Abstract
The bacterial community is an essential component of the aquaculture pond ecosystem, which not only improves and restores the aquaculture environment but also maintains a stable ecological equilibrium with the external environment. Here, Illumina 16S rRNA sequencing was conducted to characterize the bacterial community in the ecosystem of sea cucumber Apostichopus japonicus culture ponds, as well as their correlation with overall community structures. The alpha-diversities of bacterial community among water, sediment, and the gut of A. japonicus were consistent across culture ponds from different areas. Specifically, the richness and diversity of bacterial communities were the highest in sediment, followed by the gut, and the lowest in water. The dominant bacterial community among multiple media was Proteobacteria, which occupies a large proportion of the bacterial community structure, followed by Bacteroidetes and Verrucomicrobia. Highly similar bacterial community structures were present in multiple media among different areas, which provides evidence for deterministic natural evolution. Meanwhile, there was a significant difference (p < 0.05) in the specific bacterial communities across the multiple media. The specific functions of the multiple media in the ecosystem are the main reason for the formation of different bacterial communities. This work demonstrates that bacterial communities are the result of natural evolution within the ecosystem during adaptation to the required environment.
1. Introduction
The sea cucumber (Apostichopus japonicus) is a marine organism used for medicinal foods and has high economic and medicinal value [1]. A. japonicus aquaculture has been developing rapidly with increasing consumer demand in recent years. The annual output of this industry has reached over 170 thousand tonnes, which has brought about a profit of more than 100 million pounds in China [2]. Pond culture is an important aquaculture model that has made enormous contributions to the development of A. japonicus aquaculture [3]. The expansion of the scale of pond culture practices has resulted in several prominent problems, however, such as environmental destruction and biotic diseases [4].
Bacterial communities are critical to the health of the A. japonicus aquaculture pond ecosystem [5]. Environmental bacterial communities play a role in improving and restoring aquaculture environments [6]. Kirchman et al. (2002) found that Cytophaga could hydrolyze organic matter such as carbon polymers and proteins in seawater habitats. These bacteria produce a variety of extracellular enzymes to break down or utilize complex carbon sources, providing balance to the aquaculture water ecosystem [7]. Zhang et al. (2013) suggested that Kiloniella can conduct denitrification using nitrate or nitrite, hence removing nitrite pollution from water and resulting in water bioremediation [8]. Lage O M et al. (2012) found that Rhodopirellula can perform dissimilatory nitrate reduction to ammonium (DNRA) and cause the mineralization of organic matter, which has a positive effect on improving the culture water environment [9].
There is a mutualistic symbiosis relationship between the environmental bacterial community and A. japonicus. Bacterial communities can impact a number of factors related to the cultured species, such as survival, growth, disease, and immunity [5,10,11,12]. For example, Zhang et al. (2016) found that Pseudomonas could degrade organic matter and produce a variety of digestive enzymes that promote feed utilization and digestion [13]. Ma et al. (2018) suggested that Rhodotorula and Metschnikowia enhance digestive enzyme activity and stimulate the innate immune system of sea cucumber to promote its growth [14]. Backer et al. (2004) showed that Vibrio splendidus was the main pathogenic bacteria causing skin ulceration syndrome of cultured sea cucumbers [15]. Qin et al. (2010) suggested Bacillus subtilis had a positive effect on immune response and disease resistance when fed to ovine nematodes [16].
The complex bacterial community in the gut of sea cucumber derives from the habitat and maintains a relatively stable equilibrium balance with the external environment [17]. At the same time, the gut bacterial community also plays a key role in the gut micro-ecosystem. A combined system where there is interdependence and collaboration between the bacteria and their host is formed [18]. However, little academic attention has been paid to the structural characteristics of the bacterial community in A. japonicus gut and aquaculture pond environment, for which bacterial disease prevention and control for sea cucumber aquaculture is implemented without a theoretical basis. To improve the ecological environment of aquaculture ponds and enhance the disease resistance of A. japonicus more effectively, studying the characteristics of the bacterial community in A. japonicus gut and aquaculture environment will provide a healthy aquaculture and prevent disease with theoretical references.
One of the difficulties in analyzing the structure of bacterial communities is that many microorganisms usually appear in a symbiotic system in nature, which makes it difficult to accurately reflect the truth of the microorganism community in nature using traditional culture methods [19]. By avoiding bacterial culture, molecular biological techniques are able to make up for the shortcomings of traditional analytical methods [20]. With the development of molecular sequencing technology in the past few years, 16S rRNA-based amplicon sequencing has become an important means for studying the structure of bacterial communities since the comprehensive analysis of the species and population is its greatest advantage [21].
Therefore, the bacterial community in the typical shore-based half-open A. japonicus aquaculture ponds ecosystem of northern China was characterized using 16S rRNA amplicon sequencing. In addition, the correlation and specificity between the bacterial community in multiple media were analyzed. The results provide a theoretical foundation for healthy A. japonicus aquaculture and the microecological regulation of the aquaculture environment.
2. Materials and Methods
2.1. Sample Collection
Four typical shore-based half-open A. japonicus aquaculture ponds of northern China, namely Changhai (CH) (39°16′42″ N; 122°36′5″ E), Yingkou (YK) (40°37′46″ N; 122°8′59″ E), Laoting (LT) (39°52′46″ N; 119°18′37″ E) and Rushan (RS) (36°91′4″ N; 121°53′24″ E), were selected (Figure 1). The ponds were approximately 1 km2, 2–3 m deep, mostly muddy. The water source came from costal, and the tide difference brought into water and drainage. The density of sea cucumbers is 3000 per mu, and the size is about 100 g per head. It was ensured that no feed and no probiotics were provided throughout the year, allowing A. japonicus to grow naturally. Random sampling was used in the same period to ensure the reliability of the sample information.
Figure 1.
Sampling locations in four typical shore-based half-open Apostichopus japonicus culture ponds in northern China: Changhai (CH), Yingkou (YK), Laoting (LT), and Rushan (RS).
Water (w), sediment (S), and the gut of A. japonicus (G) samples of culture ponds in different areas were collected, respectively. Four sampling points were set up in each culture pond, 2 L water samples were collected with a glass water collector, and the surface sediments at the bottom of the pond were collected with a mud collector and brought back to the laboratory immediately under low-temperature conditions. Water samples were suction filtered using a 0.8 μm sterile filter and a 0.22 μm cellulose acetate membrane. The gut of A. japonicus was washed with sterile seawater and the excess water was dried with an absorbent paper towel. The body wall of A. japonicus was cut with a sterile scalpel and the surface of the intestine was rinsed with 70% ethanol, placed in a sterile 1.5 mL centrifuge tube, and stored immediately in liquid nitrogen. The A. japonicus gut, filter membrane, and sediment samples were put into sterile centrifuge tubes and stored in a −80°C freezer for the total bacterial DNA extraction.
