
Associations between Indoor and Outdoor Size-Resolved Particulate Matter in Urban Beijing: Chemical Compositions, Sources, and Health Risks
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
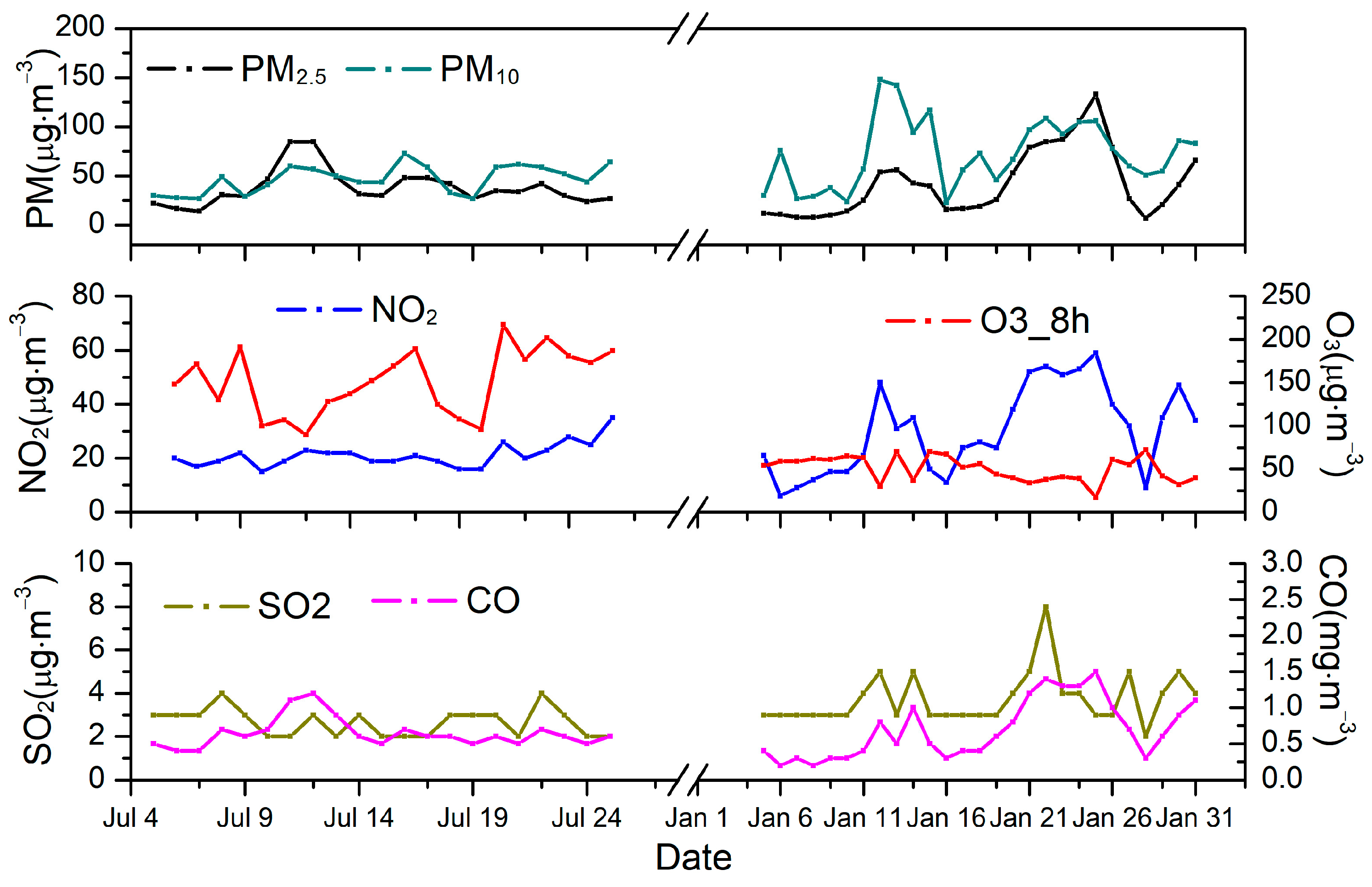
2.1. Sampling Site
2.2. Sample Collection
2.3. Chemical Analyses
2.4. Chemical Mass Closure
2.5. Acidity Evaluation
2.6. PMF Model
2.7. Health Risk Assessment
3. Results and Discussion
3.1. Mass Concentrations
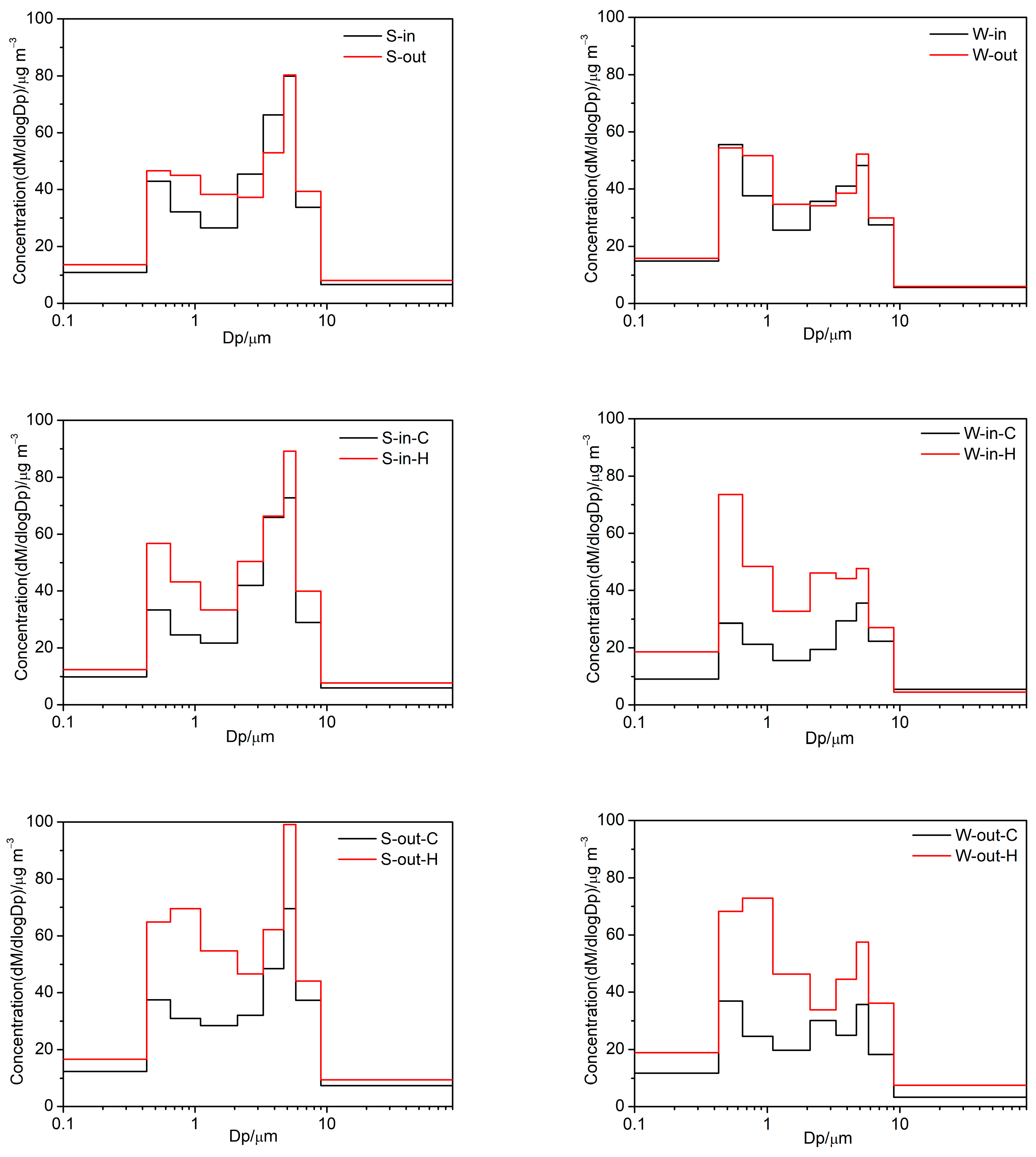
3.1.1. Size Distribution
3.1.2. Indoor/Outdoor Ratios
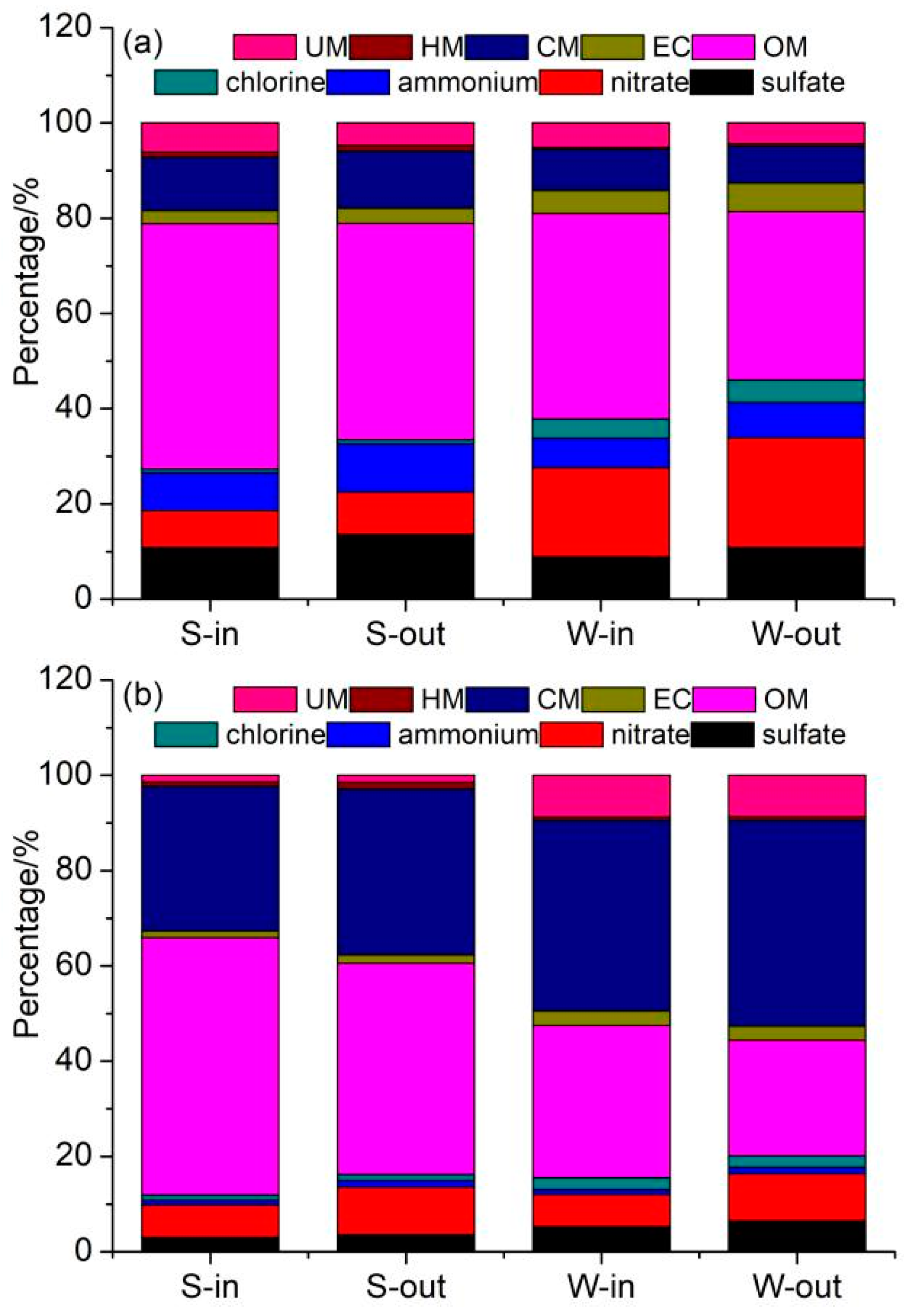
3.2. Chemical Components of PM
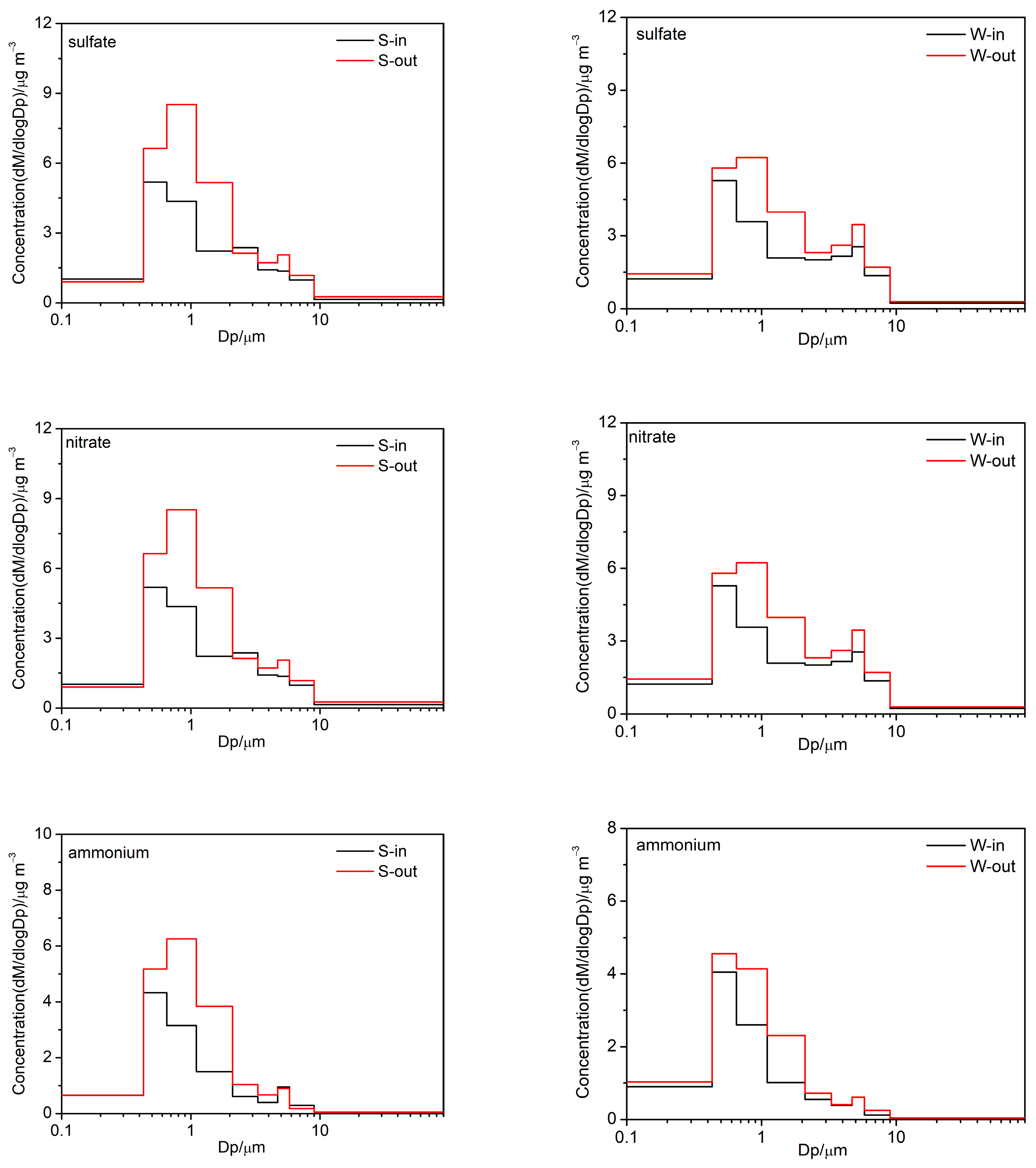
3.2.1. Water-Soluble Ions
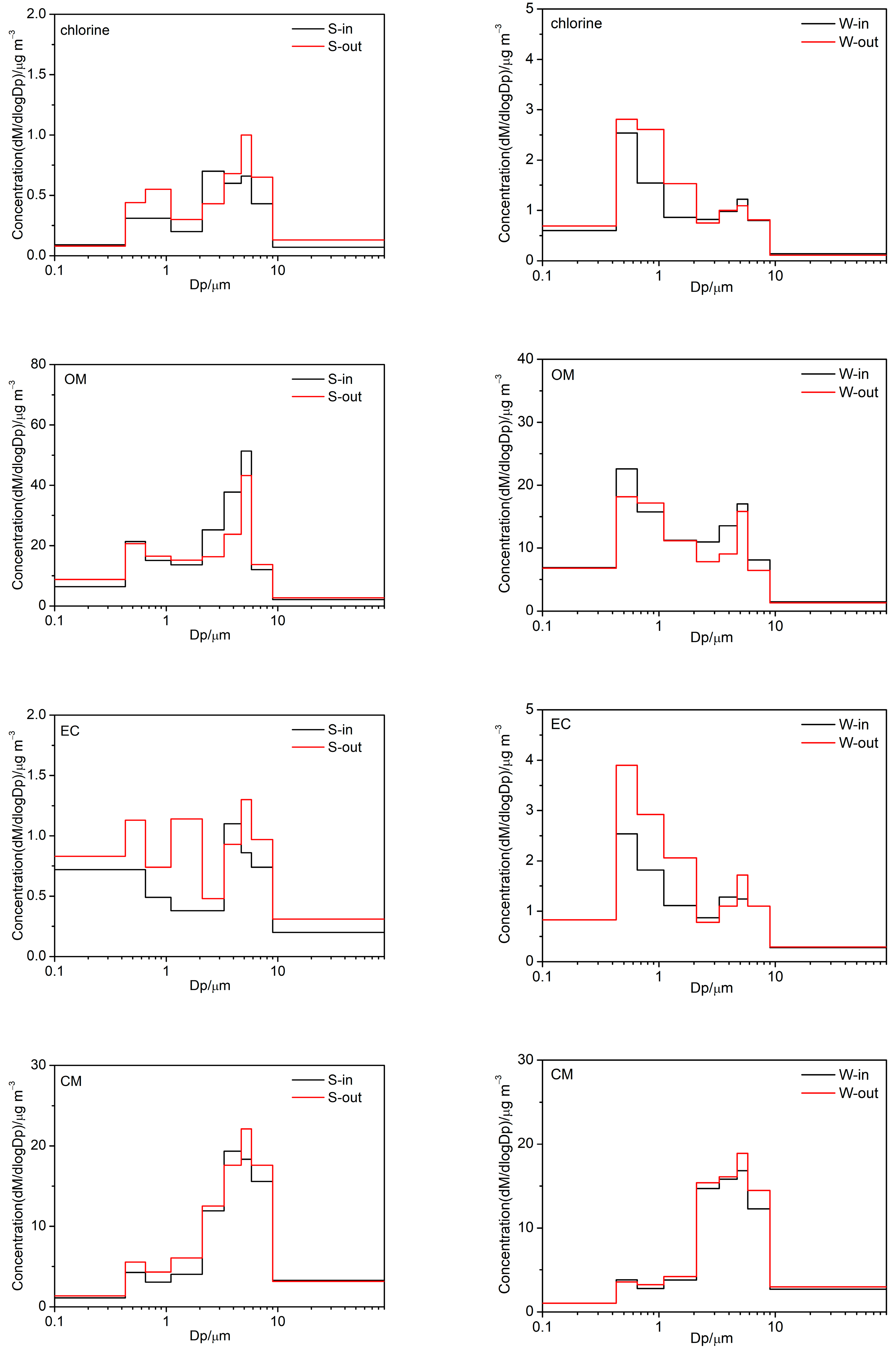
3.2.2. Carbonaceous Components
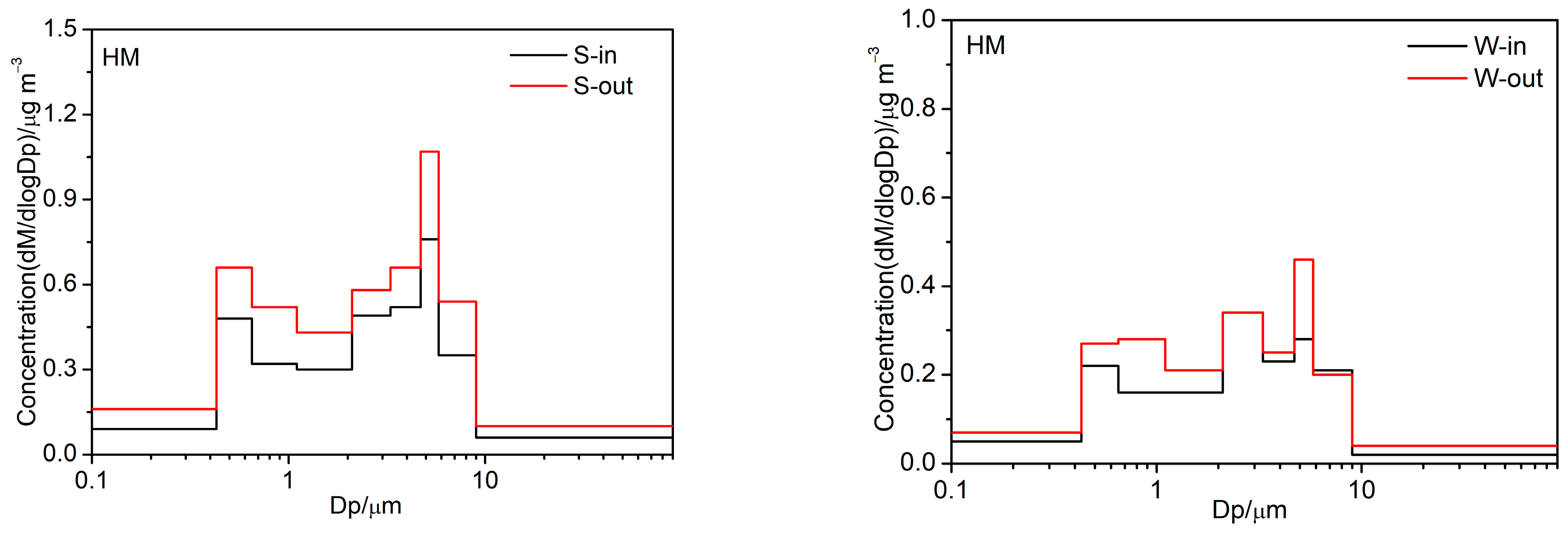
3.2.3. Trace Elements
3.3. Source Apportionment
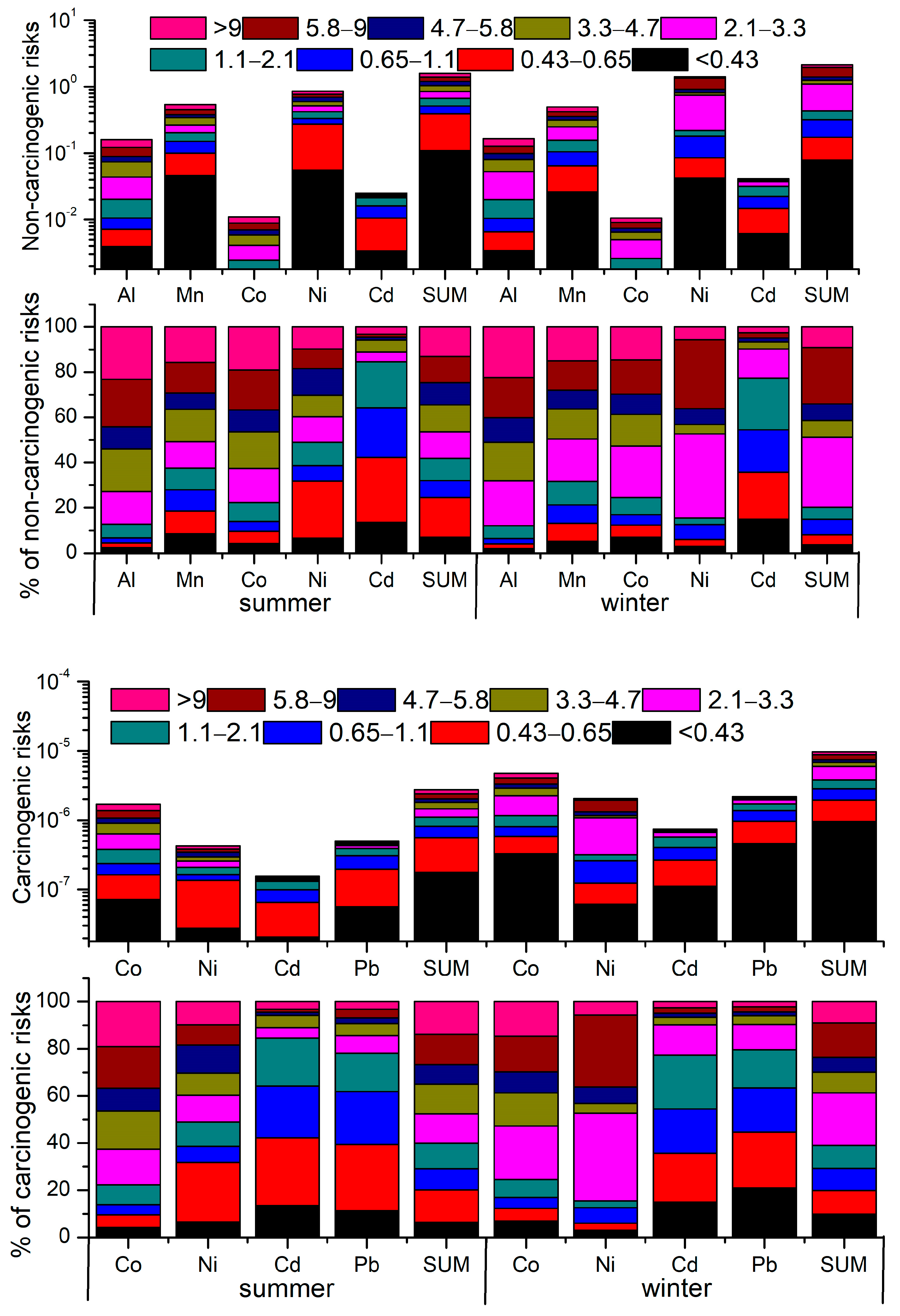
3.4. Possible Human Health Implications
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Definition | Abbreviation | Definition | Abbreviation | ||
| 1 | Particulate matter | PM | 12 | Clear days | C |
| 2 | Organic matter | OM | 13 | Summer indoor | S-in |
| 3 | Elemental carbon | EC | 14 | Summer outdoor | S-out |
| 4 | Crustal materials | CM | 15 | Winter indoor | W-in |
| 5 | Heavy metals | HM | 16 | Winter outdoor | W-out |
| 6 | Unidentified matter | UM | 17 | Ratios of indoor/outdoor | I/O |
| 7 | Positive matrix factorization | PMF | 18 | Sulfate–nitrate–ammonium | SNA |
| 8 | Reference concentration | RfC | 19 | Secondary organic carbon | SOC |
| 9 | Inhalation unit risk | IUR | 20 | Secondary inorganic aerosol | SIA |
| 10 | Hazard quotient | HQ | 21 | Carcinogenic risks | CR |
| 11 | Haze days | H | 22 | Total suspended particulates | TSP |
References
- Weber, R. A map of potentially harmful aerosols in Europe. Nature 2020, 587, 369–370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Jiang, X.J.; Tong, D.; Davis, S.J.; Zhao, H.Y.; Geng, G.N.; Feng, T.; Zheng, B.; Lu, Z.F.; Streets, D.G.; et al. Transboundary health impacts of transported global air pollution and international trade. Nature 2017, 543, 705. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J.S.; do Nascimento, R.D.S.; Cintra, J.; da Rosa, N.L.C.; Grosseli, G.M.; Fadini, P.S.; Urban, R.C. Source apportionment and health impact assessment of atmospheric particulate matter in the city of Sao Carlos, Brazil. Chemosphere 2023, 326, 138450. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, M.; Mal, P.; Saikia, N. Exploring the link between particulate matter pollution and acute respiratory infection risk in children using generalized estimating equations analysis: A robust statistical approach. Environ. Health 2024, 23, 12. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Song, X.; Xie, J.; Shi, T.; Yang, Q. Polycyclic aromatic hydrocarbons (PAHs) in ambient air of Guangzhou city: Exposure levels, health effects and cytotoxicity. Ecotoxicol. Environ. Saf. 2023, 262, 115308. [Google Scholar] [CrossRef] [PubMed]
