
Illuminating Genetic Diversity and Selection Signatures in Matou Goats through Whole-Genome Sequencing Analysis
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Approval Statement
2.2. Sampling and Illumina Whole-Genome Sequencing
2.3. Alignment and SNP Calling
2.4. Population Structure and Phylogenetic Analysis
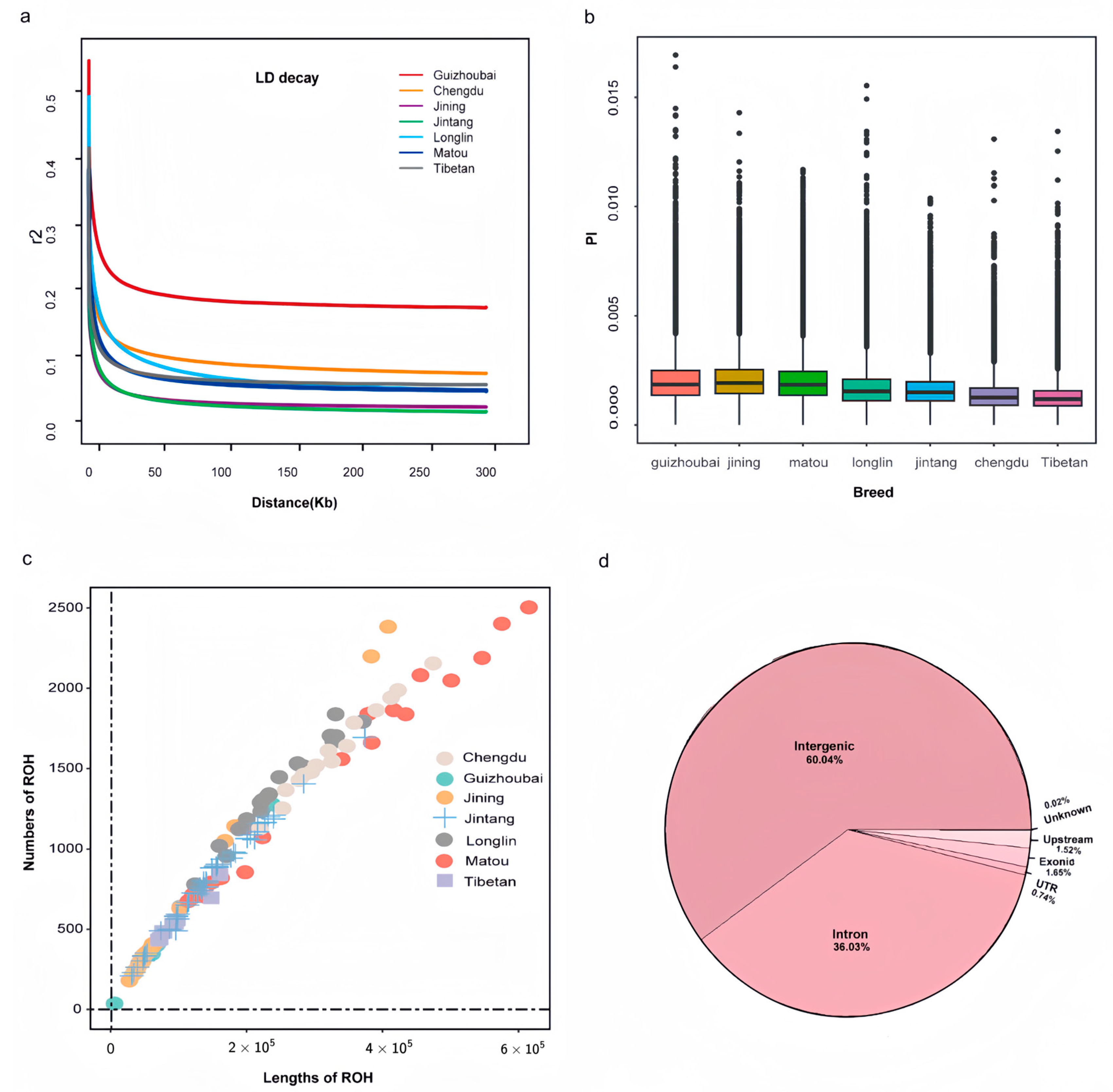
2.5. Genetic Diversity and Linkage Disequilibrium Detection
2.6. Selective Sweep Identification
2.7. Candidate Gene Enrichment Analysis
3. Results
3.1. Sequence Read Depths and Variants Landscape
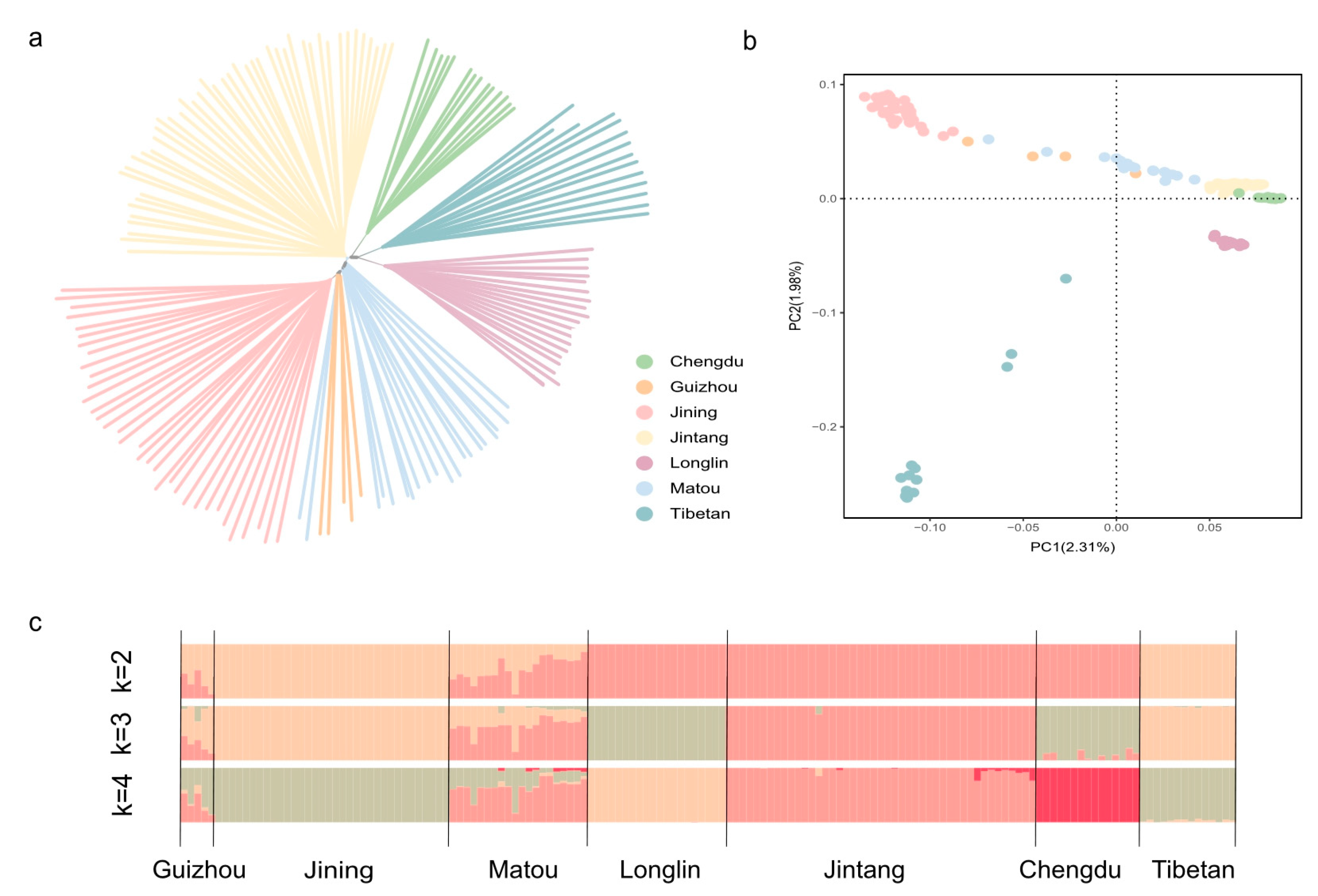
3.2. Population Structure Analysis
3.3. Population Genetic Diversity
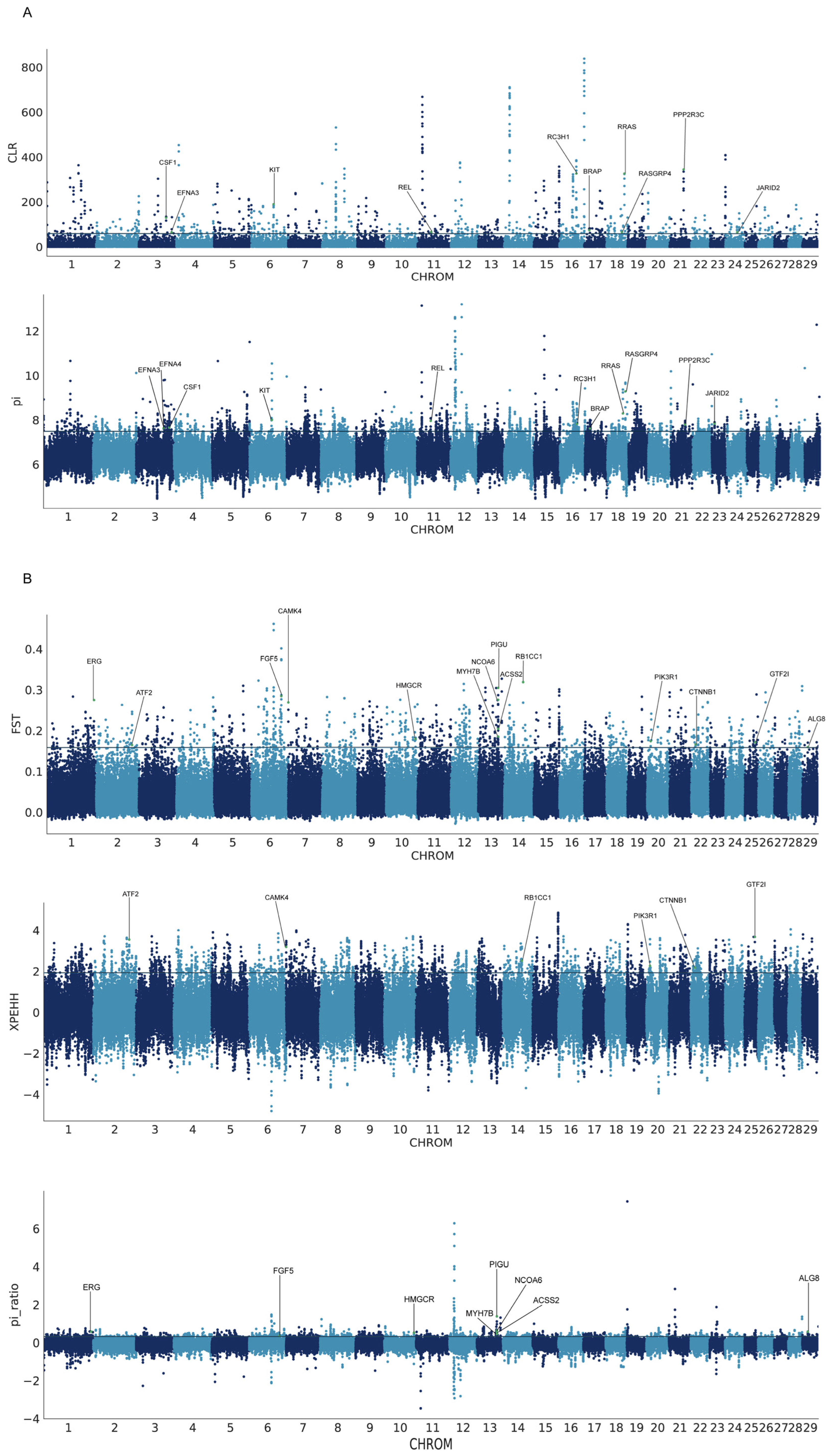
3.4. Genome-Wide Selective Sweep Test
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zeder, M.A.; Hesse, B. The initial domestication of goats (Capra hircus) in the Zagros mountains 10,000 years ago. Science 2000, 5461, 2254–2257. [Google Scholar] [CrossRef]
- Takada, T.; Kikkawa, Y.; Yonekawa, H.; Kawakami, S.; Amano, T. Bezoar (Capra aegagrus) is a matriarchal candidate for ancestor of domestic goat (Capra hircus): Evidence from the mitochondrial DNA diversity. Biochem. Genet. 1997, 35, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Wang, X.; Li, M.; Li, Y.; Yang, Z.; Wang, X.; Pan, X.; Gong, M.; Zhang, Y.; Guo, Y.; et al. The origin of domestication genes in goats. Sci. Adv. 2020, 6, eaaz5216. [Google Scholar] [CrossRef]
- Lai, F.N.; Zhai, H.L.; Cheng, M.; Ma, J.Y.; Cheng, S.F.; Ge, W.; Zhang, G.L.; Wang, J.J.; Zhang, R.Q.; Wang, X.; et al. Whole-genome scanning for the litter size trait associated genes and SNPs under selection in dairy goat (Capra hircus). Sci. Rep. 2016, 6, 38096. [Google Scholar] [CrossRef]
- Huang, Y.; Li, Y.; Wang, X.; Yu, J.; Cai, Y.; Zheng, Z.; Li, R.; Zhang, S.; Chen, N.; Asadollahpour Nanaei, H.; et al. An atlas of CNV maps in cattle, goat and sheep. Sci. China Life Sci. 2021, 64, 1747–1764. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Wang, Y.; Zhang, Y.; Zhu, J.; Lin, Y. RXRα cooperates with KLF8 to promote the differentiation of intramuscular preadipocytes in goat. Anim. Biotechnol. 2021, 32, 580–590. [Google Scholar] [CrossRef]
