Abstract
The NFIA (nuclear factor I/A) gene encodes for a transcription factor belonging to the nuclear factor I family and has key roles in various embryonic differentiation pathways. In humans, NFIA is the major contributor to the phenotypic traits of “Chromosome 1p32p31 deletion syndrome”. We report on two new cases with deletions involving NFIA without any other pathogenic protein-coding gene alterations. A cohort of 24 patients with NFIA haploinsufficiency as the sole anomaly was selected by reviewing the literature and public databases in order to analyze all clinical features reported and their relative frequencies. This process was useful because it provided an overall picture of the phenotypic outcome of NFIA haploinsufficiency and helped to define a cluster of phenotypic traits that can facilitate clinicians in identifying affected patients. NFIA haploinsufficiency can be suspected by a careful observation of the dysmorphisms (macrocephaly, craniofacial, and first-finger anomalies), and this potential diagnosis is strengthened by the presence of intellectual and developmental disabilities or other neurodevelopmental disorders. Further clues of NFIA haploinsufficiency can be provided by instrumental tests such as MRI and kidney urinary tract ultrasound and confirmed by genetic testing.
1. Introduction
“Chromosome 1p32p31 deletion syndrome”, also referred as “brain malformations with or without urinary tract defects” (BRMUTD) (OMIM # 613735), is a genomic disorder characterized by macrocephaly, CNS malformations, congenital anomalies of the kidneys and the urinary tract, developmental delay (DD), intellectual disability (ID), and facial dysmorphism. Other clinical findings may include cryptorchidism, inguinal hernia, anomalies of external genitalia, cutis marmorata, syringomyelia, congenital heart defects, and Juvenile Moyamoya [1,2,3,4,5,6,7]. The deleted region includes the NFIA gene (nuclear factor I/A) (OMIM * 600727; ENSG00000162599), which is responsible for the main phenotypic traits of this syndrome.
NFIA is a gene located on chromosome 1p31.3 that encodes for a transcription factor belonging to the nuclear factor I family of dimeric DNA binding proteins, along with NFIB, NFIC, and NFIX [8]. This protein has three domains: (1) an N-terminal highly conserved region, whose function is not known; (2) a DNA binding/dimerization domain (MH1), and (3) a C-terminal transactivation and/or repression domain (CTF_NFI). The MH1 domain binds to a nucleotide consensus sequence within the promoter region of several genes, whereas the CTF_NFI domain activates basal transcription factors at transcription start sites by displacing repressive histones from target genes and interacting with other coactivator proteins [9]. NFIA gives rise to 31 transcript variants, but the functional significance of each isoform has not been yet clarified. The MANE transcript (matched annotation from NCBI and EMBL-EBI) is NFIA-207 (ENST00000403491.8), whichis encoded by 11 exons, extending from position 61,548,233 to 61,928,460 (GRCh37/hg19), and has ubiquitous expression (https://www.gtexportal.org/, accessed on 20 September 2022).
According to in vitro analyses and studies of animal models, NFIA has a key role in various differentiation pathways during embryogenesis. It is a crucial regulator of articular cartilage differentiation [10] and controls the adipogenic and myogenic gene program to ensure brown and beige adipocyte differentiation [11]. Nfia knock-out mice show hydrocephalus and agenesis of the corpus callosum, and, at reduced penetrance, exhibit abnormalities of the ureteropelvic and ureterovesical junctions, as well as bifid and megaureters [1,8]. NFI proteins play an important role in the development of the central nervous system (CNS), including axon guidance with outgrowth and in glial or neuronal cell differentiation and migration [8,12,13,14,15].
In this paper, we focused on cases with NFIA haploinsufficiency as the sole anomaly, excluding all those patients with other pathogenic protein-coding gene alterations. We report on two new individuals and reviewed the literature and public databases to better delineate the phenotypic spectrum of NFIA haploinsufficiency and its contribution to “Chromosome 1p32p31 deletion syndrome”.