2.2. DNA Extraction, Amplification and Sequencing
All samples were set in 3 parallels, placed in pre-cooled PBS buffer, fully shaken, soaked at 4 °C for 2~4 h, and centrifuged at 800 r per minute for 10 min, and the supernatant was placed in a 1.5 mL sterile centrifuge tube. After centrifuging at 800 r/min for 10 min, the supernatant was combined, and finally centrifuged at 10,000 r/min for 10 min. The supernatant was discarded, and the precipitate was collected for DNA extraction.
The total DNA of samples was extracted using the OMEGA Soil DNA Kit (D5625), with DNA concentration and purity determined by a NanoDrop2000 spectrophotometer. Sample concentration was ≥50 ng/μL and had purity (OD260/280) ranging from 1.8 to 2.0. To confirm DNA integrity and the absence of contamination, DNA was detected by 1% agarose gel electrophoresis and showed clear and intact bands without trailing, indicating that the DNA was free from protein contamination and that no degradation occurred; hence, the quality was satisfactory. The primers for the amplification of the V3-V4 fragment of the bacterial 16S rRNA gene were 341F (5′-CCTACGGGNGGCWGCAG-3′) and 805R (5′-GACTACHVGGGTATCTAATCC-3′). The samples were performed by Sangon Biotech (Shanghai) for high-throughput sequencing using the IlluminaMiseq 2 × 300 bp sequencing platform.
2.3. Data Analysis
The following is the process of processing the raw data after sequencing.
Quality control: cutadapt software (version 1.10) was used to QC the 3′ end of the sequence, and the splice sequence was: TGGAATTCTCGGGTGCCAAGGAACTC. Prinseq-lite software (version 0.20.4) was used to excise the tail region of double-end sequences with slightly lower quality values. The double-end sequences were fused by pear software (version 1.9.4) and barcode splitting was conducted to match the sequences to their corresponding samples. Likewise, primer sequences, short fragments, low-complexity sequences, and low-quality sequences were removed from each sample using Prinseq-lite software.
The removal of chimeric and non-target region sequences: Usearch software (version 8.1.1831) was carried out to de-sequence non-specifically amplified regions after preprocessing, while correcting for sequencing errors, and uchime software (version 4.2) was used to identify and remove chimeric sequences. The chimeric sequences were then compared with representative sequences from the database using blastn, and sequences with less than 80% similarity were considered out-of-target regions; these parts of the sequences were removed.
Operational taxonomic unit (OTU) clustering was performed with a sequence similarity threshold of 97% using Usearch software (version 8.1.1831). Alpha-diversity analysis of the sample sequences was performed using Mothur software (version 1.30.1). Beta-diversity of weighted unifrac was calculated by QIIME software (version 1.9.1). PCoA analysis was displayed in R software (version 2.15.3) by the WGCNA package, the stat package and the ggplot2 package. RDP Classifier software (version 2.12) was used to perform species classification operations on sample-eligible sequences with a threshold set at 0.8, below which classification results were considered an unordered class. Based on the abundance values of each rank at different levels obtained from the species classification results, STAMP software (version 2.1.3) was used to compare the differences in abundance between samples or groups with a screening condition of p ≤ 0.05. Species comparison among groups was computed by Tukey’s HSD test and Kruskal–Wallis H test in R project Vegan package (version 2.5.3). Biomarker features in each group were screened by LEfSe software (version 1.0), randomforest package (version 4.6.12) in R project, pROC package (version 1.10.0) in R project, and labdsv package (version2.0-1) in R project.
3. Results
This section is divided by subheadings. It provides a concise and precise description of the experimental results, their interpretation, and the experimental conclusions that can be drawn.
3.1. High-Throughput Sequencing Results
With low-quality sequences, short sequences, and chimera removed by means of high-throughput sequencing, there were 41,455 ± 6248, 35,799 ± 3425, 32,569 ± 3842, and 39,413 ± 7085 valid sequences that clustered into 694 ± 10, 1584 ± 626, 1227 ± 124, and 874 ± 178 OTUs, which were obtained from the water samples of CH, YK, LT, and RS. A total of 57,680 ± 2589, 45,221 ± 6794, 42,666 ± 11,230, and 54,859 ± 12,179 valid sequences were obtained in the sediment, clustered in 3431 ± 643, 3971 ± 308, 2593 ± 943, and 2622 ± 522 OTUs, respectively. In total, 41,028 ± 2591, 63,776 ± 4608, 47,313 ± 5125, and 52,218 ± 8360 valid sequences were obtained in A. japonicus gut, clustered in 1478 ± 81, 1987 ± 286, 1443 ± 117, and 1104 ± 92 OTUs, respectively. The sequencing depth of every sample reached over 97%, which indicates that the sequencing had favorably covered the diversity of the bacterial community (Table 1).

Table 1.
High-throughput sequencing results.
3.2. Alpha-Diversity Analysis
The Shannon index and the Simpson index are used for predicting community diversity in ecology. A larger Shannon index and a lower Simpson index suggest higher community diversity. The Chao1 index and the ACE index are used to quantify the richness of species, with higher values indicating higher species abundance. The results of this study showed consistent alpha-diversity indices which was highest in sediment, followed by the gut of A. japonicus, and lowest in water. This indicates that the highest richness and diversity of the bacterial community was found in sediments, followed by the gut of A. japonicus and water (Figure 2).
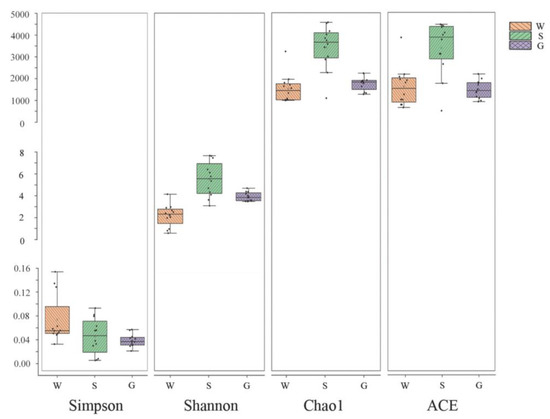
Figure 2.
Alpha-diversity indices of bacterial communities among multiple media (water (W), sediment (S), and gut of Apostichopus japonicus (G)) in different areas. The Shannon index and Simpson index show differences in species diversity in multiple media between different areas, and Chao1 index and ACE index show differences in species richness in multiple media between different areas.
3.3. Beta-Diversity Analysis
The structural diversity of the bacterial community between samples in each group was assessed by three-dimensional PCoA analysis based on weighted Unifrac distances at the OTU level. The contribution rates of the three principal component axes are PCo1 (45.0%), PCo2 (25.0%), and PCo3 (7.0%), respectively (Figure 3). Samples were distributed in different quadrants, where PCo1 showed good biological reproducibility and separation from PCo2, while PCo3 had a partial overlap but large overall differences with PCo1 and PCo2, indicating that there were some similarities between the three groups of water, sediment, and gut samples from different areas (LT, CH, RS, and YK) but large overall differences.