- Martin, P.M.; Palhière, I.; Ricard, A.; Tosser-Klopp, G.; Rupp, R. Genome Wide Association Study Identifies New Loci Associated with Undesired Coat Color Phenotypes in Saanen Goats. PLoS ONE 2016, 11, e0152426. [Google Scholar] [CrossRef]
- Hu, H.; Liu, Y.G.; Zhao, S.M.; Deng, W.D.; Gao, S.Z. Molecular characterization and tissue expression of ovine PSAM6 gene from muscle full-length cDNA library of black-boned sheep. Anim. Biotechnol. 2009, 20, 238–241. [Google Scholar] [CrossRef]
- Chen, T. Institutional Constraints and Space Expansion: A Case Study of Heifer International’s Localization Development in China. China Non-Profit Rev. 2011, 8, 11. [Google Scholar]
- Zhang, L.; Liu, Y.; Duan, C.; Zhang, Y.; Wang, Y.; Guo, Y. Analysis of Genetic Diversity and Genetic Structure in 7 Local Goat Breeds. Biotechnol. Bull. 2020, 36, 183–190. [Google Scholar]
- Yang, L.; Zhao, S.H.; Li, K.; Peng, Z.Z.; Montgomery, G.W. Determination of genetic relationships among five indigenous Chinese goat breeds with six microsatellite markers. Anim. Genet. 1999, 30, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Cai, Y.; Chen, Q.; Li, R.; Wang, K.; Huang, Y.; Hu, S.; Huang, S.; Zhang, H.; Zheng, Z.; et al. Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nat. Commun. 2018, 9, 2337. [Google Scholar] [CrossRef] [PubMed]
- Moaeen-ud-Din, M.; Yand, L.G.; Chen, S.L.; Zhang, Z.R.; Xiao, J.Z.; Wen, Q.Y.; Dai, M. Reproductive performance of Matou goat under sub-tropical monsoonal climate of Central China. Trop. Anim. Health Prod. 2008, 40, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.H.; Zhang, X.D.; Yao, N.; Ding, J.P.; Chen, H.Q.; Zhang, Z.J.; Zhang, Y.H.; Ren, C.H.; Ma, Y.H.; Zhang, X.R. Genetic differentiation of chinese indigenous meat goats ascertained using microsatellite information. Asian-Australas. J. Anim. Sci. 2012, 25, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, H.C. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Abuín, J.M.; Pichel, J.C.; Pena, T.F.; Amigo, J. BigBWA: Approaching the Burrows-Wheeler aligner to Big Data technologies. Bioinformatics 2015, 31, 4003–4005. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Qu, K.; Wang, Y.; Sinding, M.S.; Wang, F.; Hanif, Q.; Ahmed, Z.; Lenstra, J.A.; Han, J.; Lei, C.; et al. Global dispersal and adaptive evolution of domestic cattle: A genomic perspective. Stress. Biol. 2023, 3, 8. [Google Scholar] [CrossRef]