2. Materials and Methods
2.1. Molecular Analysis
Array CGH was performed. Genomic DNA of the patients was isolated from peripheral blood by standard methods. DNA from healthy subjects was used as controls. Test and reference DNA were differentially labeled with Cy5-dCTP or with Cy3-dCTP using random primer labeling and applied to 60K arrays, according to the manufacturer’s protocol (case 1: Agilent, Santa Clara, CA, USA; case 2: Technogenetics, Milan, Italy). For each case, the array-CGH was repeated twice in order to confirm the results. Since quality of the experiment influences the Copy Number Variants (CNVs) (i.e., number and type of calls and probes included/excluded in each call), we elaborated only experiments that met the ‘excellent’ criteria as determined by the QC report. The CNV analysis was performed according to the guidelines of the Italian Society of Human Genetics (https://www.sigu.net, accessed on 20 September 2022) and to the American College of Medical Genetics guidelines [16].
2.2. Bioinformatic Analyses
CNVs classification was performed using the Database of Genomic Variants (DGV) (http://projects.tcag.ca/variation, accessed on 20 September 2022) and the University of California Santa Cruz (UCSC) Genome Browser (https://genome.ucsc.edu, accessed on 20 September 2022).PubMed and Online Mendelian Inheritance in Man (OMIM) were also consulted for evaluating genotype–phenotype association.
Our search for patients with NFIA haploinsufficiency, including CNVs and SNVs (single nucleotide variants), was performed in the DECIPHER (https://www.deciphergenomics.org/, accessed on 20 September 2022), ClinVar, Leiden Open Variation, and HGMD databases.
NFIA transcripts were identified using Ensembl. Data on the expression profiles were assessed using GTEx (https://www.gtexportal.org/, accessed on 20 September 2022) and the UCSC Genome Browser.
2.3. Patients
2.3.1. Patient 1
Patient 1 (P1) is a 2-year-old male toddler who was referred to the pediatric unit at the age of 10 months for macrocephaly.
At time of referral, his height was 81 cm (>98th centile), his weight was 12.6 kg (>98th centile), his head circumference was 53 cm (>97th centile), and his BMI was 19.2 (85–97th centile). The patient was born at 37 weeks of gestation with a normal delivery following a pregnancy complicated by cholecystitis at 34 weeks of gestation. His birth weight was 3390g (90th centile), length 49 cm (64th centile), head circumference was 38 cm (>100th centile). Family history was unremarkable.
On physical examination he presented with macrocephaly, scaphocephaly, a high and wide forehead with evident frontal bossing, and low-set ears. He showed brachydactyly with bilateral proximally placed first fingers and short lower limbs. The eye examination revealed the presence of a mild strabismus, whereas hearing screening, blood tests, and metabolic and hormonal work up were all normal. EEG revealed minor, nonspecific discharges in the fronto-central-parietal regions. No epileptic seizures have been recorded. A cranial ultrasound, performed at the age of 3 months, revealed sagittal synostosis, whereas the metopic, lambdoid, and coronal sutures were still open. A brain MRI showed diffuse corpus callosum hypoplasia and a dysmorphic aspect of the cerebral ventriculus with widening of the anterior portions of the horns. No Chiari I malformation was detected.
At the first evaluation, the patient showed normal psychomotor development, but, at follow-up (age 2 years), he presented mild/moderate DD with severe delay in language (absence of speech) and hyperactivity disorder. An appearance of congestion of the optic disc was reported in anocular ultrasound, but the ophthalmological examination was negative. A brain MRI showed no alteration of the cranial nerves but confirmed the dysmorphic aspects already described previously; in particular, it confirmed the hypoplasia of the cerebral commissural system (corpus callosum and anterior commissure) with dysmorphic appearance of the lateral ventricles. An EEG showed focal abnormalities in posterior-temporal regions, with more in the right hemisphere. However, the patient did not present with epileptic seizures.
