

Structural Characteristics of Mitochondrial Genomes of Eight Treehoppers (Hemiptera: Membracidae: Centrotinae) and Their Phylogenetic Implications

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Sequencing, Assembly, Annotation, and Bioinformatic Analyses
2.3. Analysis of Codon Usage Bias
2.4. Phylogenetic Analysis
3. Results and Discussion
3.1. Genome Structure and Organization
3.2. Protein-Coding Genes (PCGs) and Codon Usage
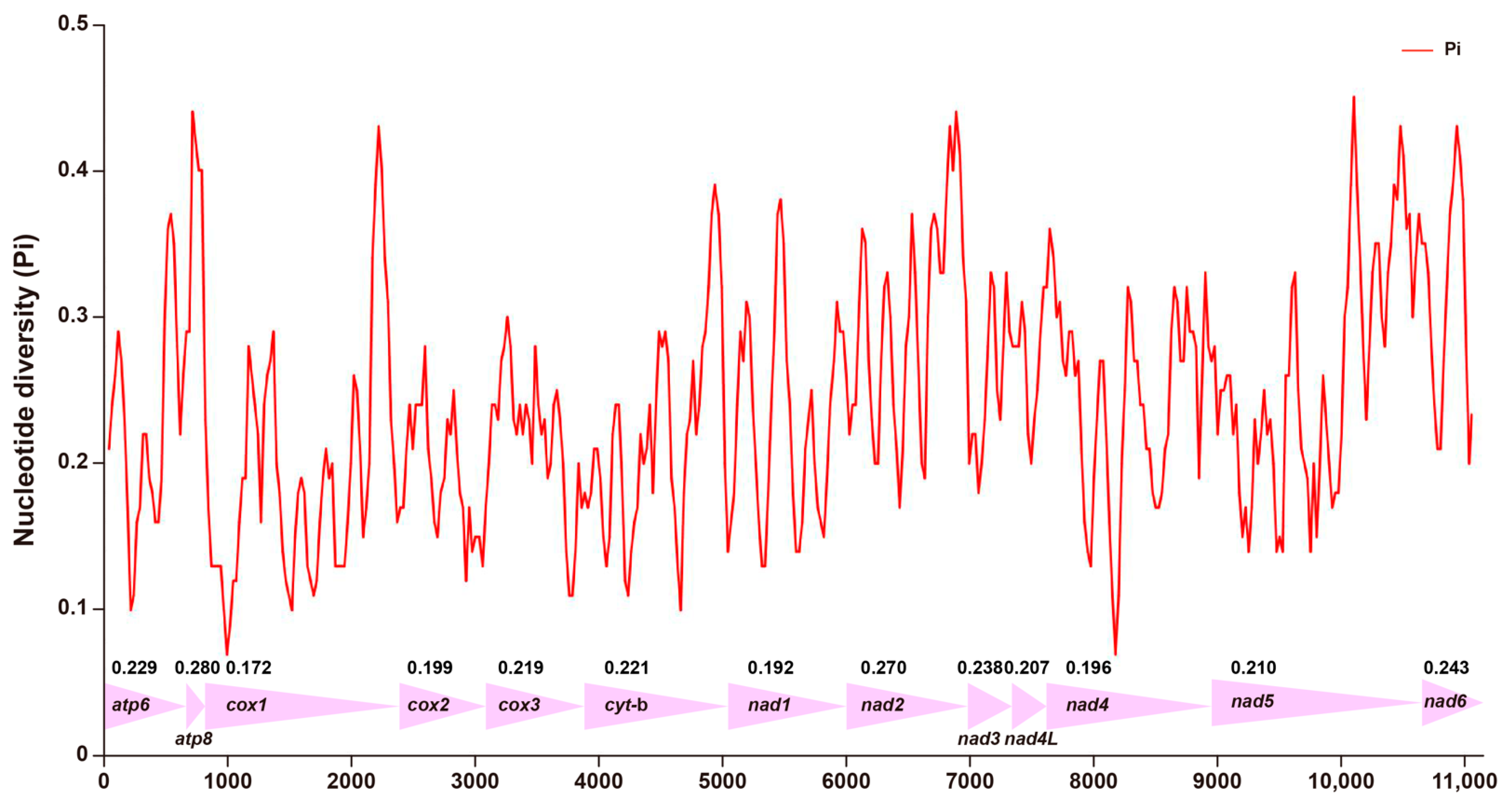
3.3. Nucleotide Diversity in PCGs of Mitogenomes
3.4. Analysis of Codon Usage Bias
3.5. Ribosomal and Transfer RNA Genes
3.6. Overlapping Sequences and Intergenic Spacers
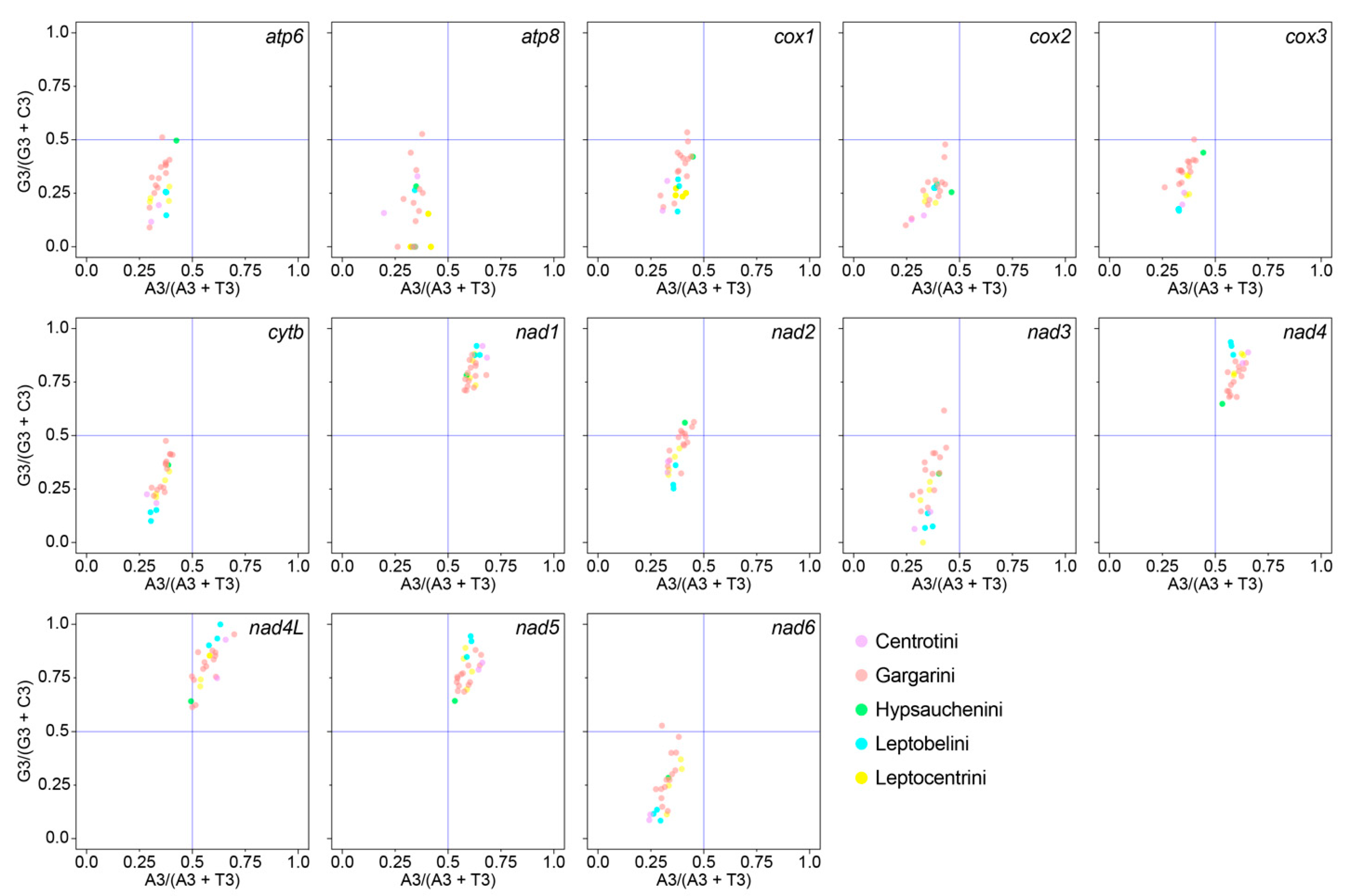
3.7. A + T-Rich Region
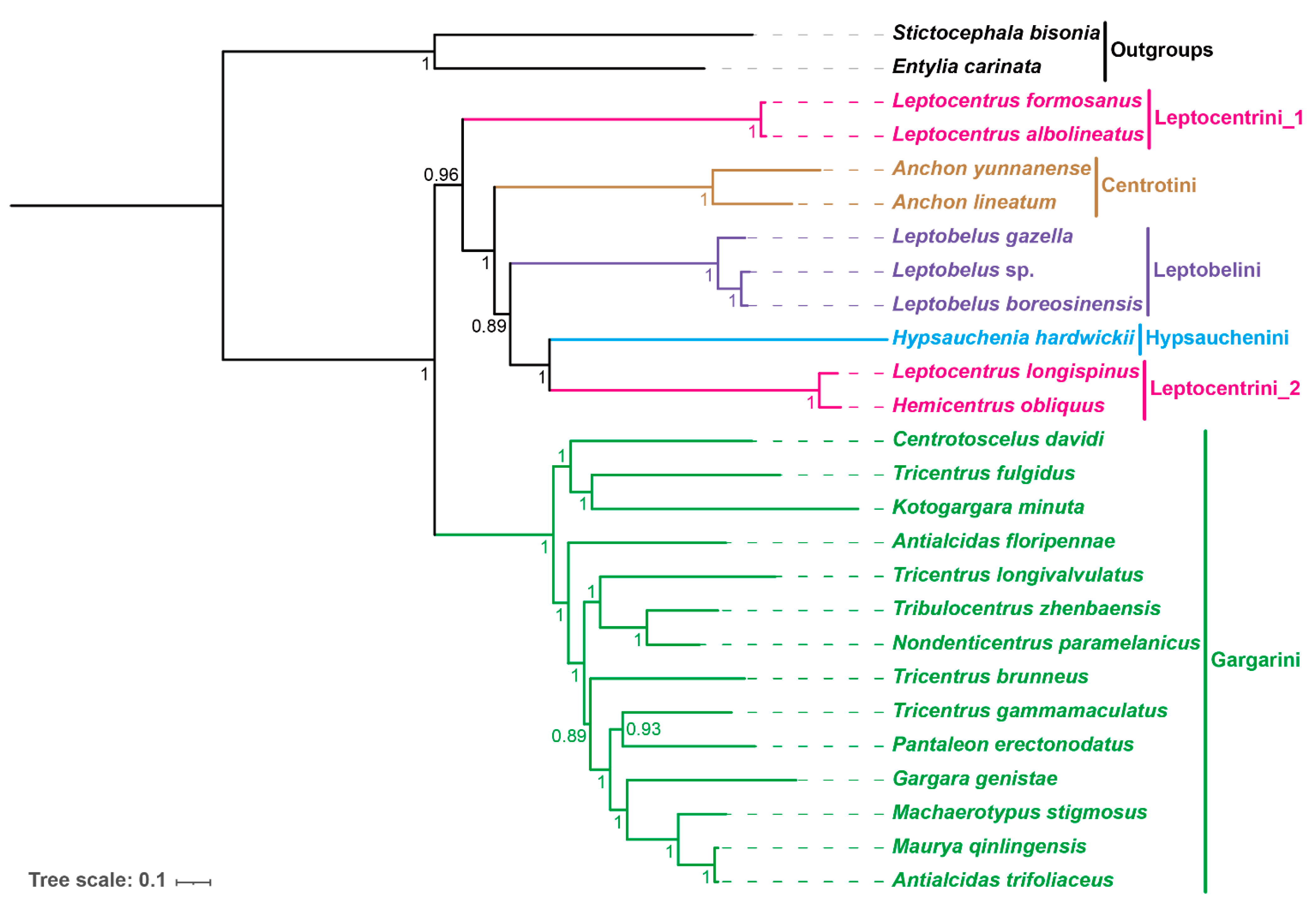