- Loupa, G.; Zarogianni, A.-M.; Karali, D.; Kosmadakis, I.; Rapsomanikis, S. Indoor/outdoor PM2.5 elemental composition and organic fraction medications, in a Greek hospital. Sci. Total Environ. 2016, 550, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yang, L.; Meng, C.; Yuan, Q.; Yan, C.; Dong, C.; Sui, X.; Yao, L.; Yang, F.; Lu, Y. Indoor/outdoor relationships and diurnal/nocturnal variations in water-soluble ion and PAH concentrations in the atmospheric PM2.5 of a business office area in Jinan, a heavily polluted city in China. Atmos. Res. 2015, 153, 276–285. [Google Scholar] [CrossRef]
- Zhu, Y.; Yang, L.; Meng, C.; Yuan, Q.; Yan, C.; Dong, C.; Sui, X.; Yao, L.; Yang, F.; Lu, Y. PM2.5 Concentrations Indoors and Outdoors in Heavy Air Pollution Days in Winter. Procedia Eng. 2015, 121, 1902–1906. [Google Scholar]
- Ni, X.; Pan, Y.P.; Shao, P.; Tian, S.L.; Zong, Z.; Gu, M.N.; Liu, B.W.; Liu, J.; Cao, J.; Sun, Q. Size distribution and formation processes of aerosol water-soluble organic carbon during winter and summer in urban Beijing. Atmos. Environ. 2021, 244, 117983. [Google Scholar] [CrossRef]
- Tian, S.; Pan, Y.; Liu, Z.; Wen, T.; Wang, Y. Size-resolved aerosol chemical analysis of extreme haze pollution events during early 2013 in urban Beijing, China. J. Hazard. Mater. 2014, 279, 452–460. [Google Scholar] [CrossRef]
- Behera, S.N.; Cheng, J.; Huang, X.; Zhu, Q.; Liu, P.; Balasubramanian, R. Chemical composition and acidity of size-fractionated inorganic aerosols of 2013-14 winter haze in Shanghai and associated health risk of toxic elements. Atmos. Environ. 2015, 122, 259–271. [Google Scholar] [CrossRef]
- Swietlik, R.; Trojanowska, M. Chemical Fractionation in Environmental Studies of Potentially Toxic Particulate-Bound Elements in Urban Air: A Critical Review. Toxics 2022, 10, 124. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.K.; Kim, C.; Cho, J.L. Association between exposure to heavy metals in atmospheric particulate matter and sleep quality: A nationwide data linkage study. Environ. Res. 2024, 247, 118217. [Google Scholar] [CrossRef] [PubMed]
- Enyoh, C.E.; Verla, A.W.; Qingyue, W.; Ohiagu, F.O.; Chowdhury, A.H.; Enyoh, E.C.; Chowdhury, T.; Verla, E.N.; Chinwendu, U.P. An overview of emerging pollutants in air: Method of analysis and potential public health concern from human environmental exposure. Trends Environ. Anal. Chem. 2020, 28, e00107. [Google Scholar] [CrossRef]