- Alexander, D.H.; Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform. 2011, 12, 246. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.; Xu, J.; He, W.; Yang, T. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Wang, D.; Wang, S.; Wang, H.; Zhou, H. Identification of key genes and pathways involved in response to pain in goat and sheep by transcriptome sequencing. Biol. Res. 2018, 51, 25. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Williamson, S.; Kim, Y.; Hubisz, M.J.; Clark, A.G.; Bustamante, C. Genomic scans for selective sweeps using SNP data. Genom. Res. 2005, 15, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Xia, X.; Hanif, Q.; Zhang, F.; Dang, R.; Huang, B.; Lyu, Y.; Luo, X.; Zhang, H.; Yan, H.; et al. Global genetic diversity, introgression, and evolutionary adaptation of indicine cattle revealed by whole genome sequencing. Nat Commun. 2023, 14, 7803. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Li, J.; Xiao, C.; Sun, L.; Xiang, W.; Chen, N.; Lei, C.; Lei, H.; Long, Y.; Long, T.; et al. Whole-Genome Resequencing of Xiangxi Cattle Identifies Genomic Diversity and Selection Signatures. Front Genet. 2022, 13, 816379. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Y.; Wang, F.; Cheng, H.; Han, J.; Dang, R.; Xia, X.; Wang, H.; Zhong, J.; Lenstra, J.A.; Zhang, H.; et al. Recent selection and introgression facilitated high-altitude adaptation in cattle. Sci. Bull. 2024; ahead of print. [Google Scholar]
- Okamura, T.; Shimizu, H.; Nagao, T.; Ueda, R.; Ishii, S. ATF-2 regulates fat metabolism in Drosophila. Mol. Biol. Cell 2007, 18, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Chandramohan, B.; Renieri, C.; La Manna, V.; La Terza, A. The alpaca agouti gene: Genomic locus, transcripts and causative mutations of eumelanic and pheomelanic coat color. Gene 2013, 521, 303–310. [Google Scholar] [CrossRef]