3.8. Phylogenetic Relationships
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, C.R.; Wegrzyn, J.L.; Jockusch, E.L. Co-option of wing-patterning genes underlies the evolution of the treehopper helmet. Nat. Ecol. Evol. 2020, 4, 250–260. [Google Scholar] [CrossRef]
- Cryan, J.R.; Wiegmann, B.M.; Deitz, L.L.; Dietrich, C.H. Phylogeny of the treehoppers (Insecta: Hemiptera: Membracidae): Evidence from two nuclear genes. Mol. Phylogenet. Evol. 2000, 17, 317–334. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, C.H.; McKamey, S.; Deitz, L. Morphology-based phylogeny of the treehopper family Membracidae (Hemiptera: Cicadomorpha: Membracoidea). Syst. Entomol. 2001, 26, 213–239. [Google Scholar] [CrossRef]
- Yuan, F.; Chou, I. Fauna Sinica: Insecta. Homoptera Membracoidea Aetalionidae Membracidae; Science Press: Beijing, China, 2002. [Google Scholar]
- Lin, C.-P.; Danforth, B.N.; Wood, T.K. Molecular phylogenetics and evolution of maternal care in membracine treehoppers. Syst. Biol. 2004, 53, 400–421. [Google Scholar] [CrossRef] [Green Version]
- Wallace, M.S.; Deitz, L.L. Australian treehoppers (Hemiptera: Membracidae: Centrotinae: Terentiini): Phylogeny and biogeography. Invertebr. Syst. 2006, 20, 163–183. [Google Scholar] [CrossRef]
- Capener, A.L. The Taxonomy of the African Membracidae. Part I. The Oxyrhachinae, Entomology Memoir; Entomological Memoir; South Africa, Department of Agriculture Technical Service: Pretoria, South Africa, 2008.
