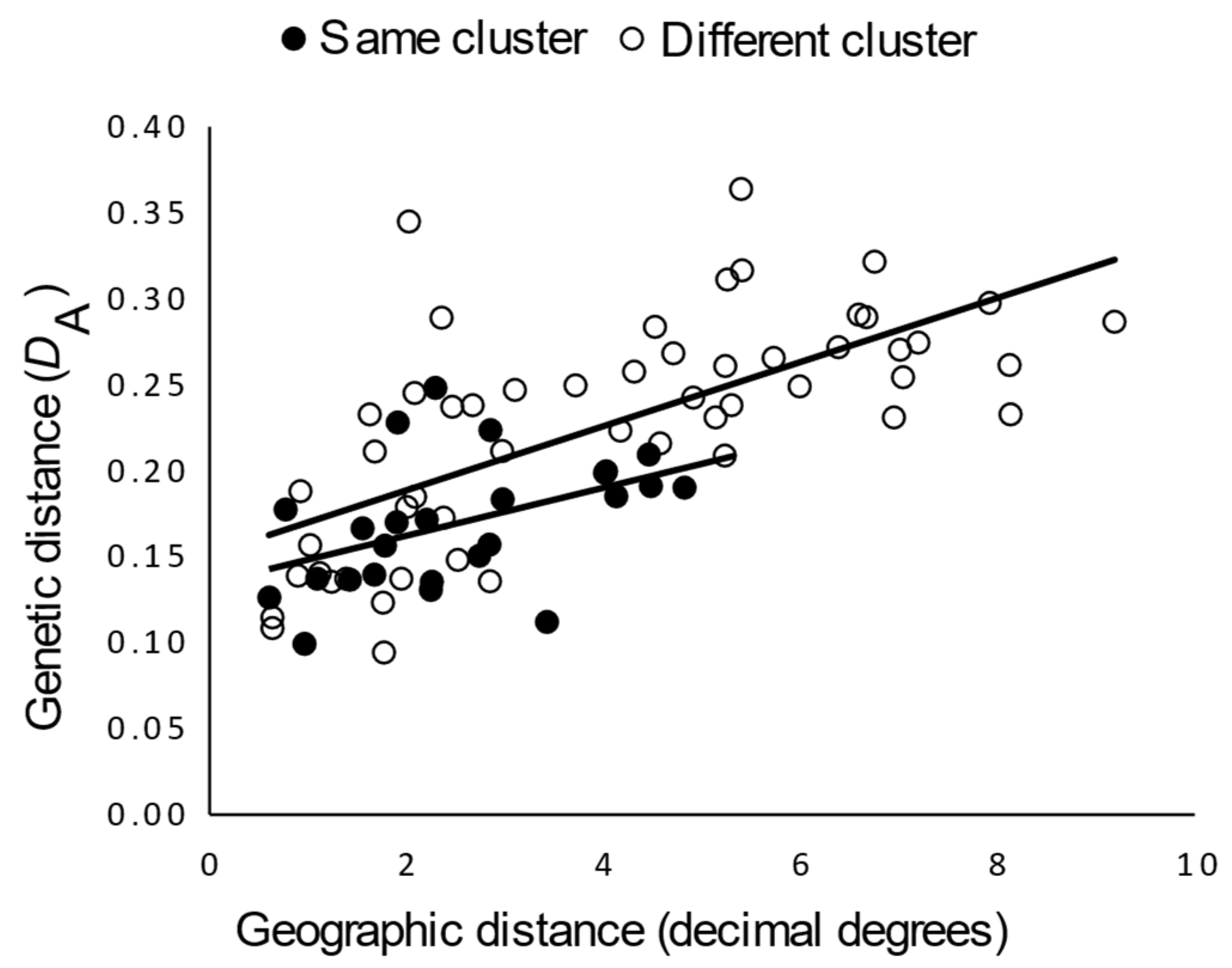
3.2.2. Genetic Structure
4. Discussion
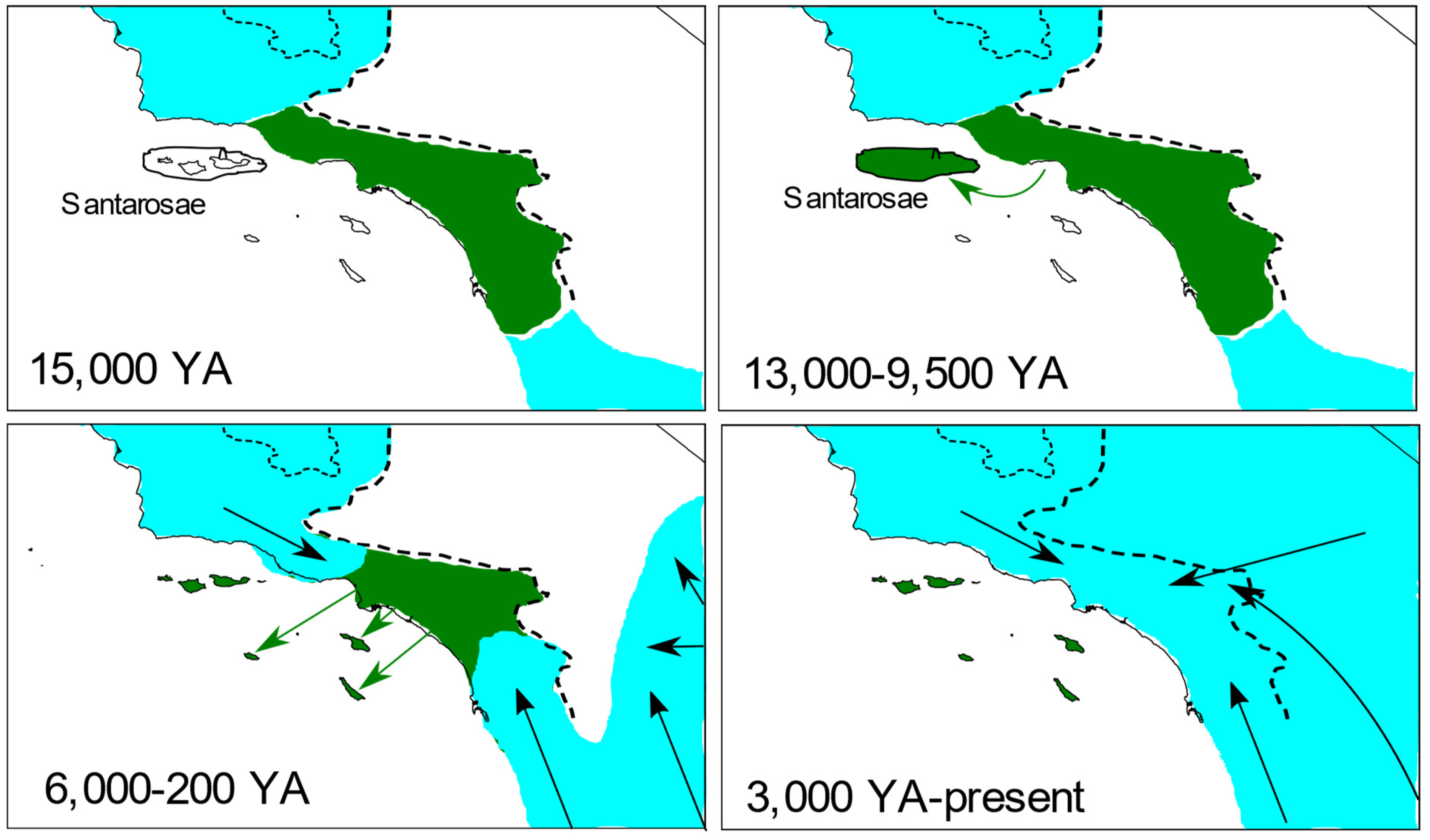
4.1. Origins of the Island Fox
4.2. Antiquity and Genetic Structure of California Gray Foxes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hewitt, G. The genetic legacy of the Quaternary ice ages. Nature 2000, 405, 907–913. [Google Scholar] [CrossRef]
- Calsbeek, R.; Thompson, J.N.; Richardson, J.E. Patterns of molecular evolution and diversification in a biodiversity hotspot: The California Floristic Province. Mol. Ecol. 2003, 12, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- Goddard, N.S.; Statham, M.J.; Sacks, B.N. Mitochondrial analysis of the most basal canid reveals deep divergence between eastern and western North American gray foxes (Urocyon spp.) and ancient roots in Pleistocene California. PLoS ONE 2015, 10, e0136329. [Google Scholar] [CrossRef] [Green Version]
- Sacks, B.N.; Bannasch, D.L.; Chomel, B.B.; Ernest, H.B. Coyotes demonstrate how habitat specialization by individuals of a generalist species can diversify populations in a heterogeneous ecoregion. Mol. Biol. Evol. 2008, 25, 1384–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacks, B.N.; Mitchell, K.J.; Quinn, C.B.; Hennelly, L.M.; Sinding, M.H.S.; Statham, M.J.; Preckler-Quisquater, S.; Fain, S.R.; Kistler, L.; Vanderzwan, S.L. Pleistocene origins, western ghost lineages, and the emerging phylogeographic history of the red wolf and coyote. Mol. Ecol. 2021, 30, 4292–4304. [Google Scholar] [CrossRef]
- Grinnell, J.; Dixon, J.S.; Linsdale, J.M. Fur-Bearing Mammals of California; University of California Press: Berkeley, CA, USA, 1937. [Google Scholar]
- Reding, D.M.; Castañeda-Rico, S.; Shirazi, S.; Hofman, C.A.; Cancellare, I.A.; Lance, S.L.; Beringer, J.; Clark, W.R.; Maldonado, J.E. Mitochondrial genomes of the United States distribution of Gray Fox (Urocyon cinereoargenteus) reveal a major phylogeographic break at the Great Plains Suture Zone. Front. Ecol. Evol. 2021, 9, 666800. [Google Scholar] [CrossRef]