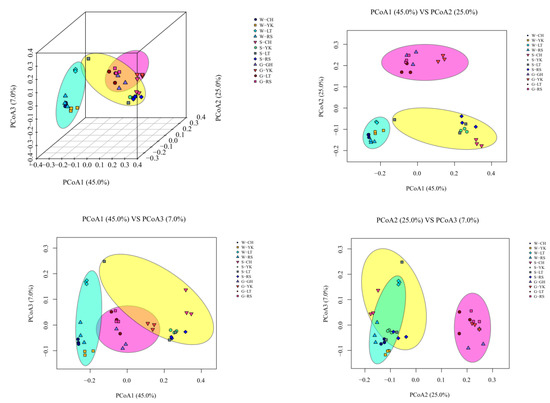
Figure 3.
Beta-diversity analyses of the bacterial communities of water (W), sediments (S), and the gut of Apostichopus japonicus (G) in different areas. Three-dimensional principal coordinate analysis (PCoA) of bacterial communities in different samples based on OTU-level weighted Unifrac distances.
The cluster tree of samples could directly reflect the similarity and difference among samples by the structure of branches. A hierarchical cluster analysis was performed based on the beta-diversity distance matrix, and then the unweighted group average method UPGMA (Unweighted pair method group with arithmetic mean) was used to construct tree structure; the tree form was obtained for visual analysis (Figure 4). When the distance was about 0.1–0.2, the samples were grouped into three groups according to the media, which showed that there were differences between multiple media, but the differences between different areas of the same media were small.
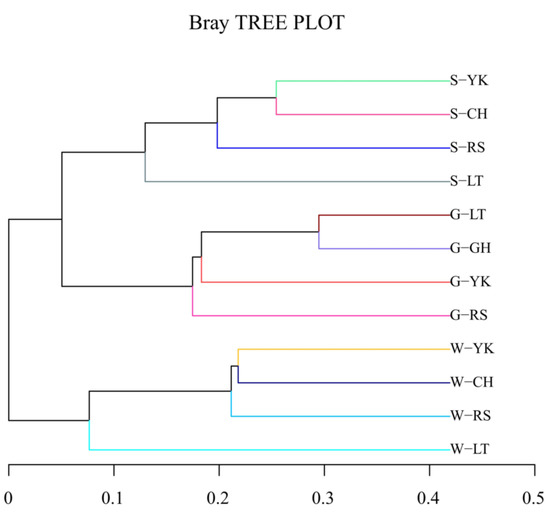
Figure 4.
Cluster dendrogram. The length of the branches represents the distance between the samples; the more similar the samples are, the closer they will be. The branches of the same color in the figure represent the same group.
3.4. Characteristics of Bacterial Communities in the Ecosystem of A. japonicus Culture Ponds
3.4.1. Water
Proteobacteria, Bacteroides, Actinobacteria, and Verrucomicrobia were the main dominant phyla in the water in different areas (Figure 5), with their relative abundance reaching 97.53–99.69% in total. The most predominant phylum of different areas was Proteobacteria, with relative abundances of 49.54%, 51.99%, 64.84%, and 49.43% in CH, YK, LT, and RS, respectively. The second most abundant phylum, Bacteroidetes, had values of 27.28%, 22.45%, and 24.22% for CH, YK, and RS, respectively. The third and fourth abundant phylum showed significant differences (p < 0.05). The relative abundance of Actinobacteria was significantly higher in YK (16.17%) and RS (20.89%) than in CH (7.25%) and LT (3.71%), while the relative abundance of Verrucomicrobia was significantly lower in YK (7.17%) and RS (4.60%) than in CH (15.68%) and LT (18.05%).
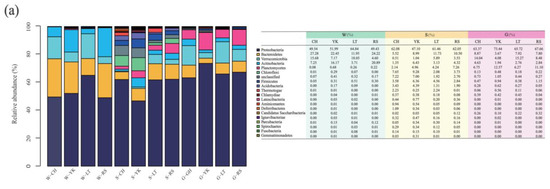
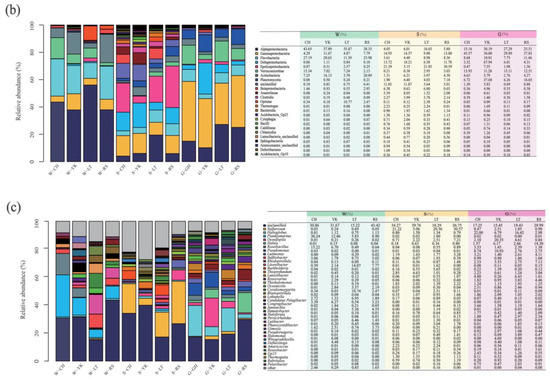
Figure 5.
The relative abundance among multiple media (water (W), sediment (S), and the gut of Apostichopus japonicus (G)) in different areas. Relative abundances of the bacterial community in multiple media were determined at the (a) phylum, (b) class, and (c) genus levels.
Alphaproteobacteria were the most predominant class in water from different areas, with relative abundances of 43.65%, 37.89%, 55.87%, and 38.35% from CH, YK, LT, and RS, respectively (Figure 5). The relative abundances of the second most abundant class Flavobacteriia were 27.19%, 20.63%, 11.39%, and 3.98%. The relative abundances of Actinobacteria were 16.15%, 20.89%, 7.25%, and 3.70% from YK, RS, CH, and LT.
At the genus level, Planktomarina, Roseibacillus, Cellulophaga, Sulfitobacter, Litoreibacter, Loktanella, and Jannaschia were dominant genera in different areas (Figure 5). The most predominant bacteria in CH, YK, and RS were Planktomarina, with relative abundances of 30.24%, 12.46%, and 8.90%, respectively. The most predominant bacteria of LT were Litoreibacter, which accounted for 14.07%. The second most abundant bacteria in CH and YK were Roseibacillus with 15.22% and 6.63%. The second most abundant bacteria in LT and RS were Coraliomargarita and Litoreibacter at 10.74% and 7.77%, respectively. Sulfitobacter was the third most abundant genus in LT and RS with 9.73% and 5.02%, respectively.
3.4.2. Sediment
At the phylum level, Proteobacteria was the most predominant phylum in the sediments of different areas, with 47.10–62.08%, which was consistent with the dominant phylum in water. The second most abundant bacteria in the sediments of CH and YK aquaculture ponds were both Chloroflexi, with relative abundances of 7.65% and 9.28%, respectively. The third most abundant bacterial phylum was both Anaplasma, with relative abundances of 8.99% and 5.51%, respectively. The second most abundant phylum of LT and RS was Bacteroidetes, with relative abundances of 11.73% and 10.50%, respectively. The third most abundant phylum in LT was Verrucomicrobia, with a relative abundance of 5.89%, while the third most abundant phylum in RS was Planctomycetess, with a relative abundance of 7.26%.