- Wang, J.; Pan, Y.P.; Tian, S.L.; Chen, X.; Wang, L.; Wang, Y.S. Size distributions and health risks of particulate trace elements in rural areas in northeastern China. Atmos. Res. 2016, 168, 191–204. [Google Scholar] [CrossRef]
- Tong, Z.; Chen, Y.; Malkawi, A.; Adamkiewicz, G.; Spengler, J.D. Quantifying the impact of traffic-related air pollution on the indoor air quality of a naturally ventilated building. Environ. Int. 2016, 89–90, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Morawska, L.; Ayoko, G.A.; Bae, G.N.; Buonanno, G.; Chao, C.Y.H.; Clifford, S.; Fu, S.C.; Hänninen, O.; He, C.; Isaxon, C.; et al. Airborne particles in indoor environment of homes, schools, offices and aged care facilities: The main routes of exposure. Environ. Int. 2017, 108, 75–83. [Google Scholar] [CrossRef]
- Chen, L.; Wang, H.; Wang, Z.; Dong, Z. Estimating the mortality attributable to indoor exposure to particulate matter of outdoor origin in mainland China. Sci. Total Environ. 2023, 872, 162286. [Google Scholar] [CrossRef]
- Liu, X.; Xue, Q.; Tian, Y.; Jia, B.; Chen, R.; Huo, R.; Wang, X.; Feng, Y. Potential toxic components in size-resolved particles and gas from residential combustion: Emission factor and health risk. Environ. Int. 2024, 185, 108551. [Google Scholar] [CrossRef]
- Nassikas, N.J.; McCormack, M.C.; Ewart, G.; Balmes, J.R.; Bond, T.C.; Brigham, E.; Cromar, K.; Goldstein, A.H.; Hicks, A.; Hopke, P.K.; et al. Indoor Air Sources of Outdoor Air Pollution: Health Consequences, Policy, and Recommendations: An Official American Thoracic Society Workshop Report. Ann. Am. Thorac. Soc. 2024, 21, 365–376. [Google Scholar] [CrossRef]
- Huang, R.J.; Zhang, Y.L.; Bozzetti, C.; Ho, K.F.; Cao, J.J.; Han, Y.M.; Daellenbach, K.R.; Slowik, J.G.; Platt, S.M.; Canonaco, F.; et al. High secondary aerosol contribution to particulate pollution during haze events in China. Nature 2014, 514, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.L.; Pan, Y.P.; Wang, Y.S. Size-resolved source apportionment of particulate matter in urban Beijing during haze and non-haze episodes. Atmos. Chem. Phys. 2016, 16, 1–19. [Google Scholar] [CrossRef]
- Huang, R.J.; Zhang, Y.L.; Bozzetti, C.; Ho, K.F.; Cao, J.J.; Han, Y.M.; Daellenbach, K.R.; Slowik, J.G.; Platt, S.M.; Canonaco, F. Source apportionment of fine particulate matter at a megacity in China, using an improved regularization supervised PMF model. Sci. Total Environ. 2023, 879, 163198. [Google Scholar]
- Dao, X.; Di, S.; Zhang, X.; Gao, P.; Wang, L.; Yan, L.; Tang, G.; He, L.; Krafft, T.; Zhang, F. Composition and sources of particulate matter in the Beijing-Tianjin-Hebei region and its surrounding areas during the heating season. Chemosphere 2022, 291, 132779. [Google Scholar] [CrossRef]