- Day, M. The genera of Australian Membracidae (Hemiptera: Auchenorrhyncha). Invertebr. Syst. 1999, 13, 629–747. [Google Scholar] [CrossRef]
- Distant, W.L. The fauna of British India including Ceylon and Burma. Rhynchota. Nature 1904, 70, 341. [Google Scholar]
- Evans, J.W. The leafhoppers and froghoppers of Australia and New Zealand (Homoptera: Cicadelloidea and Cercopoidea). Aust. Mus. 1966, 12, 1–347. [Google Scholar] [CrossRef]
- Goding, F.W. A synopsis of the subfamilies and genera of the Membracidae of North America. Trans. Am. Entomol. Soc. 1892, 19, 253–260. [Google Scholar]
- Wallace, M.S.; Deitz, L.L. Phylogeny and systematics of the treehopper subfamily Centrotinae (Hemiptera: Membracidae). In Memoirs on Entomology, International; The Associated Publishers: Washington, DC, USA, 2004; Volume 19, pp. 1–377. [Google Scholar]
- Dietrich, C.H.; Allen, J.M.; Lemmon, A.R.; Lemmon, E.M.; Takiya, D.M.; Evangelista, O.; Walden, K.K.; Grady, P.G.; Johnson, K.P. Anchored hybrid enrichment-based phylogenomics of leafhoppers and treehoppers (Hemiptera: Cicadomorpha: Membracoidea). Insect Syst. Diver. 2017, 1, 57–72. [Google Scholar] [CrossRef]
- Hu, Y.; Dietrich, C.H.; Skinner, R.K.; Zhang, Y. Phylogeny of Membracoidea (Hemiptera: Auchenorrhyncha) based on transcriptome data. Syst. Entomol. 2023, 48, 97–110. [Google Scholar] [CrossRef]
- Hu, K.; Yuan, F.; Dietrich, C.H.; Yuan, X.-Q. Structural features and phylogenetic implications of four new mitogenomes of Centrotinae (Hemiptera: Membracidae). Int. J. Biol. Macromol. 2019, 139, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Feng, L.; Dietrich, C.H.; Yuan, X. Characterization, comparison of four new mitogenomes of Centrotinae (Hemiptera: Membracidae) and phylogenetic implications supports new synonymy. Life 2022, 12, 61. [Google Scholar] [CrossRef]
- Deitz, L.L. Bibliography of the Membracoidea (Homoptera: Aetalionidae, Biturritiidae, Membracidae, and Nicomiidae) 1981–1987; North Carolina Agricultural Research Service, North Carolina State University: Raleigh, NC, USA, 1989. [Google Scholar]
- McKamey, S.H.; Deitz, L.L. Revision of the Neotropical Treehopper Genus Metcalfiella (Homoptera: Membracidae); Department of Agricultural Communications, North Carolina State University: Raleigh, NC, USA, 1991. [Google Scholar]
- Jin, J.; Yu, W.; Yang, J.; Song, Y.; DePamphilis, C.W.; Yi, T.; Li, D. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 1–31. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Sharp, P.M.; Stenico, M.; Peden, J.F.; Lloyd, A.T. Codon usage: Mutational bias, translational selection, or both? Biochem. Soc. Trans. 1993, 21, 835–841. [Google Scholar] [CrossRef] [Green Version]
- Sueoka, N. Intrastrand parity rules of DNA base composition and usage biases of synonymous codons. J. Mol. Evol. 1995, 40, 318–325. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.i.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Fukuda, Y.; Tomita, M.; Washio, T. Comparative study of overlapping genes in the genomes of Mycoplasma genitalium and Mycoplasma pneumoniae. Nucleic Acids Res. 1999, 27, 1847–1853. [Google Scholar] [CrossRef] [Green Version]