From the perspective of bacterial class, the sum of relative abundance of Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria, Deltaproteobacteria, and Epsilonproteobacteria in the sediments of A. japonicus aquaculture ponds in different areas ranged between 43.87% and 61.21%. The most predominant classes of CH, LT, and RS aquaculture ponds were all Epsilonproteobacteria, with relative abundances of 21.39%, 21.85%, and 30.59%, respectively. The most predominant class in sediments of YK was Deltaproteobacteria with a relative abundance of 18.21%.
From the perspective of genus, Sulfurovum, Thioprofundum, Burkholderia, Desulfosarcina, Desulfopila, Lutimonas, and Mesorhizobium were the dominant bacterial genera in sediments in different areas. Specifically, Sulfurovum was the most predominant genus of different areas with relative abundance ranging from 5.06% to 30.57%. Burkholderia was the second most abundant genus in CH and LT with 6.16% and 5.65%, while Thioprofundum and Desulfopila were the second most abundant genera in YK and RS with 4.63% and 7.21%, respectively.
3.4.3. Gut of A. japonicus
There were shared features and geographical differences in the structural characteristics of the bacterial community in the gut of A. japonicus in different areas at all taxonomic levels.
At the phylum level, Proteobacteria was the most predominant phylum in the gut of A. japonicus in the four areas, with relative abundance ranging from 63.37% to 73.44%. In addition, Verrucomicrobia took up 4.08–15.27%, Planctomycetes 6.2–12.57%, Bacteroidetes 3.67–8.87%, and Actinobacteria 1.94–4.63%. To be specific, the second most abundant phylum in CH and LT was Verrucomicrobia, accounting for 14.04% and 15.27%, respectively. The second most abundant phylum in YK and RS was Planctomycetess, with relative abundances of 12.57% and 11.10%. Bacteroidetes was the third most bacterial phylum in CH and LT, with relative abundances of 8.87% and 7.80%, respectively. Verrucomicrobia was the third most dominant bacterial phylum in YK and RS, with 4.08% and 8.48%, respectively.
At the class level, Gammaproteobacteria was the most predominant class in the A. japonicus gut, varying between 29.89% and 43.57% in four areas. The second most abundant phylum in CH, LT, and RS were Alphaproteobacteria with the relative abundance of 15.16%, 27.29%, and 25.51%, respectively. Deltaproteobacteria was the second dominant bacterial class in YK, accounting for 22.65%.
Considering the genus, Halioglobus, Pseudomonas, Haliea, Desulfopila, Sulfitobacter, Luteolibacter, Rhodopirellula, and Lutimonas were the dominant genera in the A. japonicus gut of different areas. Specifically, Halioglobus was the most predominant genus in the A. japonicus gut in CH and LT, with relative abundances of 22.00% and 16.42%, respectively. Desulfopila and Haliea were the most predominant genera in YK and RS, with percentages of 20.35% and 14.3p%, respectively. Pseudomonas was the second most abundant genus in CH and YK with 8.74% and 10.93%, Luteolibacter was the second most abundant genus in LT with 6.63%, and Rhodopirellula was the second most abundant genus in RS with 6.97% and 8.94%.
3.5. Specific Bacterial Community in the Ecosystem of A. japonicus Culture Ponds
LEfSe analysis was used to study the bacterial community specific to the ecosystem of A. japonicus aquaculture ponds in different areas (Figure 6). The results indicated that the bacterial community specific to water in different areas was primarily Rhodobacteraceae in Proteobacteria, followed by Flavobacteriaceae in Bacteroidetes. Specific bacterial communities of sediment were Deltaproteobacteria in Proteobacteria represented by Desulfobacterales and Desulfuromonadales. Additionally, Helicobacteraceae, Ectothiorhodospiraceae, Bacillaceae, Anaerolineaceae, and Lineaceae were also present. The gut of the A. japonicus specific bacterial community was Gammaproteobacteria in Proteobacteria, represented by Pseudomonadaceae and Alteromonadaceae. Acidimicrobiaceae and Verrucomicrobiaceae were also found. Planctomycetaceae were the bacterial community unique to A. japonicus gut.
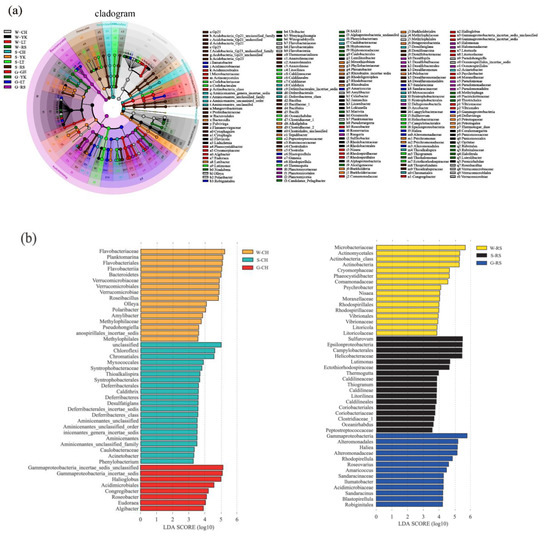
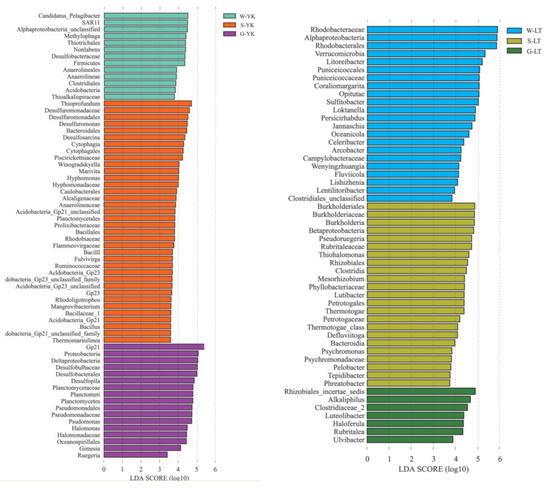
Figure 6.
LEfSE method identified the significantly specific abundant taxonomy among multiple media (water (W), sediment (S), and the gut of in Apostichopus japonicus (G)) in different areas (Changhai (CH), Rushan (RS), Yingkou (YK), and Laoting (LT), China). Biomarkers for each group are represented in different colors. The circles represent the phylogenetic level from domain to genus from inside to outside. Only taxonomies that met an LDA significance threshold of >3 are defined in this figure. (a) Taxonomic cladogram based on LEfSe analysis. (b) Histogram of LDA distribution.
4. Discussion
4.1. Dominant Bacterial Communities in the Ecosystem of A. japonicus Aquaculture Ponds
Microorganisms are major decomposers, consumers, and producers in the ecosystem of aquaculture ponds, driving the material cycle and energy flow of the ecosystem and playing a vital role in maintaining the balance of it, degrading pollutants and the health of cultured species [22]. Studying the bacterial community structure in the ecosystem of aquaculture pond provides a deeper understanding of bacterial diversity and the structure and functions of A. japonicus [23].
