
From Shell to Sequence: Optimizing DNA Extraction and PCR for Pen Shell Identification

 , , ,
, , ,  , , and
, , and 
Abstract
:1. Introduction
1.1. Pinna nobilis: Ecological Role and Conservation Challenges
1.2. DNA Extraction in Conservation Biology
1.3. COI Marker and Species Identification
2. Materials and Methods
Sample Collection and Material Preparation for DNA Extractions
- Dilute 5 g Chelex-100 in 50 mL ddH2O (always use freshly made solution).
- Place 10% Chelex-100 solution (diluted in ddH2O) on a magnetic stirrer.
- If the starting material is stored in ethanol, rehydrate the samples gradually by immersing them for 5 min each in the following ethanol (EtOH, 96–100%) and phosphate-buffered saline (PBS, 1%) solutions: (1) 75% EtOH/25% PBS, (2) 50% EtOH/50% PBS, (3) 25% EtOH/75% PBS, and (4) 100% PBS.
- Add 100 μL of well-shaken 10% Chelex-100 in your sample.
- Add 10 μL of proteinase K (20 mg/mL), stored at −20 °C and thawed on ice, to each tube.
- Vortex tubes for 15 s.
- Incubate for 1 h in 56 °C.
- Incubate in a PCR machine at 95 °C for 30 min.
- Centrifuge tubes at 4000 rpm for 3 min.
- Incubate in a PCR machine at 95 °C for 30 min.
- Centrifuge tubes at 4000 rpm for 3 min.
- Transfer 50 μL of the supernatant to a new tube.
- If the starting material is stored in ethanol, rehydrate the samples gradually by immersing them for 5 min each in the following ethanol (EtOH, 96–100%) and phosphate-buffered saline (PBS, 1%) solutions: (1) 75% EtOH/25% PBS, (2) 50% EtOH/50% PBS, (3) 25% EtOH/75% PBS, and (4) 100% PBS.
- Add 87.5 μL of 10% SDS solution to each tube.
- Add 700 μL of Extraction Buffer (10 mM Tris-HCl, 400 mM NaCl, 2 mM EDTA, pH 8.2) to each tube.
- Add 10 μL of proteinase K (20 mg/mL), stored at −20 °C and thawed on ice, to each tube. Carefully pipette.
- Briefly vortex to mix well.
- Incubate the samples at 56 °C in a water bath overnight. Occasionally vortex briefly or use a shaker water bath.
- Add 300 μL of saturated NaCl solution (6 M) to each tube and place them on a rocker for 5 min. Leave on ice for 10 min.
- Centrifuge at maximum speed for 30 min.
- Carefully transfer 700 μL of the supernatant to a new 2 mL Eppendorf tube using a pipette.
- Add an equal volume of ice-cold isopropanol or 100% ethanol to each tube and invert 20–30 times to mix well.
- Leave the tubes at −20 °C for 30 min.
- Centrifuge at maximum speed for 20 min to pellet the DNA.
- Immediately after centrifugation, carefully remove the supernatant with a pipette without losing the DNA pellet.
- Add 800 μL of 70% ice-cold ethanol to each tube and leave at room temperature for 15 min.
- Centrifuge at maximum speed for 10 min and carefully remove all the ethanol with a pipette without losing the DNA pellet.
- Repeat the previous step.
- Dry the DNA pellet at 37 °C for 10–15 min by placing the tubes with open caps on a heat block to evaporate all the ethanol. Important: check by lightly flicking the tubes for any ethanol residues. There should be no droplets on the walls.
- Resuspend the DNA in 30 μL of ultrapure water by pipetting 5–10 times and leave the samples for 10 min at room temperature. Store the samples at −20 °C.
- If the starting material is stored in ethanol, take it out and leave it on a paper towel for 15 min or wash it in water to wash out the ethanol.
- Transfer the shell material to a 1.5 mL tube.
- Add 200 μL CTAB and grind with a plastic pestle.
- Vortex for a few secs.
- Incubate at 55–60 °C for 1 h (can be extended to overnight, if convenient).
- Add an equal volume (here 200 μL) of chloroform (24:1 isoamylic alcohol used).
- Mix phases by inverting tubes many times (1–2 min).
- Centrifuge at 8000 rpm for 5 min.
- Transfer the upper, aquatic phase to a new 1,5 mL tube. Avoid debris and organic phase.
- Add equal volume of isopropanol (here: 150–200 μL) and invert tubes several times to mix.
- Place at −20 °C for 20 min (can be extended to overnight, if convenient).
- Centrifuge at maximum speed (13,000 rpm) for 15–20 min.
- Discard supernatant.
- Add 500 μL of 70% ethanol and invert tubes 5–6 times.
- Centrifuge at maximum speed (13000 rpm) for 5 min.
- Discard supernatant.
- Let it dry at 37 °C (or in R.T. overnight).
- Dilute in 50 μL of ultrapure water.
- Shells were submerged in 500 μL of PBS 1% solution 1% to hydrate (2 h).
- Extension of incubation time with proteinase K overnight.
- At the final elution step, we used 50 μL of prewarmed Buffer BE (70 °C) and incubated for 10 min before centrifugation (1 min at 11,000× g).