- Hofman, C.A.; Rick, T.C.; Hawkins, M.T.; Funk, W.C.; Ralls, K.; Boser, C.L.; Collins, P.W.; Coonan, T.; King, J.L.; Morrison, S.A. Mitochondrial genomes suggest rapid evolution of dwarf California Channel Islands foxes (Urocyon littoralis). PLoS ONE 2015, 10, e0118240. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, D.A.; Lehman, N.; O’Brien, S.J.; Wayne, R.K. Genetic fingerprinting reflects population differentiation in the California Channel Island fox. Nature 1990, 344, 764–767. [Google Scholar] [CrossRef]
- Collins, P.W. Interaction between island foxes (Urocyon littoralis) and Indians on islands off the coast of southern California: I. Morphologic and archaeological evidence of human assisted dispersal. J. Ethnobiol. 1991, 11, 51–81. [Google Scholar]
- Collins, P.W. Interaction between island foxes (Urocyon littoralis) and Native Americans on islands off the coast of southern California: II. Ethnographic, archaeological, and historical evidence. J. Ethnobiol. 1991, 11, 205–229. [Google Scholar]
- Rick, T.C.; Erlandson, J.M.; Vellanoweth, R.L.; Braje, T.J.; Collins, P.W.; Guthrie, D.A.; Stafford, T.W. Origins and antiquity of the island fox (Urocyon littoralis) on California’s Channel Islands. Quat. Res. 2009, 71, 93–98. [Google Scholar] [CrossRef]
- Stock, C. Foxes and elephants of the Channel Islands. Los Angeles Cty. Mus. Q. 1943, 3, 6–9. [Google Scholar]
- Remington, C.L. Natural history and evolutionary genetics of the California Islands. Discovery 1971, 7, 2–18. [Google Scholar]
- Von Bloeker, J.C., Jr. Land mammals of the Southern California islands. In Proceedings of the Symposium on the Biology of the California Channel Islands, 29–30 October 1965; Philbrick, R.N., Ed.; Santa Barbara Botanic Garden: Santa Barbara, CA, USA, 1967. [Google Scholar]
- Hofman, C.A.; Rick, T.C.; Maldonado, J.E.; Collins, P.W.; Erlandson, J.M.; Fleischer, R.C.; Smith, C.; Sillett, T.S.; Ralls, K.; Teeter, W. Tracking the origins and diet of an endemic island canid (Urocyon littoralis) across 7300 years of human cultural and environmental change. Quat. Sci. Rev. 2016, 146, 147–160. [Google Scholar] [CrossRef]