This study identified the dominant bacterial phyla residing in the gut of A. japonicus and aquaculture ponds from multiple areas. These bacterial communities are primarily comprised of Proteobacteria, followed by Bacteroidetes and Verrucomicrobia, which is consistent with previous studies [24,25]. As one of the largest and most diversified phyla, Proteobacteria are highly adaptive and widely distributed in aquaculture ponds. Proteobacteria have an extremely rich metabolic diversity and play an important role in the cycling of elements such as N and P in the aquatic environment [26,27]. Proteobacteria can participate in the degradation of organic matter, sulfur oxidation, and anaerobic ammonia oxidation under anaerobic conditions [28], as well as growth, metabolism, and CO2 and nitrogen fixation through photosynthesis [29]. They also play an important role in the sulfur cycle and the metabolism of metals such as iron [30,31]. Bacteroides are the dominant bacterial phylum of marine heterotrophic planktonic bacteria and are closely associated with the conversion of organic matter, such as DNA, lipids, and proteins. This is significant because the uptake and utilization of these substances is an important part of the carbon cycle in the aquatic environment [32,33]. The undigested feed and stools in sea cucumbers that are not decomposed during the culture process will settle down and cause an increase in Bacteroidetes in sediment [34]. Verrucomicrobia are widely present in various environments such as soil, sea, and animal intestines [35], and participate in the Earth’s nitrogen cycle, which can accelerate protein metabolism in water [36]. Some bacteria of the Verrucomicrobia phylum are highly specialized degraders of rockweed polysaccharides and other complex polysaccharides due to their polysaccharide hydrolase activity, which employs hundreds of enzymes to digest the algal polysaccharide fucoidan [37]. Polysaccharide substances such as algal polysaccharides are important components of carbohydrates [38]. Verrucomicrobia aids the digestion of polysaccharides that A. japonicus cannot digest alone, hence assisting in the processes of polysaccharide digestion and metabolism, participating in the nitrogen cycle of the aquatic environment, and playing a positive role in regulating A. japonicus growth.
The dominant bacterial community is influenced by the ecological environment and makes up a large proportion of the bacterial community structure. In this study, the dominant bacteria community had similarities across different areas among multiple media, which can reflect the overall function of the bacterial community.
4.2. Specific Bacterial Communities in the Ecosystem of A. japonicus Aquaculture Ponds
There are usually specific bacterial communities in different ocean environment media. In this study, the specific bacterial communities of water in different areas of sea cucumber culture ponds were mainly distributed in Alphaproteobacteria, represented by Rhodobacteraceae. Rhodobacteraceae consist of multiple branches that are highly heterogenous in metabolism, physiology and phylogeny, most of which are salt-obligate species. Not only do they play an important role in the cycling of carbon and sulfur, but they can also generate secondary metabolic active matters. To be specific, Sulfitobacter and Loktanella are represented in this study, where Sulfitobacter is an obligate aerobiotic heterotrophic genus that is able to oxidize matter such as elemental sulfur, sulfite and thiosulfate [39], participate in the sulfur cycle in sea water and degrade multiple ester compounds. Loktanella is common in sea water, moderately halophilic or salt tolerant and mostly chemoheterotrophic [40]. Water salinity is higher and more evenly distributed than sediment, so it is more suitable for Rhodobacteraceae to grow and Rhodobacteraceae also becomes a specific bacterial of water. Considering the high abundance of Rhodobacteraceae groups in A. japonicus aquaculture ponds, it is highly likely that they play a critical role in the aquaculture of A. japonicus. Therefore, the ecological functions of Rhodobacteraceae-related dominant genera or species for the water of A. japonicus aquaculture pond are worth researching further. Another specific bacterial family of water is Flavobacteriaceae that is subject to Flavobacteria of Bacteroidetes. Previous studies indicate that Flavobacteriaceae are highly abundant groups in sea water [41,42]. Most of the species in Flavobacteriaceae are able to hydrolyze organic matter such as carbon polymers and protection and decompose or utilize complex carbon sources by generating a variety of exoenzymes, which are similar to the functions of the other groups of Bacteroidetes in sea water habitat [7]. In addition, the simple carbides generated by hydrolysis provide carbon sources for these bacteria to remove excessive N and P in water [43]. Therefore, Flavobacteriaceae might play an important role in maintaining the ecosystem balance of A. japonicus aquaculture water.
In the A. japonicus aquaculture ponds from different areas, the specific bacterial communities in sediments mainly comprised Deltaproteobacteria of Proteobacteria, represented by Desulfobacterales and Desulfuromonadales, followed by Chloroflexi and Firmicutes. Desulfobacterales and Desulfuromonadales grow by utilizing the sulfur in their habitat [44,45]. The feces of cultured species and organic debris accumulate at the bottom of the aquaculture pond, and the anaerobic environment in the sediments might lead to the accumulation of substantial amounts of sulfur. Not only does this sulfur accumulation support the growth of Desulfobacterales and Desulfuromonadales, but it may also account for the differences in specific bacterial communities between sediment and water in the A. japonicus aquaculture pond. Chloroflexi can be involved in the degradation of organic matter [46]. Anaerolineaceae is a major family in the Chloroflexi phylum and is a facultative anaerobic bacterium capable of degrading other cellular materials such as carbohydrates and amino acids in the methanogenic ecosystem [47]. Chloroflexi is associated with eutrophication, where the proportion of Chloroflexi is greater in areas with higher levels of eutrophication in water [48]. Firmicutes are involved in the degradation and transformation of multiple substances and elements [49,50], while Bacillus is a probiotic bacterium, commonly used as a microecological agent with a good ability to perform heterotrophic nitrification and aerobic denitrification [51,52] and phosphorus removal [53], thus removing these nutrients from the water. From the perspective of microbial water purification, Bacillus actively purifies nitrogen, phosphorus, and organic matter [54]. Meanwhile, Bacillus can improve the environmental quality of sea cucumber aquaculture ponds, enhance the activity of digestive enzymes and non-specific immune enzymes, and thus promote the growth and immunity of the sea cucumber [55]. In this study, Bacillus was a specific bacterial community in sediment that may have positively influenced the growth and immunity of A. japonicus by promoting the evolution of nitrogen and phosphorus in the culture environment.
The specific bacterial communities in the gut of A. japonicus primarily comprised Gammaproteobacteria of Proteobacteria, specifically represented by Pseudomonadaceae and Alteromonadaceae. In addition, Pseudomonadaceae and Alteromonadaceae only existed in the gut of A. japonicus as specific bacterial communities and were not detected in water or sediments. Gammaproteobacteria are ubiquitous in high diversities and large quantities in the ocean, especially in waters with high nutrient contents [56,57]. In anaerobic environments, some species of Gammaproteobacteria can form symbiotic relationships with animals and play an important role in the carbon and sulfur cycles [58,59]. Some studies indicated that Pseudoalteromonas are highly capable of decomposing organic matter and could utilize a variety of organic materials as energy sources [60]. Similarly, Alteromonadaceae can produce a variety of digestive enzymes, such as chitinase, amylase and lipase [61]. As a benthic organism with limited feeding capacity, A. japonicus feeds on materials such as microorganisms, large algal segments, organic detritus, and bottom sediments [17], and the gut is the principal digestive organ of A. japonicus [62]. Pseudomonadaceae and Alteromonadaceae may assist A. japonicus in decomposing organic materials and converting them into energy required for growth. Over time, Pseudomonadaceae and Alteromonadaceae may thus become the specific bacteria colonizing the A. japonicus gut.