3. Results
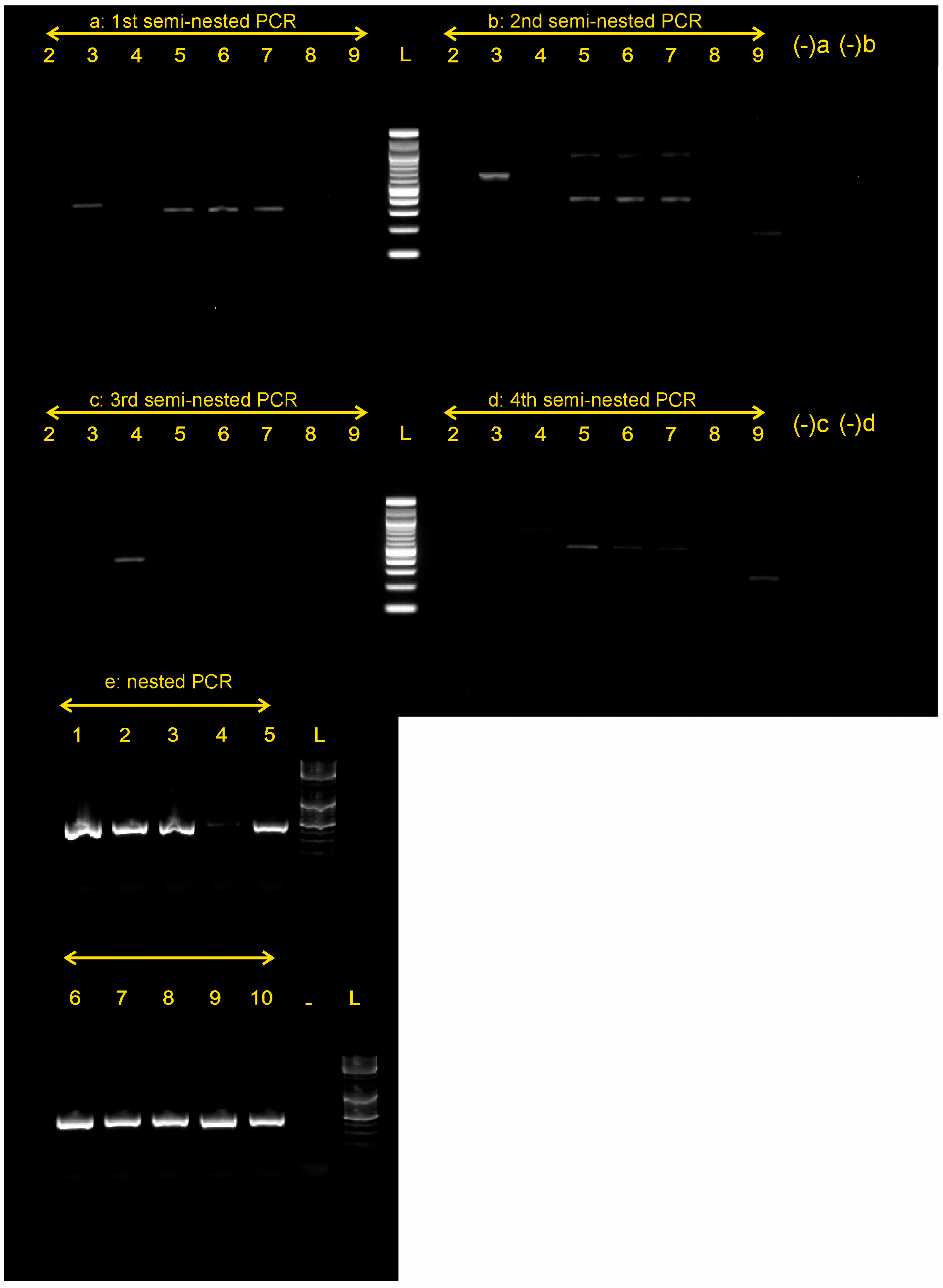
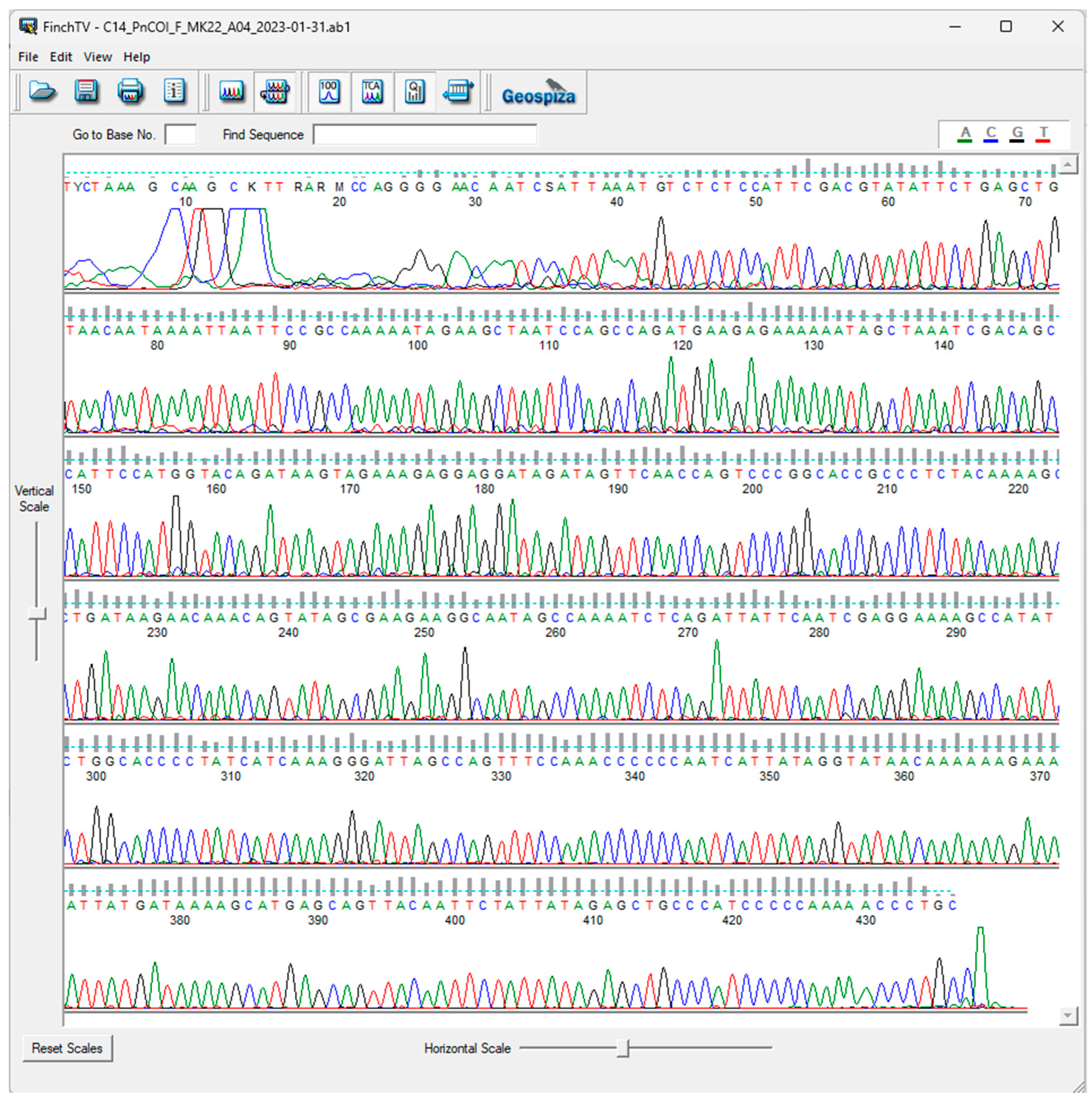
DNA Extraction, PCR Amplification, and Sanger Sequencing
4. Discussion
4.1. DNA Extraction Protocols: Comparing Yield, Cost, and Time Efficiency
4.2. Conventional vs. Nested PCR: Advantages, Limitations, and Enhancements with Specific Primers
4.3. Pros and Cons of COI Gene as a Stand-Alone Marker for Species Identification
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PCR | polymerase chain reaction |
| COI | cytochrome oxidase subunit I |
References
- Addis, P.; Secci, M.; Brundu, G.; Manunza, A.; Corrias, S.; Cau, A. Density, Size Structure, Shell Orientation and Epibiontic Colonization of the Fan Mussel Pinna nobilis L. 1758 (Mollusca: Bivalvia) in Three Contrasting Habitats in an Estuarine Area of Sardinia (W Mediterranean). Sci. Mar. 2009, 73, 143–152. [Google Scholar] [CrossRef]
- Chintiroglou, C.; Antoniadou, C.; Vafidis, D.; Koutsoubas, D. A Review on the Biodiversity of Hard Substrate Invertebrate Communities in the Aegean Sea. Mediterr. Mar. Sci. 2005, 6, 51–62. [Google Scholar] [CrossRef]
- Theodorou, J.A.; Spinos, E.; Ramfos, A.; Tsamadias, I.E.; Bekiari, V.; Kamilari, M.; Ntouni, M.-M.; Tsotsios, D.; Feidantsis, K.; Lattos, A.; et al. Fan Mussel (Pinna nobilis L.) Spat Collection, Monitoring of Early Growth and Conservation Implications by Deploying Conventional Aquaculture Methodology. J. Mar. Sci. Eng. 2024, 12, 2070. [Google Scholar] [CrossRef]
- Hendriks, I.; Basso, L.; Deudero, S.; Cabanellas-Reboredo, M.; Lvarez, E. Relative Growth Rates of the Noble Pen Shell Pinna Nobilis Throughout Ontogeny Around the Balearic Islands (Western Mediterranean, Spain). J. Shellfish Res. 2012, 31, 749–756. [Google Scholar] [CrossRef]
- Basso, L.; Hendriks, I.; Duarte, C.M. Juvenile Pen Shells (Pinna Nobilis) Tolerate Acidification but Are Vulnerable to Warming. Estuaries Coasts 2015, 38, 1976–1985. [Google Scholar] [CrossRef]
- Katsanevakis, S. The Cryptogenic Parasite Haplosporidium Pinnae Invades the Aegean Sea and Causes the Collapse of Pinna Nobilis Populations. AI 2019, 14, 150–164. [Google Scholar] [CrossRef]
- Box, A.; Sureda, A.; Deudero, S. Antioxidant Response of the Bivalve Pinna Nobilis Colonised by Invasive Red Macroalgae Lophocladia Lallemandii. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2009, 149, 456–460. [Google Scholar] [CrossRef]