- Yang, X.; Zheng, M.; Liu, Y.; Yan, C.; Liu, J.; Liu, J.; Cheng, Y. Exploring sources and health risks of metals in Beijing PM2.5: Insights from long-term online measurements. Sci. Total Environ. 2022, 814, 151954. [Google Scholar] [CrossRef] [PubMed]
- Karanasiou, A.A.; Siskos, P.A.; Eleftheriadis, K. Assessment of source apportionment by Positive Matrix Factorization analysis on fine and coarse urban aerosol size fractions. Atmos. Environ. 2009, 43, 3385–3395. [Google Scholar] [CrossRef]
- An, X.; Hou, Q.; Li, N.; Zhai, S. Assessment of human exposure level to PM10 in China. Atmos. Environ. 2013, 70, 376–386. [Google Scholar]
- Chen, L.; Pang, X.B.; Li, J.J.; Xing, B.; An, T.C.; Yuan, K.B.; Dai, S.; Wu, Z.T.; Wang, S.Q.; Wang, Q.; et al. Vertical profiles of O3, NO2 and PM in a major fine chemical industry park in the Yangtze River Delta of China detected by a sensor package on an unmanned aerial vehicle. Sci. Total Environ. 2022, 845, 157113. [Google Scholar] [CrossRef] [PubMed]
- Manojkumar, N.; Srimuruganandam, B. Size-segregated particulate matter characteristics in indoor and outdoor environments of urban traffic and residential sites. Urban Clim. 2022, 44, 101232. [Google Scholar] [CrossRef]
- Hassanvand, M.S.; Naddafi, K.; Faridi, S.; Nabizadeh, R.; Sowlat, M.H.; Momeniha, F.; Gholampour, A.; Arhami, M.; Kashani, H.; Zare, A.; et al. Characterization of PAHs and metals in indoor/outdoor PM10/PM2.5/PM1 in a retirement home and a school dormitory. Sci. Total Environ. 2015, 527–528, 100–110. [Google Scholar] [CrossRef]
- Hassanvand, M.S.; Naddafi, K.; Faridi, S.; Arhami, M.; Nabizadeh, R.; Sowlat, M.H.; Pourpak, Z.; Rastkari, N.; Momeniha, F.; Kashani, H.; et al. Indoor/outdoor relationships of PM10, PM2.5, and PM1 mass concentrations and their water-soluble ions in a retirement home and a school dormitory. Atmos. Environ. 2014, 82, 375–382. [Google Scholar] [CrossRef]
- Sangiorgi, G.; Ferrero, L.; Ferrini, B.; Porto, C.L.; Perrone, M.; Zangrando, R.; Gambaro, A.; Lazzati, Z.; Bolzacchini, E. Indoor airborne particle sources and semi-volatile partitioning effect of outdoor fine PM in offices. Atmos. Environ. 2013, 65, 205–214. [Google Scholar] [CrossRef]
- Andretta, M.; Coppola, F.; Seccia, L. Investigation on the interaction between the outdoor environment and the indoor microclimate of a historical library. J. Cult. Herit. 2016, 17, 75–86. [Google Scholar] [CrossRef]