2.3.2. Patient 2
Patient 2 (P2) is a 22-year-old womanfollowed by an outpatient basis for ID (IQ 62), oppositional defiant disorder (ODD), and trichotillomania. Physical examination showed a height of 158 cm (25th centile), weight of 47 kg (10th centile), and a normal head circumference at the upper limits (56.5 cm, 75th centile).
Slight facial dysmorphisms were shown, such as a high forehead, downslanting palpebral fissures, low-set and posteriorly rotated ears, a small and poorly structured philtrum, an open bite, hypotrophy of thenar and hypothenar eminences, and bilateral proximally placed first fingers. No skin dyschromia was revealed. An MRI showed a thin corpus callosum, Chiari malformation type I, hypoplastic rectus sinus, and a vicariant vessel along the right profile of the trunk. Neither ventricles anomies nor craniosynostosis were detected. Heart, lung, and abdomen ultrasound was normal. No epilepsy or EEG anomalies were reported. The patient was no longer available for follow up.
3. Results
Array CGH detected in P1 a de novo 1.484 kb deletion, starting from position 60,568,797 and ending at position 62,052,980 (GRCh37/hg19). The deleted region includes LINC01748, LOC101926964, NFIA-AS2, NFIA-AS1, and NFIA.
P2 showed a 27 kb deletion, starting from position 61,818,169 and ending at position 61,845,524 (GRCh37/hg19). The deleted region includes part of exon 5 and the entirety of exon 6 of NFIA (ENST00000403491.8; RefSeq ID NM_001134673). A segregation pattern could not beestablished.
These results are reported in Table 1, along with the genetic alterations of the selected patients [17,18,19,20,21,22,23,24,25].

Table 1.
Patients with solely NFIA haploinsufficiency.
The pathological traits of these patients have been grouped into macroareas: dysmorphisms (Table 2), neurological/behavioral abnormalities, renal/urinary trait alterations (Table 3), and CNS malformations (Table 4). In each table are listed only those patients where the clinical traits were evaluated. The few additional defects not included in these categories are reported in Table S1.
Table 2.
Dysmorphisms.
Table 3.
Neurological/behavioral abnormalities and renal/urinary tract defects.
Table 4.
CNS defects.
4. Discussion
Here, we report on two new individuals carrying microdeletions solely affecting NFIA without any other flanking protein-coding sequences. In order to further define the phenotypic effects of NFIA haploinsufficiency, we reviewed the literature and public databases excluding those patients without detailed clinical information or individuals with pathogenic deletions, translocations, inversions, or genetic variants disrupting additional protein-coding sequences [1,2,3,4,5,6,7,23,26,27,28,29] (Table S2). This careful selection allowed for the identification of a cohort of 24 patients with pathogenic alterations solely involving NFIA.
In Table 1 are listed our two cases along with the other 24 selected patients; their age ranges from 1- to 42-years-old; the prevalence of males and females is comparable (12 males versus 14 females); in four cases, the variants have been inherited, and twelve were de novo [17,18,19,20,21,22,23,24,25].
The precise localization of NFIA pathogenic variants is also reported. Since alteration of this gene hasoften been referred to alternative transcripts, we have converted each of them according to the NFIA-207 (ENST00000403491.8; RefSeq ID NM_001134673) (Table S3). As shown in Figure 1, SNVs do not have one hotspot and are spread over various functional domains; some microdeletions are intragenic and involve only few exons, while others disrupt all of the coding sequence with theirregulatory elements (NFIA-AS2, NFIA-AS1) and further noncoding RNAs (LINC01748, LOC101926964).
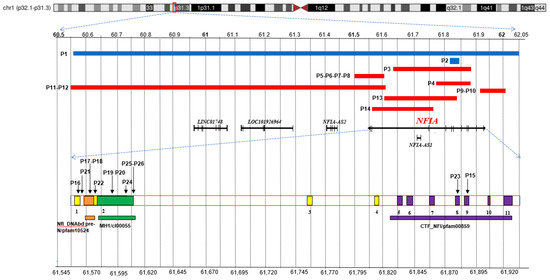
Figure 1.