- Grau, A.; Villalba, A.; Navas, J.I.; Hansjosten, B.; Valencia, J.M.; García-March, J.R.; Prado, P.; Follana-Berná, G.; Morage, T.; Vázquez-Luis, M.; et al. Wide-Geographic and Long-Term Analysis of the Role of Pathogens in the Decline of Pinna Nobilis to Critically Endangered Species. Front. Mar. Sci. 2022, 9, 666640. [Google Scholar] [CrossRef]
- Daugherty, C.H.; Cree, A.; Hay, J.M.; Thompson, M.B. 2007 IUCN Red List of Threatened Species Neglected Taxonomy and Continuing Extinctions of Tuatara (Sphenodon). Nature 1990, 347, 177–179. [Google Scholar] [CrossRef]
- Scarpa, F.; Sanna, D.; Azzena, I.; Mugetti, D.; Cerruti, F.; Hosseini, S.; Cossu, P.; Pinna, S.; Grech, D.; Cabana, D.; et al. Multiple Non-Species-Specific Pathogens Possibly Triggered the Mass Mortality in Pinna Nobilis. Life 2020, 10, 238. [Google Scholar] [CrossRef]
- Polgar, G.; Jaafar, Z. Endangered Forested Wetlands of Sundaland; Springer International Publishing: Cham, Switzerland, 2018; ISBN 978-3-319-52415-3. [Google Scholar]
- Shuttleworth, L.; Oosthuizen, C.J. Comparing DNA Yield from Fish Scales Following Different Extraction Protocols. Sci. Rep. 2022, 12, 2836. [Google Scholar] [CrossRef] [PubMed]
- Molbert, N.; Ghanavi, H.R.; Johansson, T.; Mostadius, M.; Hansson, M.C. An Evaluation of DNA Extraction Methods on Historical and Roadkill Mammalian Specimen. Sci. Rep. 2023, 13, 13080. [Google Scholar] [CrossRef] [PubMed]
- Geist, J.; Wunderlich, H.; Kuehn, R. Use of Mollusc Shells for DNA-Based Molecular Analyses. J. Molluscan Stud. 2008, 74, 337–343. [Google Scholar] [CrossRef]
- Ferreira, S.; Ashby, R.; Jeunen, G.-J.; Rutherford, K.; Collins, C.; Todd, E.V.; Gemmell, N.J. DNA from Mollusc Shell: A Valuable and Underutilised Substrate for Genetic Analyses. PeerJ 2020, 8, e9420. [Google Scholar] [CrossRef]
- Martin, K.R.; Waits, L.P.; Parent, C.E. Teaching an Old Shell New Tricks: Extracting DNA from Current, Historical, and Ancient Mollusk Shells. BioScience 2021, 71, 235–248. [Google Scholar] [CrossRef]
- Katsares, V.; Tsiora, A.; Galinou-Mitsoudi, S.; Imsiridou, A. Genetic Structure of the Endangered Species Pinna Nobilis (Mollusca: Bivalvia) Inferred from mtDNA Sequences. Biologia 2008, 63, 412–417. [Google Scholar] [CrossRef]
- Papadakis, O.; Mamoutos, I.; Ramfos, A.; Catanese, G.; Papadimitriou, E.; Theodorou, J.; Batargias, C.; Papaioannou, C.; Kamilari, M.; Tragou, E.; et al. Status, Distribution, and Threats of the Last Surviving Fan Mussel Populations in Greece. Mediterr. Mar. Sci. 2023, 24, 679–708. [Google Scholar] [CrossRef]
- Sarafidou, G.; Tsaparis, D.; Issaris, Y.; Chatzigeorgiou, G.; Grigoriou, P.; Chatzinikolaou, E.; Pavloudi, C. Insights on Pinna Nobilis Population Genetic Structure in the Aegean and Ionian Sea. PeerJ 2023, 11, e16491. [Google Scholar] [CrossRef]