- Mendoza, M.N.; Raudsepp, T.; Alshanbari, F.; Gutiérrez, G.; Ponce de León, F.A. Chromosomal Localization of Candidate Genes for Fiber Growth and Color in Alpaca (Vicugna pacos). Front. Genet. 2019, 10, 583. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhong, J.; Li, L.; Zhong, T.; Wang, L.; Song, T.; Zhang, H. Comparative genome analyses reveal the unique genetic composition and selection signals underlying the phenotypic characteristics of three Chinese domestic goat breeds. Genet. Sel. Evol. 2019, 51, 70. [Google Scholar] [CrossRef]
- Johnson, G.L.; Dohlman, H.G.; Graves, L.M. MAPK kinase kinases (MKKKs) as a target class for small-molecule inhibition to modulate signaling networks and gene expression. Curr. Opin. Chem. Biol. 2005, 9, 325–331. [Google Scholar] [CrossRef]
- Ishii, K.; Kanatsu-Shinohara, M.; Toyokuni, S.; Shinohara, T. FGF2 mediates mouse spermatogonial stem cell self-renewal via upregulation of Etv5 and Bcl6b through MAP2K1 activation. Development 2012, 139, 1734–1743. [Google Scholar] [CrossRef] [PubMed]
- Volland, C.; Schott, P.; Didié, M.; Männer, J.; Unsöld, B.; Toischer, K.; Schmidt, C.; Urlaub, H.; Nickels, K.; Knöll, R.; et al. Control of p21Cip by BRCA1-associated protein is critical for cardiomyocyte cell cycle progression and survival. Cardiovasc. Res. 2020, 116, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yao, L.; Su, C. Signal transduction pathways and functions of Eph receptor family and its ligands. Prog. Biochem. Biophys. 2001, 28, 498–501. [Google Scholar]
- Ada-Nguema, A.S.; Xenias, H.; Hofman, J.M.; Wiggins, C.H.; Sheetz, M.P.; Keely, P.J. The small GTPase R-Ras regulates organization of actin and drives membrane protrusions through the activity of PLCepsilon. J. Cell Sci. 2006, 119 Pt 7, 1307–1319. [Google Scholar] [CrossRef] [PubMed]
- Ghigo, C.; Mondor, I.; Jorquera, A.; Nowak, J.; Wienert, S.; Zahner, S.P.; Clausen, B.E.; Luche, H.; Malissen, B.; Klauschen, F.; et al. Multicolor fate mapping of langerhans cell homeostasis. J. Exp. Med. 2013, 210, 1657–1664. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, M.; Brennan, F.M.; Maini, R.N. Role of cytokines in rheumatoid arthritis. Annu. Rev. Immunol. 1996, 14, 397–440. [Google Scholar] [CrossRef] [PubMed]
- Arijs, I.; Li, K.; Toedter, G.; Quintens, R.; Van Lommel, L.; Van Steen, K.; Leemans, P.; De Hertogh, G.; Lemaire, K.; Ferrante, M.; et al. Mucosal gene signatures to predict response to infliximab in patients with ulcerative colitis. Gut 2009, 58, 1612–1619. [Google Scholar] [CrossRef] [PubMed]