- Wayne, R.K.; George, S.B.; Gilbert, D.; Collins, P.W.; Kovach, S.D.; Girman, D.; Lehman, N. A morphologic and genetic study of the island fox, Urocyon littoralis. Evolution 1991, 45, 1849–1868. [Google Scholar] [CrossRef] [PubMed]
- Funk, W.C.; Lovich, R.E.; Hohenlohe, P.A.; Hofman, C.A.; Morrison, S.A.; Sillett, T.S.; Ghalambor, C.K.; Maldonado, J.E.; Rick, T.C.; Day, M.D. Adaptive divergence despite strong genetic drift: Genomic analysis of the evolutionary mechanisms causing genetic differentiation in the island fox (Urocyon littoralis). Mol. Ecol. 2016, 25, 2176–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.A.; Ortega-Del Vecchyo, D.; Fan, Z.; Kim, B.Y.; Marsden, C.D.; Lohmueller, K.E.; Wayne, R.K. Genomic flatlining in the endangered island fox. Curr. Biol. 2016, 26, 1183–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.A.; Brown, C.; Kim, B.Y.; Lohmueller, K.E.; Wayne, R.K. Purging of strongly deleterious mutations explains long-term persistence and absence of inbreeding depression in island foxes. Curr. Biol. 2018, 28, 3487–3494.e3484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacks, B.N.; Brazeal, J.L.; Lewis, J.C. Landscape genetics of the nonnative red fox of California. Ecol. Evol. 2016, 6, 4775–4791. [Google Scholar] [CrossRef] [Green Version]
- Miles, K.A.; Holtz, M.N.; Lounsberry, Z.T.; Sacks, B.N. A paired comparison of scat-collecting versus scat-swabbing methods for noninvasive recovery of mesocarnivore DNA from an arid environment. Wildl. Soc. Bull. 2015, 39, 797–803. [Google Scholar] [CrossRef]
- Saillard, J.; Forster, P.; Lynnerup, N.; Bandelt, H.-J.; Nørby, S. mtDNA variation among Greenland Eskimos: The edge of the Beringian expansion. Am. J. Hum. Genet. 2000, 67, 718–726. [Google Scholar] [CrossRef] [Green Version]