The bacterial community in multiple media showed unique characteristics due to specific biological functions. In this study, the ecological functions among media were correlated with their bacterial community structure, and specific bacterial communities have differential expressions in different regions among media.
5. Conclusions
The characteristics of the bacterial communities in the ecosystem of A. japonicus aquaculture ponds in different areas of northern China were analyzed using 16S rRNA sequencing technology. The results suggested that the dominant bacterial community comprised a larger proportion of the bacterial community structure and tended to reflect the overall function, while the formation of specific bacterial communities in multiple media in the ecosystem is caused by the pivotal ecological functions of bacteria. The dominant and specific bacterial communities are distinct and related, but the mechanisms of their interactions remain to be further investigated.
Author Contributions
Y.Z. designed experiments, analyzed experimental results and wrote the manuscript; J.Z. participated in experiments; L.W. drafted the paper and analyzed data; H.X. participated in microbiology analysis; Z.L. carried out experiments; Y.L. analyzed experimental results; Z.H. participated in designing experiments; Y.C. revised the manuscript; J.D. designed and supported the study. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (31902395), the National Key Research and Development Program of China (2018YFD0901604), Youth Science and Technology Star Project of Dalian (2020RQ115), Liaoning Province “Xingliao Talents Plan” project (XLYC2002107), Key Special Project for Introduced Talents Team of Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou)(GML2019ZD0402).
Institutional Review Board Statement
The rearing and treatment of laboratory animals is based on the principles of animal experimentation welfare and ethical management.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Thanks to Key Laboratory of Mariculture & Stock Enhancement in North China Sea, Ministry of Agriculture and Rural Affairs of Dalian Ocean University and Southern Marine Science and Engineering Guangdong Laboratory for their support of this research.
Conflicts of Interest
The authors declare that they have no competing interests.
References
- Bordbar, S.; Anwar, F.; Saari, N. High-Value Components and Bioactives from Sea Cucumbers for Functional Foods—A Review. Mar. Drugs 2011, 9, 1761–1805. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cui, L.; Li, S.; Liu, X.; Han, X. China Fishery Statistical Yearbook; China Agriculture Press: Beijing, China, 2020. [Google Scholar]
- Yuan, X.; Yang, H.; Wang, L.; Zhou, Y.; Gabr, H.R. Effects of salinity on energy budget in pond-cultured sea cucumber Apos-tichopus japonicus (Selenka) (Echinodermata: Holothuroidea). Aquaculture 2010, 306, 348–351. [Google Scholar] [CrossRef]
- Deng, H.; He, C.; Zhou, Z.; Liu, C.; Jiang, B.; Gao, X.; Liu, W.; Geng, H.; Liu, Y. Isolation and pathogenicity of pathogens from skin ulceration and viscera ejection syndrome of sea cucumber. J. Biotechnol. 2008, 136, S551. [Google Scholar] [CrossRef]
- Deng, H.; He, C.; Zhou, Z.; Liu, C.; Tan, K.; Wang, N.; Jiang, B.; Gao, X.; Liu, W. Isolation and pathogenicity of pathogens from skin ulceration disease and viscera ejection syndrome of the sea cucumber Apostichopus japonicus. Aquaculture 2009, 287, 18–27. [Google Scholar] [CrossRef]
- Wada, M.; Zhang, D.; Do, H.-K.; Nishimura, M.; Tsutsumi, H.; Kogure, K. Co-inoculation of Capitella sp. I with its synergistic bacteria enhances degradation of organic matter in organically enriched sediment below fish farms. Mar. Pollut. Bull. 2008, 57, 86–93. [Google Scholar] [CrossRef]
- Kirchman, D.L. The ecology of Cytophaga–Flavobacteria in aquatic environments. FEMS Microbiol. Ecol. 2002, 39, 91–100. [Google Scholar] [CrossRef]
- Zhang, R.; Weinbauer, M.G.; Tam, Y.K.; Qian, P.-Y. Response of bacterioplankton to a glucose gradient in the absence of lysis and grazing. FEMS Microbiol. Ecol. 2013, 85, 443–451. [Google Scholar] [CrossRef][Green Version]
- Lage, O.M.; Bondoso, J.; Viana, F. Isolation and characterization of Planctomycetes from the sediments of a fish farm wastewater treatment tank. Arch. Microbiol. 2012, 194, 879–885. [Google Scholar] [CrossRef]
- Verschuere, L.; Rombaut, G.; Sorgeloos, P.; Verstraete, W. Probiotic Bacteria as Biological Control Agents in Aquaculture. Microbiol. Mol. Biol. Rev. 2000, 64, 655–671. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Z. Effect of probiotics for common carp (Cyprinus carpio) based on growth performance and digestive enzyme activities. Anim. Feed Sci. Technol. 2006, 127, 283–292. [Google Scholar]
- Wang, Y.-B. Effect of probiotics on growth performance and digestive enzyme activity of the shrimp Penaeus vannamei. Aquaculture 2007, 269, 259–264. [Google Scholar] [CrossRef]
- Zhang, D.; Li, H.; Liu, Y.; Qiao, G.; Chi, S.; Song, J. Screening and identification of organics-degrading bacteria from the sediment of sea cucumber Apostichopus japonicus ponds. Aquac. Int. 2016, 24, 373–384. [Google Scholar] [CrossRef]
- Ma, Y.-X.; Li, L.-Y.; Li, M.; Chen, W.; Bao, P.-Y.; Yu, Z.-C.; Chang, Y.-Q. Effects of dietary probiotic yeast on growth parameters in juvenile sea cucumber, Apostichopus japonicus. Aquaculture 2019, 499, 203–211. [Google Scholar] [CrossRef]
- Becker, P.; Gillan, D.; Lanterbecq, D.; Jangoux, M.; Rasolofonirina, R.; Rakotovao, J.; Eeckhaut, I. The skin ulceration disease in cultivated juveniles of Holothuria scabra (Holothuroidea, Echinodermata). Aquaculture 2004, 242, 13–30. [Google Scholar] [CrossRef]