Overview of NFIA pathogenic variants. Chromosome 1 ideogram with highlight on p32.1-p31.3 region. Top: the blue bars indicate the deletions detected in the patients here reported (P1-P2), and the red ones correspond to cases described in literature and public databases (P3-P14). Middle: coding sequences, including NFIA-207 (ENST00000403491.8; RefSeq ID NM_001134673) and other non-coding RNAs, are represented by dark bars. Bottom: a NFIA-207 zoom in that highlights the exons (1-11) along with the SNVs of patients P15-P26. The conserved domains are also depicted: NfI_DNAbd_pre-N/pfam10524 (orange), MH1/cI00055 (green), CTF_NFI/pfam00859 (purple). The top and bottom scales refer to GRCh37/hg19. Relative exons lengths are not to scale.
To better evaluate the phenotypic outcome of NFIA haploinsufficiency, the pathological traits of the selected patients have been grouped into macroareas: dysmorphisms (Table 2), neurological/behavioral abnormalities, renal/urinary trait alterations (Table 3), and CNS malformations (Table 4). In each table are listed only those patients where the clinical traits were evaluated. The few additional defects not included in these categories are reported in Table S3 and CNS malformations (Table 4).
The major clinical characteristics are summarized in Table 5 along with their frequency. The prevalence of each single trait was calculated both by the ratio between the presence of a clinical sign on the number of patients where this clinical sign has been evaluated and the ratio of all the 26 selected patients.
Table 5.
Major clinical features.
Dysmorphisms (Table 2). Among physical anomalies, macrocephaly is a trait almost invariably present (except P2); high forehead and a low-set ears are other recurrent signs, often associated with nonspecific facial-dysmorphic features.
Craniofacial anomalies, including asymmetries and craniosynostosis, represent a distinctive trait described in most patients. Metopic, lambdoid, or sagittal craniosynostosis were not observed in four cases (P4, P5, P6, P8). Three of them are members of the same family; thus, it was hypothesized that these anomalies could be the consequence of additional familiar genetic modifiers; our report of an additional case with craniosynostosis (P1) strengthens this correlation with haploinsufficiency. It is worth noting that in chicken, NFIA participates in osteoblast differentiation by interacting with the Wnt and Ihh pathways [10].
Bilateral proximally placed first fingers seem to be another characteristic dysmorphism, present in our patients (P1, P2) and in four other cases (P6, P8, P11, P12). This sign may have been overlooked, and it is possible that its frequency is underestimated.
Macrocephaly, craniofacial abnormalities, and first-finger anomalies are quite peculiar traits, and their concurrence can represent a first diagnostic clue for NFIA haploinsufficiency.
Neurological/behavioral abnormalities (Table 3). DD and/or ID are invariably present in this disorder, ranging from very mild to severe (details are available in Table S4). Beside ID and DD, this cohort shows the concurrence of other NDDs, including hyperactivity disorder (P1), ODD (P2), ADHD-Combined Type (P4), PDDNOS (P4), as well as neurological signs such as facial asymmetry (P9), mild spasticityat lower limbs (P9), extremely low PSI (P16), and epilepsy (P16, P17, P18, P21).It is worth noting that the comorbidity is an almost constant feature of NDDs because different NDDs can represent the variable expressivity of the same genetic alteration [30]. The comorbidity of this cohort strengthens this hypothesis. Hypotonia, exotropia, and hearing impairment are other neurological signs reported.
Renal/urinary trait and CNS defects (Table 3 and Table 4). Renal/urinary tract and CNS defects are considered to bethe key features of “Chromosome 1p32-p31 deletion syndrome”, whichis also referred to as “brain malformations with or without urinary tract defects” (BRMUTD) (OMIM # 613735).