- Sasmono, R.T.; Oceandy, D.; Pollard, J.W.; Tong, W.; Pavli, P.; Wainwright, B.J.; Ostrowski, M.C.; Himes, S.R.; Hume, D.A. A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood 2003, 101, 1155–1163. [Google Scholar] [CrossRef]
- Nakamichi, Y.; Udagawa, N.; Takahashi, N. IL-34 and CSF-1: Similarities and differences. J. Bone Miner. Metab. 2013, 31, 486–495. [Google Scholar] [CrossRef]
- Botchkareva, N.V.; Khlgatian, M.; Longley, B.J.; Botchkarev, V.A.; Gilchrest, B.A. SCF/c-kit signaling is required for cyclic regeneration of the hair pigmentation. FASEB J. 2001, 15, 645–658. [Google Scholar] [CrossRef]
- Parichy, D.M.; Rawls, J.F.; Pratt, S.J.; Whitfield, T.T.; Johnson, S.L. Zebrafish sparse corresponds to an orthologue of c-kit and is required for the morphogenesis of a subpopulation of melanocytes,but is not essential for hematopoiesis or primordial germ cell development. Development 1999, 126, 3425–3436. [Google Scholar] [CrossRef]
- Nishikawa, S.; Kusakabe, M.; Yoshinaga, K.; Ogawa, M.; Hayashi, S.; Kunisada, T.; Era, T.; Sakakura, T. In utero manipulation of coat color formation by a monoclonal anti-c-kit antibody: Two distinct waves of c-kit-dependency during melanocyte development. EMBO J. 1991, 10, 2111–2118. [Google Scholar] [CrossRef]
- Deng, S.; Huang, L.; Ren, J.; Chen, K.; Ding, N. Study on porcine dominant white hair regulatory gene (KIT). Heredity 2000, 22, 434–436. [Google Scholar]
- Zhang, Q.; Wang, S.; Jiang, X.; Wang, S.; Lai, Q. A preliminary study on the relationship between dominant white hair regulatory gene (KIT) and sheep wool color. J. Anhui Agric. Sci. 2009, 37, 12433–12434. [Google Scholar]
- Wei, Y.; Zhou, R.; Zhang, J.; Li, X.; Xi, J.; Li, L.; Chen, H.; Zhang, Z. Expression of c-KIT in the skin of goats of different coat colors. Jiangsu Agric. Sci. 2016, 44, 38–41. [Google Scholar]
- Stevens, R.L.; Morokawa, N.; Wang, J.; Krilis, S.A. RasGRP4 in mast cell signalling and disease susceptibility. Novartis Found. Symp. 2005, 271, 54–68. [Google Scholar] [PubMed]
- Zhang, Y.; Kuang, S. The rise and fall and development prospect of Jining green goat. J. Anhui Agric. Sci. 2017, 45, 99–100+105. [Google Scholar]