- Rédei, G.P. Encyclopedia of Genetics, Genomics, Proteomics, and Informatics; Springer Science & Business Media: Berlin, Germany, 2008. [Google Scholar]
- Sudbery, P. Human Molecular Genetics, 2nd ed.; Benjamin Cummings: San Francisco, CA, USA, 2002. [Google Scholar]
- Pick, K.; Philippe, H.; Schreiber, F.; Erpenbeck, D.; Jackson, D.; Wrede, P.; Wiens, M.; Alié, A.; Morgenstern, B.; Manuel, M. Improved phylogenomic taxon sampling noticeably affects nonbilaterian relationships. Mol. Biol. Evol. 2010, 27, 1983–1987. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.W.; Hejnol, A.; Matus, D.Q.; Pang, K.; Browne, W.E.; Smith, S.A.; Seaver, E.; Rouse, G.W.; Obst, M.; Edgecombe, G.D. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature 2008, 452, 745–749. [Google Scholar] [CrossRef]
- Li, F.-E.; Yang, L.; Long, J.-K.; Chang, Z.-M.; Chen, X.-S. Revisiting the phylogenetic relationship and evolution of Gargarini with mitochondrial genome (Hemiptera: Membracidae: Centrotinae). Int. J. Mol. Sci. 2023, 24, 694. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liang, A. Complete DNA sequence of the mitochondrial genome of the treehopper Leptobelus gazella (Membracoidea: Hemiptera). Mitochondrial DNA A 2016, 27, 3318–3319. [Google Scholar] [CrossRef]
- Li, H.; Leavengood, J.M., Jr.; Chapman, E.G.; Burkhardt, D.; Song, F.; Jiang, P.; Liu, J.; Zhou, X.; Cai, W. Mitochondrial phylogenomics of Hemiptera reveals adaptive innovations driving the diversification of true bugs. Proc. R. Soc. B-Biol. Sci. 2017, 284, 20171223. [Google Scholar] [CrossRef]
- Mao, M.; Yang, X.; Bennett, G. The complete mitochondrial genome of Entylia carinata (Hemiptera: Membracidae). Mitochondrial DNA B 2016, 1, 662–663. [Google Scholar] [CrossRef] [Green Version]
- Yu, R.; Feng, L.; Yuan, X. Complete mitochondrial genome sequence of the global invasive species Stictocephala bisonia (Hemiptera: Membracidae: Smiliinae). Mitochondrial DNA B 2021, 6, 1601–1602. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, H.; Zhang, J.; Dietrich, C.H.; Li, Y.; Yuan, X. Structural Characteristics of Mitochondrial Genomes of Eight Treehoppers (Hemiptera: Membracidae: Centrotinae) and Their Phylogenetic Implications. Genes 2023, 14, 1510. https://doi.org/10.3390/genes14071510
Bai H, Zhang J, Dietrich CH, Li Y, Yuan X. Structural Characteristics of Mitochondrial Genomes of Eight Treehoppers (Hemiptera: Membracidae: Centrotinae) and Their Phylogenetic Implications. Genes. 2023; 14(7):1510. https://doi.org/10.3390/genes14071510
Chicago/Turabian StyleBai, Haijun, Jinrui Zhang, Christopher H. Dietrich, Yiping Li, and Xiangqun Yuan. 2023. "Structural Characteristics of Mitochondrial Genomes of Eight Treehoppers (Hemiptera: Membracidae: Centrotinae) and Their Phylogenetic Implications" Genes 14, no. 7: 1510. https://doi.org/10.3390/genes14071510
APA StyleBai, H., Zhang, J., Dietrich, C. H., Li, Y., & Yuan, X. (2023). Structural Characteristics of Mitochondrial Genomes of Eight Treehoppers (Hemiptera: Membracidae: Centrotinae) and Their Phylogenetic Implications. Genes, 14(7), 1510. https://doi.org/10.3390/genes14071510