- Catanese, G.; Vázquez-Luis, M.; Giacobbe, S.; García-March, J.R.; Zotou, M.; Patricia, P.; Papadakis, O.; Tena-Medialdea, J.; Katsanevakis, S.; Grau, A. Internal Transcribed Spacer as Effective Molecular Marker for the Detection of Natural Hybridization between the Bivalves Pinna Nobilis and Pinna Rudis. Ecol. Evol. 2024, 14, e70227. [Google Scholar] [CrossRef]
- Martínez-Martínez, P.; López-Nuñez, R.; Fernández-Torquemada, Y.; Cortés-Melendreras, E.; Valverde-Urrea, M.; Lopez-Moya, F.; Giménez-Casalduero, F. Identifying Surviving Pinna Nobilis after the Mass Mortality Event (MME) in the Mediterranean: Proposal of a Low-Risk Methodology for Collecting Genetic Samples. Mar. Environ. Res. 2025, 205, 107006. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Ratnasingham, S.; de Waard, J.R. Barcoding Animal Life: Cytochrome c Oxidase Subunit 1 Divergences among Closely Related Species. Proc. R. Soc. Lond. B Biol. Sci. 2003, 270, S96–S99. [Google Scholar] [CrossRef] [PubMed]
- Savolainen, V.; Cowan, R.S.; Vogler, A.P.; Roderick, G.K.; Lane, R. Towards Writing the Encyclopaedia of Life: An Introduction to DNA Barcoding. Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 1805–1811. [Google Scholar] [CrossRef]
- Ratnasingham, S.; Hebert, P.D.N. Bold: The Barcode of Life Data System (http://www.barcodinglife.org). Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef]
- Ward, R.D.; Hanner, R.; Hebert, P.D.N. The Campaign to DNA Barcode All Fishes, FISH-BOL. J. Fish. Biol. 2009, 74, 329–356. [Google Scholar] [CrossRef]
- Mallo, D.; Posada, D. Multilocus Inference of Species Trees and DNA Barcoding. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371, 20150335. [Google Scholar] [CrossRef]
- Klymus, K.E.; Merkes, C.M.; Allison, M.J.; Goldberg, C.S.; Helbing, C.C.; Hunter, M.E.; Jackson, C.A.; Lance, R.F.; Mangan, A.M.; Monroe, E.M.; et al. Reporting the Limits of Detection and Quantification for Environmental DNA Assays. Environ. DNA 2020, 2, 271–282. [Google Scholar] [CrossRef]
- Langlois, V.S.; Allison, M.J.; Bergman, L.C.; To, T.A.; Helbing, C.C. The Need for Robust qPCR-Based eDNA Detection Assays in Environmental Monitoring and Species Inventories. Environ. DNA 2021, 3, 519–527. [Google Scholar] [CrossRef]