- Shen, Y. Discussion on longevity signaling pathways and their transmission between different tissues. Chin. J. Cell Biol. 2015, 37, 7. [Google Scholar]
- Klass, M.R. A method for the isolation of longevity mutants in the nematode Caenorhabditis elegans and initial results. Mech. Ageing Dev. 1983, 22, 279–286. [Google Scholar] [CrossRef]
- Morris, J.Z.; Ruvkun, G.; Tissenbaum, H.A. A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature 1996, 382, 536. [Google Scholar] [CrossRef]
- Hsin, H.; Kenyon, C. Signals from the reproductive system regulate the lifespan of C. elegans. Nature 1999, 399, 362. [Google Scholar] [CrossRef] [PubMed]
- Dell’Agnello, C.; Leo, S.; Agostino, A.; Szabadkai, G.; Tiveron, C.; Zulian, A.; Prelle, A.; Roubertoux, P.; Rizzuto, R.; Zeviani, M. Increased longevity and refractoriness to Ca2+-dependent neurodegeneration in Surf1 knockout mice. Hum. Mol. Genet. 2007, 16, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Weimer, S.; Priebs, J.; Kuhlow, D.; Groth, M.; Priebe, S.; Mansfeld, J.; Merry, T.L.; Dubuis, S.; Laube, B.; Pfeiffer, A.F.; et al. D-Glucosamine supplementation extends life span of nematodes and of ageing mice. Nat. Commun. 2014, 5, 3563. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Smits, R.; Hao, H.; He, C. Wnt/β-Catenin Signaling in Liver Cancers. Cancers. 2019, 11, 926. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, N.; Kurzrock, R. Targeting the Wnt/β-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 2018, 62, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.J. Wnt signaling pathway in non-small cell lung cancer. J. Natl. Cancer Inst. 2014, 106, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yan, H.; Wang, K.; Zhou, T.; Chen, M.; Zhu, H.; Pan, C.; Zhang, E. Goat CTNNB1: mRNA expression profile of alternative splicing in testis and association analysis with litter size. Gene 2018, 679, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Fiset, A.; Xu, E.; Bergeron, S.; Marette, A.; Pelletier, G.; Siminovitch, K.A.; Olivier, M.; Beauchemin, N.; Faure, R.L. Compartmentalized CDK2 is connected with SHP-1 and β-catenin and regulates insulin internalization. Cell Signal 2011, 23, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef]