- Tian, S.; Pan, Y.; Wang, Y. Ion balance and acidity of size-segregated particles during haze episodes in urban Beijing. Atmos. Res. 2018, 201, 159–167. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, X.; Ding, X.; He, Q.; Zhang, Z.; Liu, T.; Fu, X.; Gao, B.; Wang, Y.; Zhang, Y.; et al. Compositions and sources of organic acids in fine particles (PM2.5) over the Pearl River Delta region, south China. J. Environ. Sci. 2014, 26, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Kong, L.; Cheng, T.; Chen, J.; Du, J.; Li, L.; Xia, X.; Leng, C.; Huang, G. Insights into summertime haze pollution events over Shanghai based on online water-soluble ionic composition of aerosols. Atmos. Environ. 2011, 45, 5131–5137. [Google Scholar] [CrossRef]
- Cheng, Y.; Engling, G.; He, K.-b.; Duan, F.-k.; Du, Z.-y.; Ma, Y.-l.; Liang, L.-l.; Lu, Z.-f.; Liu, J.-m.; Zheng, M.; et al. The characteristics of Beijing aerosol during two distinct episodes: Impacts of biomass burning and fireworks. Environ. Pollut. 2014, 185, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Gupta, L.; Bansal, M.; Nandi, P.; Habib, G.; Raman, R.S. Source apportionment and potential source regions of size-resolved particulate matter at a heavily polluted industrial city in the Indo-Gangetic Plain. Atmos. Environ. 2023, 298, 119614. [Google Scholar] [CrossRef]
- Luo, L.; Bai, X.; Liu, S.; Wu, B.; Liu, W.; Lv, Y.; Guo, Z.; Lin, S.; Zhao, S.; Hao, Y.; et al. Fine particulate matter (PM2.5/PM1.0) in Beijing, China: Variations and chemical compositions as well as sources. J. Environ. Sci. 2022, 121, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Si, R.; Xin, J.; Zhang, W.; Wen, T.; Li, S.; Ma, Y.; Wu, X.; Cao, Y.; Xu, X.; Tang, H.; et al. Environmental and health benefits of establishing a coal banning area in the Beijing-Tianjin-Hebei region of China. Atmos. Environ. 2021, 247, 118191. [Google Scholar] [CrossRef]
- Tian, Y.; Li, Y.; Liang, Y.; Xue, Q.; Feng, X.; Feng, Y. Size distributions of source-specific risks of atmospheric heavy metals: An advanced method to quantify source contributions to size-segregated respiratory exposure. J. Hazard. Mater. 2020, 407, 124355. [Google Scholar] [CrossRef]
- Dai, Q.L.; Bi, X.H.; Huangfu, Y.Q.; Yang, J.M.; Li, T.K.; Khan, J.Z.; Song, C.B.; Xu, J.; Wu, J.H.; Zhang, Y.F.; et al. A size-resolved chemical mass balance (SR-CMB) approach for source apportionment of ambient particulate matter by single element analysis. Atmos. Environ. 2019, 197, 45–52. [Google Scholar] [CrossRef]
- Pan, Y.P.; Liu, J.; Zhang, L.; Cao, J.; Hu, J.B.; Tian, S.L.; Li, X.Y.; Xu, W. Bulk Deposition and Source Apportionment of Atmospheric Heavy Metals and Metalloids in Agricultural Areas of Rural Beijing during 2016–2020. Atmosphere 2021, 12, 13. [Google Scholar] [CrossRef]