- Forster, P.; Röhl, A.; Lünnemann, P.; Brinkmann, C.; Zerjal, T.; Tyler-Smith, C.; Brinkmann, B. A short tandem repeat–based phylogeny for the human Y chromosome. Am. J. Hum. Genet. 2000, 67, 182–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandelt, H.-J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, S.T. hp-rare 1.0: A computer program for performing rarefaction on measures of allelic richness. Mol. Ecol. Notes 2005, 5, 187–189. [Google Scholar] [CrossRef]
- Park, S. The Excel Microsatellite Toolkit. Trypanotolerance in West African Cattle and the Population Genetic Effects of Selection. Ph.D. Thesis, University of Dublin, Dublin, Ireland, 2001. [Google Scholar]
- Goudet, J. FSTAT (version 1.2): A computer program to calculate F-statistics. J. Hered. 1995, 86, 485–486. [Google Scholar] [CrossRef]
- Goudet, J. FSTAT, a Program to Estimate and Test Gene Diversities and Fixation Indices (Version 2.9. 3). Available online: http://www./unil.ch/izea/softwares/fstat.html 2001 (accessed on 5 February 2002).
- Rice, W.R. Analyzing tables of statistical tests. Evolution 1989, 43, 223–225. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Sacks, B.N.; Brown, S.K.; Ernest, H.B. Population structure of California coyotes corresponds to habitat-specific breaks and illuminates species history. Mol. Ecol. 2004, 13, 1265–1275. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [CrossRef]
- Smouse, P.E.; Long, J.C.; Sokal, R.R. Multiple regression and correlation extensions of the Mantel test of matrix correspondence. Syst. Zool. 1986, 35, 627–632. [Google Scholar] [CrossRef]
- Rosenberg, M.S.; Anderson, C.D. PASSaGE: Pattern analysis, spatial statistics and geographic exegesis. Version 2. Methods Ecol. Evol. 2011, 2, 229–232. [Google Scholar] [CrossRef]
- Takezaki, N.; Nei, M. Genetic distances and reconstruction of phylogenetic trees from microsatellite DNA. Genetics 1996, 144, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Langella, O. Populations, 1.2. 30. Available online: http://bioinformatics.org/~tryphon/populations/1999 (accessed on 7 February 2003).