In cases with NFIA haploinsufficiency, the frequency of renal/urinary tract abnormalities is probably lower than previously supposed [1], and in this cohort, they were excluded in twelve cases (Table 3). The defects described are heterogeneous, even when intrafamilial (i.e., P5 versus P7). Hydronephrosis and renal cysts are the most common clinical signs, even if each of them is reported only in three patients (P4, P7, P15 and P5, P9, P17, respectively). The absence of recurrent clinical features, along with their low frequency, make the renal/urinary tract anomalies not very helpful for the identification of this disorder. As experiments in knocked-out mice highlighted the involvement of NFIA in the embryonic development of the excretory apparatus [1], a surveillance of kidneys and urinary trait is mandatory when haploinsufficiency of this gene is detected.
On the other hand, CNS alterations represent a key trait of NFIA haploinsufficiency. Corpus callosum and cerebral ventricles anomalies are the most distinctive signs: the corpus callosum can be thin, hypoplastic, absent, or dysgenetic, and the presence of ventriculomegaly or hydrocephalus is quite constant. Along with these defects, a constellation of less common and heterogeneous CNS anomalies has been described, such as cortical malformations (P11, P12, P15, P21, P26), hypoplasia or atrophy of brain areas (P5, P11, P12, P19), decreased periventricular white matter (P3, P11, P25), partial incomplete inversion of the left hippocampi (P5, P11), and cyst presence (P11, P15, P25). P2 shows the presence of cerebral vascular malformations that have never been reported before. All these anomalies confirm the key role of NFIA in the differentiation of multiple brain areas during embryonic development [12,13,14,15].
From Table 1, Table 2, Table 3 and Table 4, a strict correlation between the genetic variants and clinical signs does not emerge: greater deletions do not correspond to a more severe phenotype; moreover, familialcases that share the same anomaly show a clinical spectrum, confirming a variable expressivity of some phenotypic traits.
5. Conclusions
NFIA is a gene with a key role in various differentiation pathways during embryogenesis; therefore, its haploinsufficiency is thought to determine a pleiotropic effect.
In this article, two new cases with deletions solely involving NFIA are reported, and the literature and public databases were reviewed in order to evaluate the phenotypic outcome of NFIA haploinsufficiency. First, all those patients with NFIA alterations and pathogenic genetic variants disrupting additional protein-coding sequences were excluded, since a thoughtful selection of cases is the most important step to accurately delineate genotype–phenotype correlations of a genetic disorder. Second, all the clinical signs and their relative frequencies in the 24 selected cases were analyzed. This process was useful to have an overall picture of phenotypic outcome of NFIA haploinsufficiency and to define a cluster of phenotypic traits that can facilitate clinicians in identifying the affected patients (Table 5). The potential diagnosis of NFIA haploinsufficiency can be advanced by a careful observation of the dysmorphisms (macrocephaly, craniofacial, and first-finger anomalies) and be strengthened by the presence of ID/DD or other NDDs. Instrumental tests such as MRI and kidney urinary tract ultrasound can provide key elements to further strengthen this hypothesis, which can be confirmed by genetic testing. Additional cases with well-described phenotypes will be helpful to better elucidate the full clinical spectrum.
The present analysis confirms that NFIA is responsible for the main phenotypic traits of Chromosome 1p32p31 deletion syndrome, even if other genes can contribute to modify the severity of some signs, such as ID/DD [6]. Less common traits of this syndrome, such as cryptorchidism, inguinal hernia, anomalies of external genitalia, syringomyelia, congenital heart defects, and Juvenile Moyamoya [1,2,3,4,5,6,7], were never reported in this selected cohort with only NFIA haploinsufficiency, and they are probably caused by dosage alterations of different deleted genes.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/genes13122249/s1. Table S1: Excluded patients; Table S2: Comparison between NFIA-207 and NFIA-205 transcripts; Table S3: Other defects described; Table S4: Neurodevelopmental details.