- Tsay, A.; Wang, J.C. The Role of PIK3R1 in Metabolic Function and Insulin Sensitivity. Int. J. Mol. Sci. 2023, 24, 12665. [Google Scholar] [CrossRef]
- Fatoki, T.H. Effect of pH on structural dynamics of HMG-CoA reductase and binding affinity to β-sitosterol. J. Biomol. Struct. Dyn. 2023, 41, 4398–4404. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhang, M.; Plec, A.A.; Estill, S.J.; Cai, L.; Repa, J.J.; McKnight, S.L.; Tu, B.P. ACSS2 promotes systemic fat storage and utilization through selective regulation of genes involved in lipid metabolism. Proc. Natl. Acad. Sci. USA 2018, 115, E9499–E9506. [Google Scholar] [CrossRef] [PubMed]
- Mascarello, F.; Toniolo, L.; Cancellara, P.; Reggiani, C.; Maccatrozzo, L. Expression and identification of 10 sarcomeric MyHC isoforms in human skeletal muscles of different embryological origin. Diversity and similarity in mammalian species. Ann. Anat. 2016, 207, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Resnicow, D.; Deacon, J.C.; Warrick, H.M.; Spudich, J.A.; Leinwand, L.A. Functional diversity among a family of human skeletal muscle myosin motors. Proc. Natl. Acad. Sci. USA 2010, 107, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.A.; Karabina, A.; Broadwell, L.J.; Leinwand, L.A. The ancient sarcomeric myosins found in specialized muscles. Skelet. Muscle 2019, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Cui, A.; Ding, D.; Li, Y. Regulation of Hepatic Metabolism and Cell Growth by the ATF/CREB Family of Transcription Factors. Diabetes 2021, 70, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Delliaux, C.; Tian, T.V.; Bouchet, M.; Fradet, A.; Vanpouille, N.; Flourens, A.; Deplus, R.; Villers, A.; Leroy, X.; Clézardin, P.; et al. TMPRSS2:ERG gene fusion expression regulates bone markers and enhances the osteoblastic phenotype of prostate cancer bone metastases. Cancer Lett. 2018, 438, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Jiang, R.; Mao, A.; Liu, G.E.; Zhan, S.; Li, L.; Zhong, T.; Wang, L.; Cao, J.; Chen, Y.; et al. Genome-wide association study reveals 14 new SNPs and confirms two structural variants highly associated with the horned/polled phenotype in goats. BMC Genom. 2021, 22, 769. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Tian, H.; Li, X. Research Progress of Fibroblast Growth Factor 5. China Biotechnol. 2020, 40, 117–124. [Google Scholar]