- Zhu, Y.; Huang, L.; Li, J.; Ying, Q.; Zhang, H.; Liu, X.; Liao, H.; Li, N.; Liu, Z.; Mao, Y.; et al. Sources of particulate matter in China: Insights from source apportionment studies published in 1987–2017. Environ. Int. 2018, 115, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liu, Y.; Yang, Z.; Qi, X.; Schauer, J.J. High loadings of carbonaceous aerosols from wood smoke in the atmosphere of Beijing from 2015 to 2017: Implications for energy transition policy. Environ. Pollut. 2024, 344, 123240. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S-in | S-out | W-in | W-out | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PM2.1 | PM2.1-9 | PM2.1 | PM2.1-9 | PM2.1 | PM2.1-9 | PM2.1 | PM2.1-9 | |||||||||
| Mean | Range | Mean | Range | Mean | Range | Mean | Range | Mean | Range | Mean | Range | Mean | Range | Mean | Range | |
| PM | 29.4 | 19.1–44.7 | 32.8 | 24.4–43.0 | 38.1 | 23.7–64.1 | 30.3 | 21.8–43.4 | 35.2 | 16.1–54.2 | 23.0 | 12.7–30.5 | 41.4 | 20.2–64.7 | 23.1 | 13.2–30.8 |
| OC | 9.47 | 6.98–12.25 | 11.08 | 8.42–13.84 | 10.81 | 8.06–14.78 | 8.38 | 6.23–11.50 | 9.48 | 4.67–13.94 | 4.59 | 2.83–6.10 | 9.14 | 4.91–13.85 | 3.50 | 2.38–4.71 |
| EC | 0.80 | 0.50–1.14 | 0.46 | 0.33–0.56 | 1.22 | 0.88–1.48 | 0.54 | 0.30–0.86 | 1.71 | 1.13–2.89 | 0.69 | 0.56–1.03 | 2.47 | 1.64–4.24 | 0.69 | 0.65–1.06 |
| Na+ | 0.25 | 0.16–0.40 | 0.37 | 0.26–0.49 | 0.31 | 0.20–0.50 | 0.34 | 0.19–0.61 | 0.50 | 0.32–0.68 | 0.64 | 0.42–0.85 | 0.63 | 0.42–0.84 | 0.67 | 0.42–0.94 |
| NH4+ | 2.34 | 0.95–3.88 | 0.32 | 0.20–0.47 | 3.85 | 1.57–6.48 | 0.42 | 0.22–0.67 | 2.18 | 0.78–3.71 | 0.25 | 0.15–0.35 | 3.07 | 1.11–5.29 | 0.31 | 0.17–0.46 |
| K+ | 0.14 | 0.11–0.21 | 0.11 | 0.09–0.16 | 0.22 | 0.16–0.33 | 0.10 | 0.07–0.14 | 0.46 | 0.34–0.78 | 0.18 | 0.14–0.29 | 0.58 | 0.41–0.98 | 0.20 | 0.16–0.33 |
| Mg2+ | 0.44 | 0.35–0.63 | 0.75 | 0.59–1.02 | 0.48 | 0.39–0.65 | 0.58 | 0.42–0.87 | 0.36 | 0.23–0.47 | 0.68 | 0.42–0.95 | 0.33 | 0.25–0.42 | 0.66 | 0.37–0.99 |
| Ca2+ | 1.61 | 1.25–1.88 | 3.14 | 2.35–3.91 | 1.89 | 1.44–2.45 | 3.41 | 2.56–4.48 | 2.47 | 1.66–3.67 | 6.57 | 4.61–9.24 | 3.19 | 2.64–4.03 | 6.99 | 4.72–9.96 |
| Cl− | 0.24 | 0.17–0.35 | 0.37 | 0.28–0.48 | 0.34 | 0.24–0.51 | 0.40 | 0.21–0.81 | 1.42 | 0.71–2.53 | 0.57 | 0.39–0.91 | 1.97 | 1.06–3.40 | 0.56 | 0.41–0.87 |
| NO3− | 2.26 | 1.02–4.54 | 2.24 | 1.18–3.96 | 3.39 | 1.17–7.85 | 3.01 | 1.63–5.54 | 6.59 | 1.89–10.14 | 1.53 | 0.89–1.83 | 9.51 | 2.98–15.43 | 2.29 | 1.05–3.15 |
| SO42− | 3.20 | 1.33–8.09 | 0.99 | 0.95–1.44 | 5.16 | 2.47–13.63 | 1.10 | 0.73–2.59 | 3.13 | 1.10–5.28 | 1.22 | 0.60–1.75 | 4.49 | 1.64–7.50 | 1.50 | 0.72–2.27 |
| Na | 0.5525 | 0.4259–0.8061 | 0.6736 | 0.5185–0.9847 | 0.6821 | 0.4883–1.0687 | 0.6843 | 0.5281–1.0098 | 0.3865 | 0.1945–0.5496 | 0.4833 | 0.2653–0.6550 | 0.4209 | 0.2496–0.5349 | 0.5135 | 0.2761–0.6949 |