- Qin, Z.; Hong, M.; Kang, M.; Wen, Z.; Zhi, L.; Wei, X. Interaction of dietary Bacillus subtilis and fructooligosaccharide on the growth performance, non-specific immunity of sea cucumber, Apostichopus japonicus. Fish Shellfish. Immunol. 2010, 29, 204–211. [Google Scholar]
- Liao, Y. Fauna Sinica: Phylum Echinodermata: Class Holothuroidea; Science Press: Beijing, China, 1997. [Google Scholar]
- Zhou, M.; Hernandez-Sanabria, E.; Guan, L.L. Assessment of the Microbial Ecology of Ruminal Methanogens in Cattle with Different Feed Efficiencies. Appl. Environ. Microbiol. 2009, 75, 6524–6533. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4516–4522. [Google Scholar] [CrossRef]
- Youssef, N.; Sheik Cody, S.; Krumholz Lee, R.; Najar Fares, Z.; Roe Bruce, A.; Elshahed Mostafa, S. Comparison of species richness estimates obtained using nearly complete fragments and simulated pyrosequencing-generated fragments in 16S rRNA gene-based environmental surveys. Appl. Environ. Microbiol. 2009, 75, 5227–5236. [Google Scholar] [CrossRef]
- Sun, X.; Gao, Y.; Yang, Y. Recent advancement in microbial environmental research using metagenomics tools. Biodivers. Sci. 2013, 21, 393. [Google Scholar]
- Zhou, Q.; Li, K.; Jun, X.; Bo, L. Role and functions of beneficial microorganisms in sustainable aquaculture. Bioresour. Technol. 2009, 100, 3780–3786. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, Y.; Juck, D.; Fortin, N.; Greer, C.W.; Tang, Q. Phylogenetic analysis of bacterial communities in the shrimp and sea cucumber aquaculture environment in northern China by culturing and PCR–DGGE. Aquac. Int. 2010, 18, 977–990. [Google Scholar] [CrossRef]
- Xiong, J.; Wang, K.; Wu, J.; Qiuqian, L.; Yang, K.; Qian, Y.; Zhang, D. Changes in intestinal bacterial communities are closely associated with shrimp disease severity. Appl. Microbiol. Biotechnol. 2015, 99, 6911–6919. [Google Scholar] [CrossRef] [PubMed]
- Rungrassamee, W.; Klanchui, A.; Maibunkaew, S.; Chaiyapechara, S.; Jiravanichpaisal, P.; Karoonuthaisiri, N. Characteriza-tion of Intestinal Bacteria in Wild and Domesticated Adult Black Tiger Shrimp (Penaeus monodon). PLoS ONE 2014, 9, e91853. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-L.; Liu, W.-T.; Chong, M.-L.; Wong, M.-T.; Ong, S.L.; Seah, H.; Ng, W.J. Community structure of microbial biofilms associated with membrane-based water purification processes as revealed using a polyphasic approach. Appl. Microbiol. Biotechnol. 2004, 63, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.-Y.; Xu, Y.; Liu, Y.; Liu, Z.-P. Bacterial diversity, community structure and function associated with biofilm development in a biological aerated filter in a recirculating marine aquaculture system. Mar. Biodivers. 2011, 42, 1–11. [Google Scholar] [CrossRef]
- Freitag, T.E.; Prosser, J.I. Community Structure of Ammonia-Oxidizing Bacteria within Anoxic Marine Sediments. Appl. Environ. Microbiol. 2003, 69, 1359–1371. [Google Scholar] [CrossRef] [PubMed]
- Imhoff, J.F. The Phototrophic Alpha-Proteobacteria. In The Prokaryotes: Volume 5: Proteobacteria: Alpha and Beta Subclasses; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; pp. 41–64. [Google Scholar]
- Watsuji, T.-O.; Nishizawa, M.; Morono, Y.; Hirayama, H.; Kawagucci, S.; Takahata, N.; Sano, Y.; Takai, K. Cell-Specific Thioautotrophic Productivity of Epsilon-Proteobacterial Epibionts Associated with Shinkaia crosnieri. PLoS ONE 2012, 7, e46282. [Google Scholar] [CrossRef]
- Murillo, A.A.; Ramírez-Flandes, S.; DeLong, E.F.; Ulloa, O. Enhanced metabolic versatility of planktonic sulfur-oxidizing γ-proteobacteria in an oxygen-deficient coastal ecosystem. Front. Mar. Sci. 2014, 1, 18. [Google Scholar] [CrossRef]
- Cottrell, M.T.; Kirchman, D.L. Natural Assemblages of Marine Proteobacteria and Members of the Cytophaga-Flavobacter Cluster Consuming Low- and High-Molecular-Weight Dissolved Organic Matter. Appl. Environ. Microbiol. 2000, 66, 1692–1697. [Google Scholar] [CrossRef]
- O’Sullivan Louise, A.; Weightman Andrew, J.; Fry John, C. New degenerate Cytophaga-Flexibacter-Bacteroides-specific 16S ribosomal DNA-targeted oligonucleotide probes reveal high bacterial diversity in River Taff epilithon. Appl. Env. Ment. Microbiol. 2002, 68, 201–210. [Google Scholar] [CrossRef]
- Rosselló-Mora, R.; Thamdrup, B.; Schäfer, H.; Weller, R.; Amann, R. The Response of the Microbial Community of Marine Sediments to Organic Carbon Input under Anaerobic Conditions. Syst. Appl. Microbiol. 1999, 22, 237–248. [Google Scholar] [CrossRef]
- Hou, S.; Makarova, K.S.; Saw, J.H.; Senin, P.; Ly, B.V.; Zhou, Z.; Ren, Y.; Wolf, Y.; Yutin, N.; Koonin, E.; et al. Complete genome sequence of the extremely acidophilic methanotroph isolate V4, Methylacidiphilum infernorum, a representative of the bacterial phylum Verrucomicrobia. Biol. Direct 2008, 3, 1–25. [Google Scholar] [CrossRef]
- Freitas, S.; Hatosy, S.; Fuhrman, J.; Huse, S.M.; Welch, D.M.; Sogin, M.L.; Martiny, A.C. Global distribution and diversity of marine Verrucomicrobia. ISME J. 2012, 6, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Sichert, A.; Corzett, C.H.; Schechter, M.S.; Unfried, F.; Markert, S.; Becher, D.; Fernandez-Guerra, A.; Liebeke, M.; Schweder, T.; Polz, M.F.; et al. Verrucomicrobia use hundreds of enzymes to digest the algal polysaccharide fucoidan. Nat. Microbiol. 2020, 5, 1026–1039. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.B.A.; Adel, M.; Talati, A.; Kumar, M.S.; Abdulrahim, K.; AbdulHameed, M.M. Seaweed Polysaccharides and Their Production and Applications. In Seaweed Polysaccharides; Elsevier: Amsterdam, The Netherlands, 2017; pp. 369–382. [Google Scholar] [CrossRef]
- Bouchard, B.; Beaudet, R.; Villemur, R.; McSween, G.; Lepine, F.; Bisaillon, J.G. Isolation and characterization of Desulfitobac-terium frappieri sp. nov.; an anaerobic bacterium which reductively dechlorinates pentachlorophenol to 3-chlorophenol. Int. J. Syst. Evol. Microbiol. 1996, 46, 1010–1015. [Google Scholar]