Author Contributions
Conceptualization, V.B. and A.V.; methodology, V.B. and F.C.; software, V.B., F.C. and A.V.; investigations, A.O., A.B., A.F., A.S., M.S., D.P. and M.E.; validation, V.B., F.C. and A.V.; formal analysis, F.C.; resources, V.B. and F.C.; data curation, O.G., F.C., A.F., A.S. and M.S.; writing—original draft preparation, V.B., A.O. and A.V.; writing—review and editing, V.B., A.V. and F.C.; supervision, D.P., M.E. and A.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Publicly available datasets were analyzed in this study. Thesedata can be found at: https://www.deciphergenomics.org/ (accessed on 20 September 2022); https://www.genome.ucsc.edu/ (accessed on 5 April 2022); https://omim.org/ (accessed on 20 September 2022); https://www.ensembl.org (accessed on 20 September 2022); https://www.gtexportal.org/home (accessed on 20 September 2022); http://projects.tcag.ca/variation (accessed on 20 September 2022); http://www.ncbi.nlm.nih.gov/pubmed (accessed on 20 September 2022); https://www.sigu.net (accessed on 20 September 2022);ClinVar (https://www.ncbi.nlm.nih.gov/clinvar, accessed on 20 September 2022); Leiden Open Variation Database (https://www.lovd.nl, accessed on 20 September 2022); the Human Gene Mutation Database (https://www.hgmd.cf.ac.uk/ac/index.php, accessed on 20 September 2022); and Varsome (https:varsome.com, accessed on 16 November 2022).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lu, W.; Quintero-Rivera, F.; Fan, Y.; Alkuraya, F.S.; Donovan, D.J.; Xi, Q.; Turbe-Doan, A.; Li, Q.G.; Campbell, C.G.; Shanske, A.L.; et al. NFIA haploinsufficiency is associated with a CNS malformation syndrome and urinary tract defects. PLoS Genet. 2007, 3, 830–843. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.G.N.; Wang, H.; Hunter, G.W. Interstitial microdeletion of Chromosome 1p in two siblings. Am. J. Med. Genet. 2002, 111, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Koehler, U.; Holinski-Feder, E.; Ertl-Wagner, B.; Kunz, J.; von Moers, A.; von Voss, H.; Schell-Apacik, C. A novel 1p31.3p32.2 deletion involving the NFIA gene detected by array CGH in a patient with macrocephaly and hypoplasia of the corpus callosum. Case Rep. Eur. J. Pediatr. 2010, 169, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.P.; Su, Y.N.; Chen, Y.Y.; Chern, S.R.; Liu, Y.P.; Wu, P.C.; Lee, C.C.; Chen, Y.T.; Wang, W. Chromosome 1p32-p31 deletion syndrome: Prenatal diagnosis by array comparative genomic hybridization using uncultured amniocytes and association with NFIA haploinsufficiency, ventriculomegaly, corpus callosum hypogenesis, abnormal external genitalia, and intrauterine growth restriction. Case Rep. Taiwan J. Obstet. Gynecol. 2011, 50, 345–352. [Google Scholar]
- Ji, J.; Salamon, N.; Quintero-Rivera, F. Microdeletion of 1p32-p31 involving NFIA in a patient with hypoplastic corpus callosum, ventriculomegaly, seizures and urinary tract defects. Comment Eur. J. Med. Genet. 2014, 57, 267–268. [Google Scholar] [CrossRef]
- Labonne, J.D.J.; Shen, Y.; Kong, I.K.; Diamond, M.P.; Layman, L.C.; Kim, H.G. Comparative deletion mapping at 1p31.3-p32.2 implies NFIA responsible for intellectual disability coupled with macrocephaly and the presence of several other genes for syndromic intellectual disability. Mol. Cytogenet. 2016, 17, 9–24. [Google Scholar] [CrossRef]