- Goldstein, D.B.; Roemer, G.W.; Smith, D.A.; Reich, D.E.; Bergman, A.; Wayne, R.K. The use of microsatellite variation to infer population structure and demographic history in a natural model system. Genetics 1999, 151, 797–801. [Google Scholar] [CrossRef]
- Bolas, E.C.; Quinn, C.B.; Van Vuren, D.H.; Lee, A.; Vanderzwan, S.L.; Floyd, C.H.; Jones, K.L.; Shaskey, L.; Sacks, B.N. Pattern and timing of mitochondrial divergence of island spotted skunks on the California Channel Islands. J. Mammal. 2022, 103, 231–242. [Google Scholar] [CrossRef]
- Hendricks, S.A.; King, J.L.; Duncan, C.L.; Vickers, W.; Hohenlohe, P.A.; Davis, B.W. Genomic Assessment of Cancer Susceptibility in the Threatened Catalina Island Fox (Urocyon littoralis catalinae). Genes 2022, 13, 1496. [Google Scholar] [CrossRef]
- Farías, V.; Fuller, T.K.; Sauvajot, R.M. Activity and distribution of gray foxes (Urocyon cinereoargenteus) in southern California. Southwest. Nat. 2012, 57, 176–181. [Google Scholar] [CrossRef]
- Fuller, T.K. Variable home-range sizes of female gray foxes. J. Mammal. 1978, 59, 446–449. [Google Scholar] [CrossRef]
- Davis, F.; Stoms, D.; Hollander, A.; Thomas, K.; Stine, P.; Odion, D.; Borchert, M.; Thorne, J.; Gray, M.; Walker, R. The California Gap Analysis Project—Final Report; University of California: Santa Barbara, CA, USA, 1998. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | n | He (SE) | Ho (SE) | NA (SE) | AR (SE) | FIS (SE) |
|---|---|---|---|---|---|---|
| Island foxes (all) 1 | 20 | 0.43 (0.07) | 0.15 (0.02) | 3.15 (0.36) | 2.21 (0.19) | 0.59 (0.08) * |
| S. Clemente Island | 11 | 0.27 (0.06) | 0.19 (0.03) | 2 (0.21) | 0.29 (0.09) * | |
| S. Nicolas Island | 6 | 0.04 (0.02) | 0.04 (0.02) | 1.16 (0.09) | −0.14 (0.1) | |
| Deserts | 13 | 0.70 (0.06) | 0.58 (0.03) | 6.6 (0.63) | 3.51 (0.25) | 0.17 (0.04) * |
| North Coast | 42 | 0.61 (0.05) | 0.52 (0.02) | 5.7 (0.65) | 2.89 (0.22) | 0.17 (0.04) * |
| Cascades/Sierras | 26 | 0.61 (0.06) | 0.57 (0.02) | 6.4 (0.70) | 3.04 (0.26) | 0.07 (0.03) |
| Southern California (all) | 38 | 0.68 (0.05) | 0.58 (0.02) | 6.9 (0.63) | 3.32 (0.22) | 0.17 (0.05) * |
| Santa Monica Mtns | 24 | 0.64 (0.05) | 0.55 (0.02) | 5.6 (0.50) | 0.13 (0.05) * | |
| Southern subpopulation | 14 | 0.70 (0.05) | 0.61 (0.03) | 6.2 (0.73) | 0.12 (0.05) * | |
| Central Coast | 11 | 0.63 (0.06) | 0.62 (0.03) | 5.1 (0.48) | 3.16 (0.24) | 0.02 (0.05) |
| Central Valley | 38 | 0.63 (0.05) | 0.56 (0.02) | 6.7 (0.67) | 3.07 (0.24) | 0.12 (0.03) * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sacks, B.N.; Statham, M.J.; Serieys, L.E.K.; Riley, S.P.D. Population Genetics of California Gray Foxes Clarify Origins of the Island Fox. Genes 2022, 13, 1859. https://doi.org/10.3390/genes13101859
Sacks BN, Statham MJ, Serieys LEK, Riley SPD. Population Genetics of California Gray Foxes Clarify Origins of the Island Fox. Genes. 2022; 13(10):1859. https://doi.org/10.3390/genes13101859
Chicago/Turabian StyleSacks, Benjamin N., Mark J. Statham, Laurel E. K. Serieys, and Seth P. D. Riley. 2022. "Population Genetics of California Gray Foxes Clarify Origins of the Island Fox" Genes 13, no. 10: 1859. https://doi.org/10.3390/genes13101859
APA StyleSacks, B. N., Statham, M. J., Serieys, L. E. K., & Riley, S. P. D. (2022). Population Genetics of California Gray Foxes Clarify Origins of the Island Fox. Genes, 13(10), 1859. https://doi.org/10.3390/genes13101859