- Aljanabi, S.M.; Martinez, I. Universal and Rapid Salt-Extraction of High Quality Genomic DNA for PCR-Based Techniques. Nucleic Acids Res. 1997, 25, 4692–4693. [Google Scholar] [CrossRef]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A Simple Salting out Procedure for Extracting DNA from Human Nucleated Cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A Rapid DNA Isolation Procedure for Small Quantities of Fresh Leaf Tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Boom, R.; Sol, C.J.; Salimans, M.M.; Jansen, C.L.; Wertheim-van Dillen, P.M.; van der Noordaa, J. Rapid and Simple Method for Purification of Nucleic Acids. J. Clin. Microbiol. 1990, 28, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Panda, B.B.; Meher, A.S.; Hazra, R.K. Comparison between Different Methods of DNA Isolation from Dried Blood Spots for Determination of Malaria to Determine Specificity and Cost Effectiveness. J. Parasit. Dis. 2019, 43, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Walsh, P.S.; Metzger, D.A.; Higuchi, R. Chelex 100 as a Medium for Simple Extraction of DNA for PCR-Based Typing from Forensic Material. Biotechniques 1991, 10, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Karp, A.; Isaac, P.G.; Ingram, D.S. Isolation of Nucleic Acids Using Silica-Gel Based Membranes: Methods Based on the Use of QIAamp Spin Columns. In Molecular Tools for Screening Biodiversity: Plants and Animals; Karp, A., Isaac, P.G., Ingram, D.S., Eds.; Springer: Dordrecht, The Netherlands, 1998; pp. 59–63. ISBN 978-94-009-0019-6. [Google Scholar]
- Esser, K.-H.; Marx, W.H.; Lisowsky, T. Nucleic Acid-Free Matrix: Regeneration of DNA Binding Columns. BioTechniques 2005, 39, 270–271. [Google Scholar] [CrossRef]
- Folmer, O. DNA Primers for Amplification of Mitochondrial Cytochrome c Oxidase Subunit I from Diverse Metazoan Invertebrates—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/7881515/ (accessed on 20 December 2024).
- Sanna, D.; Cossu, P.; Dedola, G.L.; Scarpa, F.; Maltagliati, F.; Castelli, A.; Franzoi, P.; Lai, T.; Cristo, B.; Curini-Galletti, M.; et al. Mitochondrial DNA Reveals Genetic Structuring of Pinna Nobilis across the Mediterranean Sea. PLoS ONE 2013, 8, e67372. [Google Scholar] [CrossRef]
- Mosa, K.A.; Gairola, S.; Jamdade, R.; El-Keblawy, A.; Al Shaer, K.I.; Al Harthi, E.K.; Shabana, H.A.; Mahmoud, T. The Promise of Molecular and Genomic Techniques for Biodiversity Research and DNA Barcoding of the Arabian Peninsula Flora. Front. Plant Sci. 2019, 9, 1929. [Google Scholar] [CrossRef]
- Carlson, D.; Lazar, J.G.; Connolly, M. Extraction of Dna from Biological Samples 2005. Available online: https://patents.google.com/patent/EP1581646A2/en (accessed on 3 April 2025).