- Prontera, P.; Rogaia, D.; Mencarelli, A.; Ottaviani, V.; Sallicandro, E.; Guercini, G.; Esposito, S.; Bersano, A.; Merla, G.; Stangoni, G. Juvenile Moyamoya and Craniosynostosis in a Child with Deletion 1p32p31: Expanding the Clinical Spectrum of 1p32p31 Deletion Syndrome and a Review of the Literature. Int. J. Mol. Sci. 2017, 18, 1998. [Google Scholar] [CrossRef]
- Zenker, M.; Bunt, J.; Schanze, I.; Schanze, D.; Piper, M.; Priolo, M.; Gerkes, E.H.; Gronostajski, R.M.; Richards, L.J.; Vogt, J.; et al. Variants in nuclear factor I genes influence growth and development. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 611–626. [Google Scholar] [CrossRef]
- Gronostajski, R.M. Roles of the NFI/CTF gene family in transcription and development. Gene 2000, 249, 31–45. [Google Scholar] [CrossRef]
- Singh, P.N.P.; Yadav, U.S.; Azad, K.; Goswami, P.; Kinare, V.; Bandyopadhyay, A. NFIA and GATA3 are crucial regulators of embryonic articular cartilage differentiation. Development 2018, 145, dev156554. [Google Scholar]
- Hiraike, Y.; Waki, H.; Miyake, K.; Wada, T.; Oguchi, M.; Saito, K.; Tsutsumi, S.; Aburatani, H.; Yamauchi, T.; Kadowaki, T. NFIA differentially controls adipogenic and myogenic gene program through distinct pathways to ensure brown and beige adipocyte differentiation. PLoS Genet. 2020, 16, e1009044. [Google Scholar] [CrossRef] [PubMed]
- Tchieu, J.; Calder, E.L.; Guttikonda, S.R.; Gutzwiller, E.M.; Aromolaran, K.A.; Steinbeck, J.A.; Goldstein, P.A.; Studer, L. NFIA is a gliogenic switch enabling rapid derivation of functional human astrocytes from pluripotent stem cells. Nat. Biotechnol. 2019, 37, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Sagner, A.; Zhang, I.; Watson, T.; Lazaro, J.; Melchionda, M.; Briscoe, J. A shared transcriptional code orchestrates temporal patterning of the central nervous system. PLoS Biol. 2021, 19, e3001450. [Google Scholar] [CrossRef] [PubMed]
- Santo, M.; Rigoldi, L.; Falcone, C.; Tuccillo, M.; Calabrese, M.; Martínez-Cerdeño, V.; Mallamaci, A. Spatial control of astrogenesis progression by cortical arealization genes. Cereb. Cortex. 2022, 12, mikabhac264. [Google Scholar] [CrossRef] [PubMed]
- Quist, E.; Trovato, F.; Avaliani, N.; Zetterdahl, O.G.; Gonzalez-Ramos, A.; Hansen, M.G.; Kokaia, M.; Canals, I.; Ahlenius, H. Transcription factor-based direct conversion of human fibroblasts to functional astrocytes. Stem Cell Rep. 2022, 17, 1620–1635. [Google Scholar] [CrossRef]
- Kearney, H.M.; Thorland, E.C.; Brown, K.K.; Quintero-Rivera, F.; South, S.T.; Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet. Med. 2011, 13, 680–685. [Google Scholar] [CrossRef]
- Mikhail, F.M.M.; Lose, E.J.; Robin, N.H.; Descartes, M.D.; Rutledge, K.D.; Rutledge, S.L.; Korf, B.R.; Carroll, A.J. Clinically relevant single gene or intragenic deletions encompassing critical neurodevelopmental genes in patients with developmental delay, mental retardation, and/or autism spectrum disorders. Am. J. Med. Genet. A 2011, 155, 2386–2396. [Google Scholar] [CrossRef]
- Hollenbeck, D.; Williams, C.L.; Drazba, K.; Descartes, M.; Korf, B.R.; Rutledge, S.L.; Lose, E.J.; Robin, N.H.; Carroll, A.J.; Mikhail, F.M. Clinical relevance of small copy-number variants in chromosomal microarray clinical testing. Genet. Med. 2016, 19, 377–385. [Google Scholar] [CrossRef]