| Mg | 0.1756 | 0.1179–0.2186 | 0.5426 | 0.3582–0.6880 | 0.1888 | 0.1106–0.2887 | 0.4452 | 0.3269–0.5324 | 0.1503 | 0.0684–0.2181 | 0.4234 | 0.2043–0.5927 | 0.1247 | 0.0723–0.1608 | 0.3486 | 0.1760–0.4889 |
| Al | 0.0937 | 0.0616–0.1360 | 0.5062 | 0.3871–0.6480 | 0.1392 | 0.0775–0.2542 | 0.5427 | 0.4272–0.6712 | 0.0996 | 0.0383–0.1907 | 0.5309 | 0.2666–0.8567 | 0.0986 | 0.0722–0.1264 | 0.5769 | 0.3239–0.8573 |
| K | 0.1852 | 0.1165–0.2542 | 0.2128 | 0.1482–0.2706 | 0.2446 | 0.1405–0.3742 | 0.2235 | 0.1617–0.2723 | 0.2845 | 0.0991–0.5148 | 0.2358 | 0.1298–0.3352 | 0.3155 | 0.1558–0.4946 | 0.2737 | 0.1333–0.4311 |
| Ca | 0.6098 | 0.4822–0.7709 | 1.8422 | 1.4379–2.3587 | 0.9028 | 0.5331–1.4901 | 2.0038 | 1.6071–2.5326 | 0.4989 | 0.3128–0.7386 | 1.2514 | 0.8256–1.7930 | 0.5665 | 0.4675–0.7178 | 1.3817 | 0.9664–1.9301 |
| V | 0.0003 | 0.0001–0.0005 | 0.0008 | 0.0005–0.0013 | 0.0005 | 0.0002–0.0013 | 0.0008 | 0.0006–0.0011 | 0.0003 | 0.0001–0.0006 | 0.0010 | 0.0005–0.0013 | 0.0005 | 0.0003–0.0007 | 0.0011 | 0.0006–0.0015 |
| Cr | 0.0039 | 0.0021–0.0059 | 0.0042 | 0.0025–0.0056 | 0.0048 | 0.0027–0.0073 | 0.0068 | 0.0035–0.0108 | 0.0051 | 0.0028–0.0082 | 0.0066 | 0.0032–0.0114 | 0.0038 | 0.0031–0.0058 | 0.0118 | 0.0040–0.0217 |
| Mn | 0.0095 | 0.0056–0.0158 | 0.0123 | 0.0083–0.0181 | 0.0128 | 0.0073–0.0224 | 0.0138 | 0.0091–0.0202 | 0.0078 | 0.0038–0.0139 | 0.0127 | 0.0089–0.0187 | 0.0082 | 0.0067–0.0110 | 0.0158 | 0.0098–0.0243 |
| Fe | 0.2663 | 0.1659–0.3538 | 0.6707 | 0.3937–0.9452 | 0.3849 | 0.1825–0.6526 | 0.7874 | 0.4789–1.0579 | 0.2553 | 0.1350–0.4349 | 0.8596 | 0.5417–1.3227 | 0.2868 | 0.2437–0.3758 | 1.0331 | 0.6716–1.5598 |
| Co | 0.0001 | 0.0001–0.0002 | 0.0003 | 0.0002–0.0004 | 0.0002 | 0.0001–0.0003 | 0.0004 | 0.0002–0.0006 | 0.0001 | 0.0001–0.0002 | 0.0003 | 0.0002–0.0004 | 0.0002 | 0.0001–0.0002 | 0.0004 | 0.0002–0.0006 |
| Ni | 0.0025 | 0.0018–0.0037 | 0.0021 | 0.0013–0.0038 | 0.0027 | 0.0017–0.0049 | 0.0024 | 0.0017–0.0042 | 0.0008 | 0.0007–0.0026 | 0.0072 | 0.0028–0.0135 | 0.0038 | 0.0015–0.0067 | 0.0045 | 0.0010–0.0125 |
| Cu | 0.0117 | 0.0079–0.0204 | 0.0074 | 0.0049–0.0131 | 0.0172 | 0.0084–0.0346 | 0.0095 | 0.0085–0.0133 | 0.0066 | 0.0060–0.0167 | 0.0285 | 0.0125–0.0559 | 0.0251 | 0.0217–0.0340 | 0.0153 | 0.0059–0.0461 |
| Zn | 0.0487 | 0.0351–0.0946 | 0.0266 | 0.0205–0.0490 | 0.0599 | 0.0428–0.1181 | 0.0248 | 0.0232–0.0352 | 0.0444 | 0.0273–0.0632 | 0.0232 | 0.0139–0.0333 | 0.0490 | 0.0371–0.0612 | 0.0328 | 0.0149–0.0530 |
| As | 0.0012 | 0.0005–0.0023 | 0.0006 | 0.0004–0.0010 | 0.0021 | 0.0008–0.0037 | 0.0003 | 0.0004–0.0007 | 0.0013 | 0.0006–0.0019 | 0.0005 | 0.0003–0.0007 | 0.0013 | 0.0007–0.0019 | 0.0007 | 0.0003–0.0011 |