- Pujalte, M.J.; Lucena, T.; Ruvira, M.A.; Arahal, D.R.; Macián, M.C. The Family Rhodobacteraceae. In The Prokaryotes: Alphaproteobacteria and Betaproteobacteria; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 439–512. [Google Scholar]
- Mcilroy, S.; Nielsen, P. The Family Saprospiraceae. In The Prokaryotes; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- McBride, M.J. The Family Flavobacteriaceae. In The Prokaryotes; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Nielsen, P.H.; Mielczarek, A.T.; Kragelund, C.; Nielsen, J.L.; Saunders, A.; Kong, Y.; Hansen, A.A.; Vollertsen, J. A conceptual ecosystem model of microbial communities in enhanced biological phosphorus removal plants. Water Res. 2010, 44, 5070–5088. [Google Scholar] [CrossRef] [PubMed]
- Adrados, B.; Sánchez, O.; Arias, C.; Becares, E.; Garrido, L.; Mas, J.; Brix, H.; Morató, J. Microbial communities from different types of natural wastewater treatment systems: Vertical and horizontal flow constructed wetlands and biofilters. Water Res. 2014, 55, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.-F.; Ling, J.; Dong, J.-D.; Chen, B.; Zhang, Y.-Y.; Zhang, Y.-Z.; Wang, Y.-S. Illumina-based analysis the microbial diversity associated with Thalassia hemprichii in Xincun Bay, South China Sea. Ecotoxicology 2015, 24, 1548–1556. [Google Scholar] [CrossRef] [PubMed]
- Daniel, L.M.C.; Pozzi, E.; Foresti, E.; Chinalia, F.A. Removal of ammonium via simultaneous nitrification–denitrification nitrite-shortcut in a single packed-bed batch reactor. Bioresour. Technol. 2009, 100, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Sekiguchi, Y.; Imachi, H.; Kamagata, Y.; Ohashi, A.; Harada, H. Diversity, localization, and physiological prop-erties of filamentous microbes belonging to Chloroflexi subphylum I in mesophilic and thermophilic methanogenic sludge granules. Appl. Environ. Microbiol. 2005, 71, 7493–7503. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Watanabe, A.Y.; Okabe, S. Significance of Chloroflexi in Performance of Submerged Membrane Bioreactors (MBR) Treating Municipal Wastewater. Environ. Sci. Technol. 2007, 41, 7787–7794. [Google Scholar] [CrossRef] [PubMed]
- Goffredi, S.K.; Orphan, V.J. Bacterial community shifts in taxa and diversity in response to localized organic loading in the deep sea. Environ. Microbiol. 2010, 12, 344–363. [Google Scholar] [CrossRef]
- Ward, N.; Challacombe, J.; Janssen, P.; Henrissat, B.; Coutinho, P.; Wu, M.; Bruce, D.; Creasy, T.; Daugherty, S.; Davidsen, T.; et al. Three Genomes from the Phylum Acidobacteria Provide Insight into the Lifestyles of These Microorganisms in Soils. Appl. Environ. Microbiol. 2009, 75, 2046–2056. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-P.; Wang, S.-M.; Zhang, D.-W.; Zhou, L.-X. Isolation and nitrogen removal characteristics of an aerobic heterotrophic nitrifying–denitrifying bacterium, Bacillus subtilis A1. Bioresour. Technol. 2011, 102, 854–862. [Google Scholar] [CrossRef]
- Zhang, Q.L.; Liu, Y.; Ai, G.M.; Miao, L.L.; Zheng, H.Y.; Liu, Z.P. The characteristics of a novel heterotrophic nitrifica-tion-aerobic denitrification bacterium, Bacillus methylotrophicus strain L7. Bioresour. Technol. 2012, 108, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, P.; Holguin, G.; Puente, M.E.; Lopez-Cortes, A.; Bashan, Y. Phosphate-solubilizing microorganisms associated with the rhizosphere of mangroves in a semiarid coastal lagoon. Biol. Fertil. Soils 2000, 30, 460–468. [Google Scholar] [CrossRef]
- Wang, F.; Bai, Y.; Yang, F.; Zhu, Q.; Zhao, Q.; Zhang, X.; Wei, Y.; Liao, H. Degradation of Nitrogen, Phosphorus, and Organic Matter in Urban River Sediments by Adding Microorganisms. Sustainability 2021, 13, 2580. [Google Scholar] [CrossRef]
- Lu, Z.; Yang, Q.; Lin, Q.; Wu, J.; Du, H.; Zhou, C. Effects of Bacillus spp. on growth, gut microbiota compositions and immunity of sea cucumber (Apostichopus japonicus). Aquac. Res. 2021, 52, 3978–3990. [Google Scholar] [CrossRef]
- Ivanova, E.P.; Sawabe, T.; Zhukova, N.; Gorshkova, N.M.; Nedashkovskaya, O.I.; Hayashi, K.; Frolova, G.M.; Sergeev, A.F.; Pavel, K.G.; Mikhailov, V.V.; et al. Occurrence and Diversity of Mesophilic Shewanella Strains Isolated from the North-West Pacific Ocean. Syst. Appl. Microbiol. 2003, 26, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Payne, M.S.; Hall, M.R.; Bannister, R.; Sly, L.; Bourne, D.G. Microbial diversity within the water column of a larval rearing system for the ornate rock lobster (Panulirus ornatus). Aquaculture 2006, 258, 80–90. [Google Scholar] [CrossRef]
- Bakunina, I.I.; Shevchenko, L.S.; Nedashkovskaia, O.I.; Shevchenko, N.M.; Alekseeva, S.A.; Mikhaĭlov, V.V.; Zviagintseva, T.N. Screening of marine bacteria for fucoidan hydrolases. Muкpoбuoлoгuя 2000, 69, 370–376. [Google Scholar]
- Huber, I.; Spanggaard, B.; Appel, K.; Rossen, L.; Nielsen, T.; Gram, L. Phylogenetic analysis and in situ identification of the intestinal microbial community of rainbow trout (Oncorhynchus mykiss, Walbaum). J. Appl. Microbiol. 2004, 96, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Ribas, F.; Perramon, J.; Terradillos, A.; Frias, J.; Lucena, F. The Pseudomonas group as an indicator of potential regrowth in water distribution systems. J. Appl. Microbiol. 2000, 88, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Tzuc, J.T.; Escalante, D.R.; Herrera, R.R.; Cortés, G.G.; Ortiz, M.L.A. Microbiota from Litopenaeus vannamei: Digestive tract microbial community of Pacific white shrimp (Litopenaeus vannamei). SpringerPlus 2014, 3, 280. [Google Scholar] [CrossRef] [PubMed]
- Ward-Rainey, N.; Rainey, F.A.; Stackebrandt, E. A study of the bacterial flora associated with Holothuria atra. J. Exp. Mar. Biol. Ecol. 1996, 203, 11–26. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).