- Ota, Y.; Saitoh, Y.; Suzuki, S.; Ozawa, K.; Kawano, M.; Imamura, T. Fibroblast growth factor 5 inhibits hair growth by blocking dermal papilla cell activation. Biochem. Biophys. Res. Comm. 2002, 290, 169–176. [Google Scholar] [CrossRef]
- Wang, X.; Yu, H.; Lei, A.; Zhou, J.; Zeng, W.; Zhu, H.; Dong, Z.; Niu, Y.; Shi, B.; Cai, B. Generation of gene-modified goats targeting MSTN and FGF5 via zygote injection of CRISPR/Cas9 system. Sci. Rep. 2015, 5, 13878. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cai, B.; Zhou, J.; Zhu, H.; Niu, Y.; Ma, B.; Yu, H.; Lei, A.; Yan, H.; Shen, Q. Disruption of FGF5 in cashmere goats using CRISPR/Cas9 results in more secondary hair follicles and longer fibers. PLoS ONE 2016, 11, e0164640. [Google Scholar]
- Szpiech, Z.A.; Hernandez, R.D. Selscan: An efficient multithreaded program to perform EHH-based scans for positive selection. Mol. Biol. Evol. 2014, 31, 2824–2827. [Google Scholar] [CrossRef] [PubMed]
- Oh, G.S.; Kim, S.R.; Lee, E.S.; Yoon, J.; Shin, M.K.; Ryu, H.K.; Kim, D.S.; Kim, S.W. Regulation of Hepatic Gluconeogenesis by Nuclear Receptor Coactivator 6. Mol. Cells 2022, 45, 180–192. [Google Scholar] [CrossRef]
- Ikezawa, H. Glycosylphosphatidylinositol (GPI)-anchored proteins. Biol. Pharm. Bull. 2002, 25, 409–417. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
HuangFu, R.; Li, H.; Luo, Y.; He, F.; Huan, C.; Ahmed, Z.; Zhang, B.; Lei, C.; Yi, K. Illuminating Genetic Diversity and Selection Signatures in Matou Goats through Whole-Genome Sequencing Analysis. Genes 2024, 15, 909. https://doi.org/10.3390/genes15070909
HuangFu R, Li H, Luo Y, He F, Huan C, Ahmed Z, Zhang B, Lei C, Yi K. Illuminating Genetic Diversity and Selection Signatures in Matou Goats through Whole-Genome Sequencing Analysis. Genes. 2024; 15(7):909. https://doi.org/10.3390/genes15070909
Chicago/Turabian StyleHuangFu, Ruiyao, Haobang Li, Yang Luo, Fang He, Cheng Huan, Zulfiqar Ahmed, Baizhong Zhang, Chuzhao Lei, and Kangle Yi. 2024. "Illuminating Genetic Diversity and Selection Signatures in Matou Goats through Whole-Genome Sequencing Analysis" Genes 15, no. 7: 909. https://doi.org/10.3390/genes15070909