| Mo | 0.0003 | 0.0002–0.0004 | 0.0003 | 0.0002–0.0004 | 0.0006 | 0.0003–0.0010 | 0.0005 | 0.0004–0.0006 | 0.0002 | 0.0001–0.0004 | 0.0003 | 0.0002–0.0004 | 0.0003 | 0.0002–0.0004 | 0.0005 | 0.0002–0.0007 |
| Cd | 0.0002 | 0.0001–0.0004 | 0.0000 | 0.0000–0.0001 | 0.0003 | 0.0002–0.0006 | 0.0000 | 0.0000–0.0001 | 0.0003 | 0.0001–0.0005 | 0.0001 | 0.0000–0.0001 | 0.0003 | 0.0002–0.0004 | 0.0001 | 0.0000–0.0002 |
| Sb | 0.2177 | 0.1558–0.3093 | 0.2500 | 0.1794–0.3545 | 0.3435 | 0.2377–0.5302 | 0.3476 | 0.2442–0.5308 | 0.0707 | 0.0426–0.0903 | 0.0836 | 0.0486–0.1087 | 0.1004 | 0.0585–0.1318 | 0.0967 | 0.0547–0.1269 |
| Ba | 0.0045 | 0.0036–0.0054 | 0.0156 | 0.0119–0.0193 | 0.0074 | 0.0042–0.0122 | 0.0184 | 0.0137–0.0232 | 0.0047 | 0.0028–0.0072 | 0.0126 | 0.0076–0.0194 | 0.0064 | 0.0035–0.0104 | 0.0130 | 0.0094–0.0186 |
| Pb | 0.0130 | 0.0080–0.0200 | 0.0033 | 0.0019–0.0054 | 0.0204 | 0.0109–0.0353 | 0.0036 | 0.0026–0.0049 | 0.0214 | 0.0111–0.0329 | 0.0044 | 0.0027–0.0064 | 0.0225 | 0.0136–0.0350 | 0.0075 | 0.0026–0.0136 |
| Summer | Winter | ||||||
|---|---|---|---|---|---|---|---|
| PM2.1 | PM9 | TSP | PM2.1 | PM9 | TSP | ||
| HQ | Al | 0.02 | 0.12 | 0.16 | 0.02 | 0.13 | 0.16 |
| Mn | 0.20 | 0.45 | 0.54 | 0.16 | 0.42 | 0.50 | |
| Co | 0.00 | 0.01 | 0.01 | 0.00 | 0.01 | 0.01 | |
| Ni | 0.42 | 0.77 | 0.86 | 0.22 | 1.34 | 1.42 | |
| Cd | 0.02 | 0.02 | 0.03 | 0.03 | 0.04 | 0.04 | |
| Sum | 0.67 | 1.38 | 1.59 | 0.43 | 1.94 | 2.14 | |
| CR | Co | 3.78 × 10−7 | 1.37 × 10−6 | 1.69 × 10−6 | 1.17 × 10−6 | 4.05 × 10−6 | 4.75 × 10−6 |
| Ni | 2.07 × 10−7 | 3.82 × 10−7 | 4.24 × 10−7 | 3.18 × 10−7 | 1.93 × 10−6 | 2.05 × 10−6 | |
| Cd | 1.31 × 10−7 | 1.50 × 10−7 | 1.55 × 10−7 | 5.73 × 10−7 | 7.22 × 10−7 | 7.40 × 10−7 | |
| Pb | 3.90 × 10−7 | 4.82 × 10−7 | 4.99 × 10−7 | 1.73 × 10−6 | 2.12 × 10−6 | 2.17 × 10−6 | |
| Sum | 1.11 × 10−6 | 2.38 × 10−6 | 2.77 × 10−6 | 3.78 × 10−6 | 8.83 × 10−6 | 9.71 × 10−6 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, S.; Wang, L.; Liu, Q.; Luo, L.; Qian, C.; Wang, B.; Liu, Y. Associations between Indoor and Outdoor Size-Resolved Particulate Matter in Urban Beijing: Chemical Compositions, Sources, and Health Risks. Atmosphere 2024, 15, 721. https://doi.org/10.3390/atmos15060721
Tian S, Wang L, Liu Q, Luo L, Qian C, Wang B, Liu Y. Associations between Indoor and Outdoor Size-Resolved Particulate Matter in Urban Beijing: Chemical Compositions, Sources, and Health Risks. Atmosphere. 2024; 15(6):721. https://doi.org/10.3390/atmos15060721
Chicago/Turabian StyleTian, Shili, Liming Wang, Qingyang Liu, Liang Luo, Chunyan Qian, Baocheng Wang, and Yanju Liu. 2024. "Associations between Indoor and Outdoor Size-Resolved Particulate Matter in Urban Beijing: Chemical Compositions, Sources, and Health Risks" Atmosphere 15, no. 6: 721. https://doi.org/10.3390/atmos15060721