- Efremenkov, V.; Drace, Z. Considerations for Waste Minimization at the Design Stage of Nuclear Facilities; Technical Reports Series/International Atomic Energy Agency; International Atomic Energy Agency: Vienna, Austria, 2007; ISBN 978-92-0-105407-4. [Google Scholar]
- Hoy, M.A. Insect Molecular Genetics: An Introduction to Principles and Applications; Academic Press: Cambridge, MA, USA, 2013; ISBN 978-0-240-82131-3. [Google Scholar]
- Conlon, B.H.; Schmidt, S.; Poulsen, M.; Shik, J.Z. Orthogonal Protocols for DNA Extraction from Filamentous Fungi. STAR Protoc. 2022, 3, 101126. [Google Scholar] [CrossRef]
- Adema, C.M. Sticky Problems: Extraction of Nucleic Acids from Molluscs. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2021, 376, 20200162. [Google Scholar] [CrossRef]
- Schiebelhut, L.M.; Abboud, S.S.; Gómez Daglio, L.E.; Swift, H.F.; Dawson, M.N. A Comparison of DNA Extraction Methods for High-Throughput DNA Analyses. Mol. Ecol. Resour. 2017, 17, 721–729. [Google Scholar] [CrossRef]
- Jaksch, K.; Eschner, A.; Rintelen, T.V.; Haring, E. DNA Analysis of Molluscs from a Museum Wet Collection: A Comparison of Different Extraction Methods. BMC Res. Notes 2016, 9, 348. [Google Scholar] [CrossRef]
- Der Sarkissian, C.; Pichereau, V.; Dupont, C.; Ilsøe, P.C.; Perrigault, M.; Butler, P.; Chauvaud, L.; Eiríksson, J.; Scourse, J.; Paillard, C.; et al. Ancient DNA Analysis Identifies Marine Mollusc Shells as New Metagenomic Archives of the Past. Mol. Ecol. Resour. 2017, 17, 835–853. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Doeven, E.H.; Yuan, D.; Guijt, R.M. Method for Lysis and Paper-Based Elution-Free DNA Extraction with Colourimetric Isothermal Amplification. Sci. Rep. 2024, 14, 14479. [Google Scholar] [CrossRef] [PubMed]
- Dairawan, M.; Shetty, P.J. The Evolution of DNA Extraction Methods. AJBSR 2020, 8, 39. [Google Scholar]
- Evans, M.F. The Polymerase Chain Reaction and Pathology Practice. Diagn. Histopathol. 2009, 15, 344–356. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Habor Laboratory: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Palumbi, S.; Martin, A.; Romano, S.; McMillan, W.O.; Stice, L.; Grabowski, G. The Simple Fool’s Guide To PCR. Available online: https://stacks.stanford.edu/file/druid:yh393jm6703/Simple_Fool%27s_Master%20PCR.pdf (accessed on 3 April 2025).
- Taberlet, P.; Griffin, S.; Goossens, B.; Questiau, S.; Manceau, V.; Escaravage, N.; Waits, L.P.; Bouvet, J. Reliable Genotyping of Samples with Very Low DNA Quantities Using PCR. Nucleic Acids Res. 1996, 24, 3189–3194. [Google Scholar] [CrossRef]
- Hajibabaei, M.; Smith, M.A.; Janzen, D.H.; Rodriguez, J.J.; Whitfield, J.B.; Hebert, P.D.N. A Minimalist Barcode Can Identify a Specimen Whose DNA Is Degraded. Mol. Ecol. Notes 2006, 6, 959–964. [Google Scholar] [CrossRef]
- Wandeler, P.; Hoeck, P.E.A.; Keller, L.F. Back to the Future: Museum Specimens in Population Genetics. Trends Ecol. Evol. 2007, 22, 634–642. [Google Scholar] [CrossRef]
- Giribet, G.; Sharma, P.P.; Benavides, L.R.; Boyer, S.L.; Clouse, R.M.; de Bivort, B.L.; Dimitrov, D.; Kawauchi, G.Y.; Murienne, J.; Schwendinger, P.J. Evolutionary and Biogeographical History of an Ancient and Global Group of Arachnids (Arachnida: Opiliones: Cyphophthalmi) with a New Taxonomic Arrangement. Biol. J. Linn. Soc. 2012, 105, 92–130. [Google Scholar] [CrossRef]
- Dabney, J.; Meyer, M.; Pääbo, S. Ancient DNA Damage. Cold Spring Harb. Perspect. Biol. 2013, 5, a012567. [Google Scholar] [CrossRef]
- Tao, S.-C.; Jiang, D.; Lu, H.-L.; Xing, W.-L.; Zhou, Y.-X.; Cheng, J. One-Tube Nested RT-PCR Enabled by Using a Plastic Film and Its Application for the Rapid Detection of SARS-Virus. Biotechnol. Lett. 2004, 26, 179–183. [Google Scholar] [CrossRef]
- Ahmed, S.; Ibrahim, M.; Nantasenamat, C.; Nisar, M.F.; Malik, A.A.; Waheed, R.; Ahmed, M.Z.; Ojha, S.C.; Alam, M.K. Pragmatic Applications and Universality of DNA Barcoding for Substantial Organisms at Species Level: A Review to Explore a Way Forward. Biomed. Res. Int. 2022, 2022, 1846485. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Yuan, C.; Tao, L.; Cai, Y.; Zhang, W. Life Barcoded by DNA Barcodes. Conserv. Genet. Resour. 2022, 14, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.L.; Núñez-Rodriguez, D.; Britzke, R.; Siccha-Ramirez, R.; Ramírez, R. DNA Barcoding for Assessing Biodiversity. In Conservation Genetics in the Neotropics; Galetti, P.M., Jr., Ed.; Springer International Publishing: Cham, Switzerland, 2023; ISBN 978-3-031-34854-9. [Google Scholar]
- Vialle, A.; Feau, N.; Allaire, M.; Didukh, M.; Martin, F.; Moncalvo, J.-M.; Hamelin, R.C. Evaluation of Mitochondrial Genes as DNA Barcode for Basidiomycota. Mol. Ecol. Resour. 2009, 9 (Suppl. S1), 99–113. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Liu, Q.; Qiu, S.; Dai, J.; Gao, X. DNA Barcoding: An Efficient Technology to Authenticate Plant Species of Traditional Chinese Medicine and Recent Advances. Chin. Med. 2022, 17, 112. [Google Scholar] [CrossRef]
- Campbell, M.A.; Buser, T.J.; Alfaro, M.E.; López, J.A. Addressing Incomplete Lineage Sorting and Paralogy in the Inference of Uncertain Salmonid Phylogenetic Relationships. PeerJ 2020, 8, e9389. [Google Scholar] [CrossRef]
- Joly, S.; McLenachan, P.A.; Lockhart, P.J. A Statistical Approach for Distinguishing Hybridization and Incomplete Lineage Sorting. Am. Nat. 2009, 174, E54–E70. [Google Scholar] [CrossRef]
- Mitochondrial Capture and Incomplete Lineage Sorting in the Diversification of Balitorine Loaches (Cypriniformes, Balitoridae) Revealed by Mitochondrial and Nuclear Genes—Tang—2012—Zoologica Scripta—Wiley Online Library. Available online: https://onlinelibrary.wiley.com/doi/full/10.1111/j.1463-6409.2011.00530.x (accessed on 30 January 2025).