- Rao, A.; O’Donnell, S.; Bain, N.; Meldrum, C.; Shorter, D.; Goel, H. An intragenic deletion of the NFIA gene in a patient with a hypoplastic corpus callosum, craniofacial abnormalities and urinary tract defects. Eur. J. Med. Genet. 2014, 57, 65–70. [Google Scholar] [CrossRef]
- Nyboe, D.; Kreiborg, S.; Kirchhoff, M.; Hove, H.B. Familial craniosynostosis associated with a microdeletion involving the NFIA gene. Case Rep. Clin. Dysmorphol. 2015, 24, 109–112. [Google Scholar] [CrossRef]
- Bayat, A.; Kirchhoff, M.; Madsen, C.G.; Roos, L.; Kreiborg, S. Familial craniofacial abnormality and polymicrogyria associated with a microdeletion affecting the NFIA gene. Case Rep. Clin. Dysmorphol. 2017, 26, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Negishi, Y.; Miya, F.; Hattori, A.; Mizuno, K.; Hori, I.; Ando, N.; Okamoto, N.; Kato, M.; Tsunoda, T.; Yamasaki, M.; et al. Truncating mutation in NFIA causes brain malformation and urinary tract defects. Hum. Genome Var. 2015, 2, 15007. [Google Scholar] [CrossRef]
- Revah-Politi, A.; Ganapathi, M.; Bier, L.; Cho, M.T.; Goldstein, D.B.; Hemati, P.; Iglesias, A.; Juusola, J.; Pappas, J.; Petrovski, S.; et al. Loss-of-function variants in NFIA provide further support that NFIA is a critical gene in 1p32-p31 deletion syndrome: A four patient series. Am. J. Med. Genet. 2017, 173, 3158–3164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lin, C.M.; Zheng, X.L.; Abuduxikuer, K. A novel NFIA gene nonsense mutation in a Chinese patient with macrocephaly, corpus callosum hypoplasia, developmental delay, and dysmorphic features. Mol. Genet. Genomic. Med. 2020, 8, e1492. [Google Scholar] [CrossRef]
- Uehara, T.; Sanuki, R.; Ogura, Y.; Yokoyama, A.; Yoshida, T.; Futagawa, H.; Yoshihashi, H.; Yamada, M.; Suzuki, H.; Takenouchi, T.; et al. Recurrent NFIA K125E substitution represents a loss-of-function allele: Sensitive in vitro and in vivo assays for nontruncating alleles. Am. J. Med. Genet. A 2021, 185, 2084–2093. [Google Scholar] [CrossRef]
- Coci, E.G.; Koehler, U.; Liehr, T.; Stelzner, A.; Fink, C.; Langen, H.; Riedel, J. CANPMR syndrome and chromosome 1p32-p31 deletion syndrome coexist in two related individuals affected by simultaneous haplo-insufficiency of CAMTA1 and NIFA genes. Mol. Cytogenet. 2016, 9, 10. [Google Scholar] [CrossRef]
- Ogura, Y.; Uehara, T.; Ujibe, K.; Yoshihashi, H.; Yamada, M.; Suzuki, H.; Takenouchi, T.; Kosaki, K.; Hirata, H. The p.Thr395Met missense variant of NFIA found in a patient with intellectual disability is a defective variant. Am. J. Med. Genet. A 2022, 188, 1184–1192. [Google Scholar] [CrossRef]
- Wongkittichote, P.; Kondis, J.S.; Peglar, L.M.; Strahle, J.M.; Miller-Thomas, M.; Abell, K.B. Pathogenic variant in NFIA associated with subdural hematomas mimicking nonaccidental trauma. Am. J. Med. Genet. A 2022, 188, 1538–1544. [Google Scholar] [CrossRef]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.H.; Narzisi, G.; Leotta, A.; et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef]
- Bertini, V.; Milone, R.; Cristofani, P.; Cambi, F.; Bosetti, C.; Barbieri, F.; Bertelloni, S.; Cioni, G.; Valetto, A.; Battini, R. Enhancing DLG2 Implications in Neuropsychiatric Disorders: Analysis of a Cohort of Eight Patients with 11q14.1 Imbalances. Genes 2022, 13, 859. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).