- Weber, A.A.-T.; Stöhr, S.; Chenuil, A. Species Delimitation in the Presence of Strong Incomplete Lineage Sorting and Hybridization: Lessons from Ophioderma (Ophiuroidea: Echinodermata). Mol. Phylogenet. Evol. 2019, 131, 138–148. [Google Scholar] [CrossRef]
- Giantsis, I.A.; Abatzopoulos, T.J.; Angelidis, P.; Apostolidis, A.P. Mitochondrial Control Region Variability in Mytilus Galloprovincialis Populations from the Central-Eastern Mediterranean Sea. Int. J. Mol. Sci. 2014, 15, 11614–11625. [Google Scholar] [CrossRef]
- Sanna, D.; Azzena, I.; Locci, C.; Ankon, P.; Kružić, P.; Manfrin, C.; Pallavicini, A.; Ciriaco, S.; Segarich, M.; Batistini, E.; et al. Reconstructing the Evolutionary History of Pinna Nobilis: New Genetic Signals from the Past of a Species on the Brink of Extinction. Animals 2023, 14, 114. [Google Scholar] [CrossRef]
- Dabert, M.; Witalinski, W.; Kazmierski, A.; Olszanowski, Z.; Dabert, J. Molecular Phylogeny of Acariform Mites (Acari, Arachnida): Strong Conflict between Phylogenetic Signal and Long-Branch Attraction Artifacts. Mol. Phylogenet. Evol. 2010, 56, 222–241. [Google Scholar] [CrossRef]
- Thomas Thorpe, J.A. Phylogenomics Supports a Single Origin of Terrestriality in Isopods. Proc. R. Soc. B Biol. Sci. 2024, 291, 20241042. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-J.; Liu, Y.; Wang, Y.-L.; Xie, L.-L.; Wang, X.-Y.; Ma, Y.-S. Population Genetic Diversity and Structure of Tephritis Angustipennis and Campiglossa Loewiana (Diptera: Tephritidae) Based on COI DNA Barcodes in the Three-River Source Region, China. J. Insect Sci. 2024, 24, 7. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.J. Assessing the Value of DNA Barcodes and Other Priority Gene Regions for Molecular Phylogenetics of Lepidoptera. PLoS ONE 2010, 5, e10525. [Google Scholar] [CrossRef] [PubMed]
- Padial, J.M.; Gonzales, L.; Reichle, S.; Aguayo, R.; de la Riva, I. First Records of Five Species of the Genus Eleutherodactylus Dumeril and Bibron, 1841 (Anura, Leptodactylidae) for Bolivia. Graellsia 2004, 60, 167–174. [Google Scholar] [CrossRef]
- Zamani, A.; Dal Pos, D.; Fric, Z.F.; Orfinger, A.B.; Scherz, M.D.; Bartoňová, A.S.; Gante, H.F. The Future of Zoological Taxonomy Is Integrative, Not Minimalist. Syst. Biodivers. 2022, 20, 1–14. [Google Scholar] [CrossRef]
- Daglio, L.G.; Dawson, M.N. Integrative Taxonomy: Ghosts of Past, Present and Future. J. Mar. Biol. Assoc. UK 2019, 99, 1237–1246. [Google Scholar] [CrossRef]
- Dayrat, B. Towards Integrative Taxonomy. Biol. J. Linn. Soc. 2005, 85, 407–417. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence (5′–3′) | Reference |
|---|---|---|
| LCO1490 | GGTCAACAAATCATAAAGATATTGG | [37] |
| HCO2198 | TAAACTTXAGGGTGACCAAAAAATCA | |
| PnCOI_F_KTSR08 | CCCTGCCAAATTACACCAGT | [17] |
| PnCOI_R_KTSR08 | TTTTGGCTTTTGCCTTCTTC | |
| PnCOI_Fdg_KTSR08 | CCCTAGCCAAAATTACACCAGT | [17] |
| PnCOI_Rdg_KTSR08 | GAAGAAGGCAAWAGCCAAAA | |
| PnCOI_F_MK22 | CAACACAGGAAGAGAGACTACCA | [18] |
| PnCOI_R_MK22 | GGCAGGGTTTTTGGGGGA | |
| PnCOI_L_SAN13 | GGTTGAACTATHTATCCNCC | [38] |
| PnCOI_H_SAN13 | GAAATCATYCCAAAAGC | |
| Pmur_COIF_KTSR08 | GAAAGTGCCCGGTAACAAAA | [17] |
| Pmur_COIR_KTSR08 | TGATAGGGGTTCCGGATATG | |
| Pmur_COIFdg_KTSR08 | GAAAGTGCCCRGTWACAAART | [17] |
| Pmur_COIRdg_KTSR08 | CATATCYGGMACCCCTATCA |
| PCR Trial | 1st PCR Primer Pair | 2nd PCR Primer Pair | Amplicon Size |
|---|---|---|---|
| a: 1st semi-nested | PnCOI_F_KTSR08-PnCOI_R_KTSR08 | PnCOI_F_MK22-PnCOI_R_KTSR08 | ca. 500 bp |
| b: 2nd semi-nested | PnCOI_Fdg_KTSR08-PnCOI_Rdg_KTSR08 | PnCOI_F_MK22-PnCOI_Rdg_KTSR08 | ca. 550 bp |
| c: 3rd semi-nested | PnCOI_L_SAN13-PnCOI_H_SAN13 | PnCOI_F_MK22-PnCOI_H_SAN13 | ca. 490 bp |
| d: 4th semi-nested | Pmur_COIFdg_KTSR08-Pmur_COIR_KTSR08 | PnCOI_F_MK22-Pmur_COIR_KTSR08 | ca. 650 bp |
| e: nested | LCO1490-HCO2198 | PnCOI_F_MK22 -R_MK22 | ca. 465 bp |
| DNA Extraction Method | Chelex-100 | NaCl | Fast-CTAB | NucleoSpin | ||
| Positive indication of high-molecular-weight DNA % | 50.0 | 100.0 | 100.0 | 100.0 | ||
| Consistency * | 1 | 2 | 3 | 3 | ||
| COI amplification success rate % | Sanger Seq success rate on successful PCRs% after CTAB | Total successful taxonomic identification of samples % after CTAB | ||||
| DNA extraction method | Chelex-100 | NaCl | Fast-CTAB | NucleoSpin | ||
| LCO-HCO | 30.0 | 90.0 | 100.0 | 80.0 | 0 | 0 |
| 1st semi-nested PCR | 50.0 | 50.0 | 75 | 30.0 | ||
| 2nd semi-nested PCR | 37.5 | 37.5 | 100 | 30.0 | ||
| 3rd semi-nested PCR | 12.5 | 12.5 | 100 | 10.0 | ||
| 4th semi-nested PCR | 0.0 | 0.0 | 100 | 0.0 | ||
| Nested PCR | 100.0 | 100.0 | 100 | 100.0 | ||
| Protocol | Effectiveness (Purity and Yield) | Hands-On Time Required | Cost Per SAMPLE |
|---|---|---|---|
| Chelex100 | Moderate: Yields DNA suitable for PCR but may not be ideal for applications requiring high-purity DNA. | Very low: Quick, single-tube process (~20–30 min). | Very low: Inexpensive reagents; ideal for high-throughput and routine PCR applications. |
| NaCl Precipitation | High for routine DNA extractions; yields clean DNA but may not efficiently remove all contaminants (e.g., proteins). | Moderate: labor-intensive with multiple centrifugation and incubation steps and washing (~2–3 h and an overnight step). | Low: Requires minimal reagents and equipment; very economical for bulk extractions. |
| Fast-CTAB | High: effective for tissues with polysaccharides and ideal for minute starting material; adaptable for diverse samples. | Moderate: Several steps involving incubation, precipitation, and washing (~2–3 h). | Low: Requires basic reagents and equipment; minimal recurring costs. |
| Silica-based column (NucleoSpin Tissue) | High: Produces high-purity DNA suitable for sensitive downstream applications (e.g., qPCR, sequencing), user-friendly for non-experts. | Low to moderate: Rapid, streamlined process (1–2 h and an overnight incubation step) | High: Commercial kits are expensive; cost scales with sample volume and kit usage. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamilari, M.; Papaioannou, C.; Augustinos, A.; Spinos, E.; Giantsis, I.A.; Ramfos, A.; Theodorou, J.A.; Batargias, C. From Shell to Sequence: Optimizing DNA Extraction and PCR for Pen Shell Identification. Water 2025, 17, 1162. https://doi.org/10.3390/w17081162
Kamilari M, Papaioannou C, Augustinos A, Spinos E, Giantsis IA, Ramfos A, Theodorou JA, Batargias C. From Shell to Sequence: Optimizing DNA Extraction and PCR for Pen Shell Identification. Water. 2025; 17(8):1162. https://doi.org/10.3390/w17081162
Chicago/Turabian StyleKamilari, Maria, Charikleia Papaioannou, Antonios Augustinos, Efthimios Spinos, Ioannis A. Giantsis, Alexios Ramfos, John A. Theodorou, and Costas Batargias. 2025. "From Shell to Sequence: Optimizing DNA Extraction and PCR for Pen Shell Identification" Water 17, no. 8: 1162. https://doi.org/10.3390/w17081162
APA StyleKamilari, M., Papaioannou, C., Augustinos, A., Spinos, E., Giantsis, I. A., Ramfos, A., Theodorou, J. A., & Batargias, C. (2025). From Shell to Sequence: Optimizing DNA Extraction and PCR for Pen Shell Identification. Water, 17(8), 1162. https://doi.org/10.3390/w17081162