
The Reactivity of Human and Equine Estrogen Quinones towards Purine Nucleosides

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Computational Methods
2.1. Molecular Gibbs Free Energies
2.2. Acidity Constants and the Fraction of the Non-Dominant Protonation State
2.3. Activation Gibbs Free Energies (Barriers)
2.4. Effective Activation Gibbs Free Energies (Effective Barriers)
3. Results and Discussion
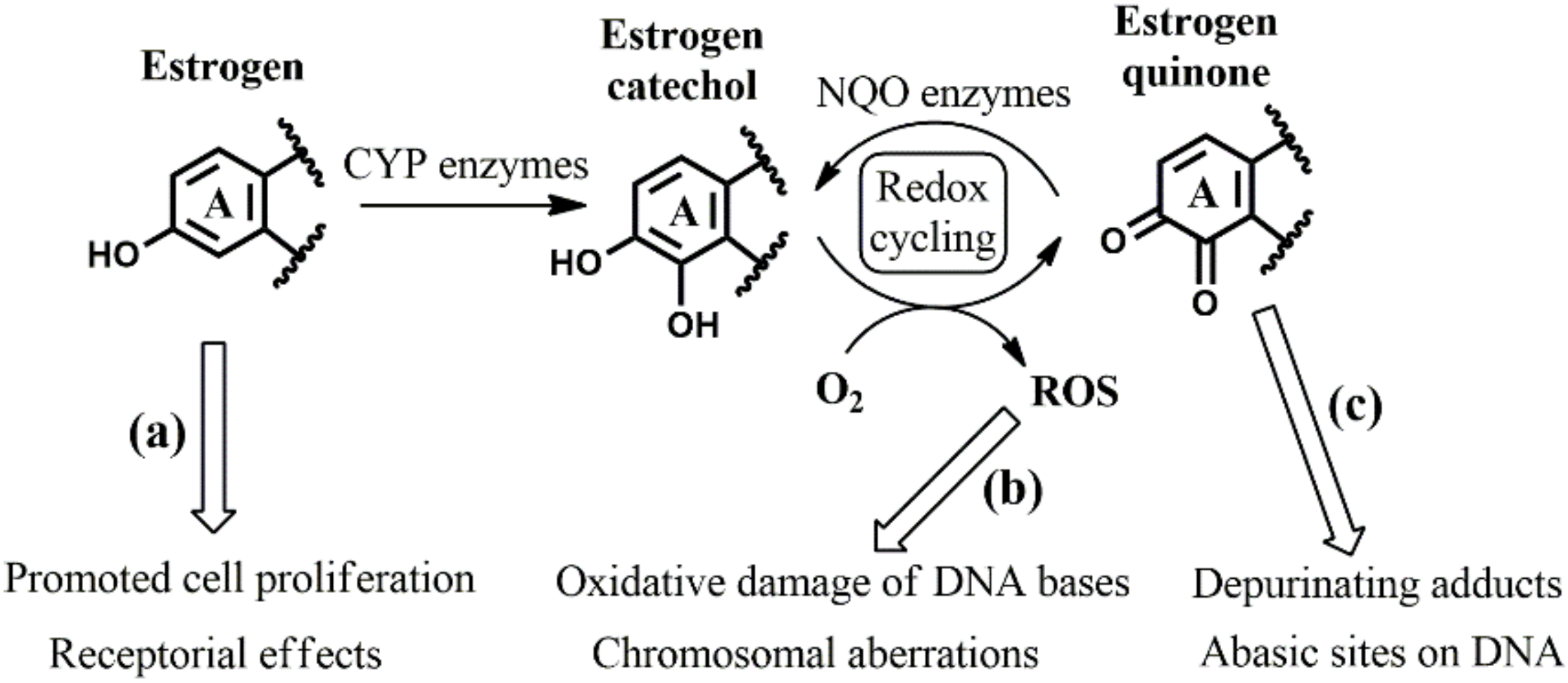
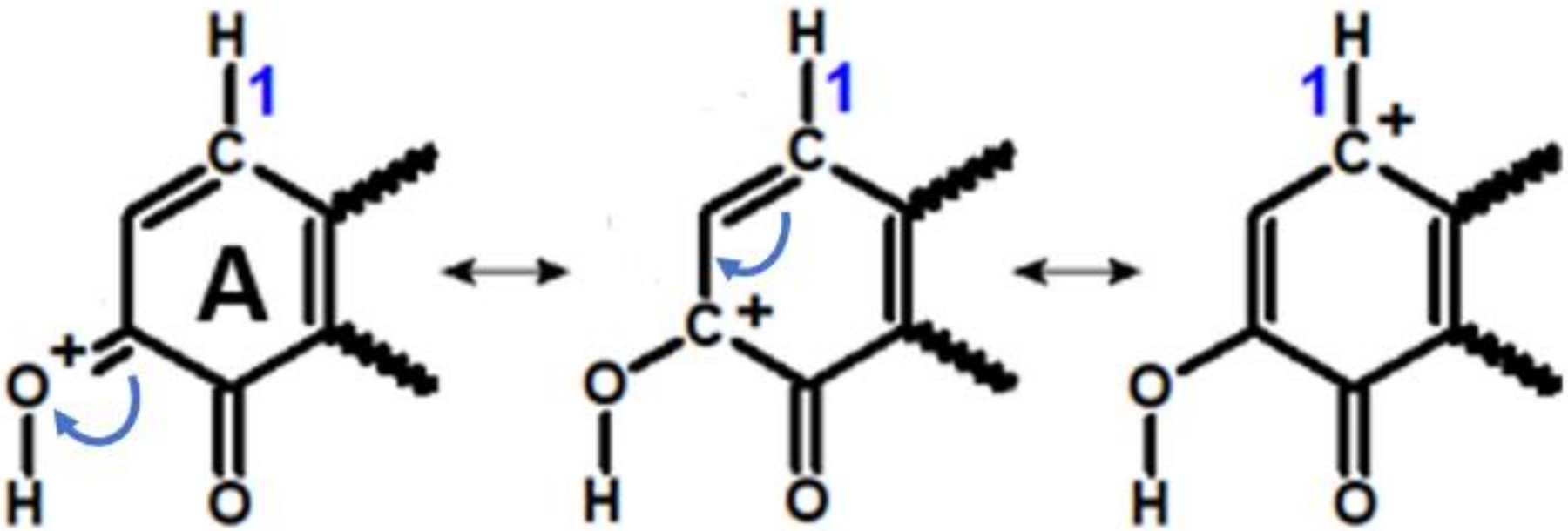
3.1. Identifying the Elementary Steps of Depurination
3.2. Accounting for the Distribution of Protonation States: Effective Activation Gibbs Free Energies
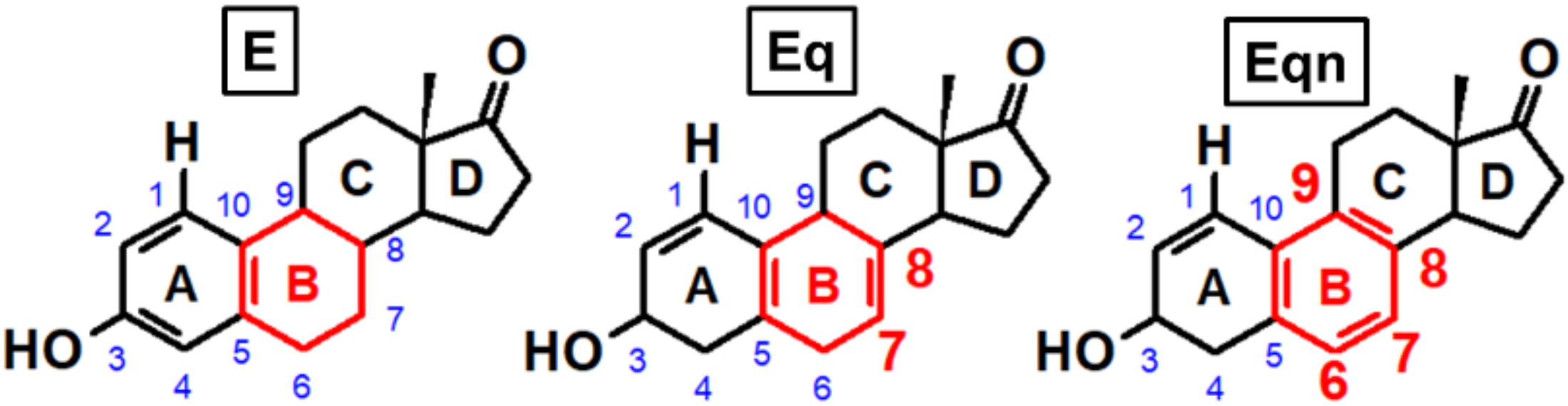
3.3. Effect of Ring “B” on Michael Addition
3.4. Effect of Ring “B” on Rearomatization
3.5. Effect of ring “B” on Gylcosidic Cleavage
4. Conclusions
- (1)
- in the case of Michael addition of the DNA nucleoside to the quinone metabolite (first elementary step), where a higher rate of orders of magnitude is expected with equilenin-3,4-quinone compared to the analogous estrone derivative. The 5 kcal/mol lower effective activation Gibbs free energy with equilenin can be explained by the relatively high stability of its protonated 3,4-quinone, which serves as the actual reaction partner of adenosine or guanosine.
- (2)
- in the case of the subsequent rearomatization step (i.e., proton loss from C1 of estrogen), where the proton release is slightly accelerated by the other equine estrogen, equilin. Here, a less significant, 1.5–2 kcal/mol decrease in the effective barrier was observed compared to that calculated for the estrone derivative.
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- ESHRE Capri Workshop Group. Hormones and breast cancer. Hum. Reprod. Update 2004, 10, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Clemons, M.; Goss, P. Estrogen and the Risk of Breast Cancer. N. Engl. J. Med. 2001, 344, 276–285. [Google Scholar] [CrossRef]
- Million Women Study Collaborators. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet 2003, 362, 419–427. [Google Scholar] [CrossRef]
- Ravdin, P.M.; Cronin, K.A.; Howlader, N.; Berg, C.D.; Chlebowski, R.T.; Feuer, E.J.; Edwards, B.K.; Berry, D.A. The Decrease in Breast-Cancer Incidence in 2003 in the United States. N. Engl. J. Med. 2007, 356, 1670–1674. [Google Scholar] [CrossRef]
- Prokai-Tatrai, K.; Prokai, L. Impact of Metabolism on the Safety of Estrogen Therapy. Ann. N. Y. Acad. Sci. 2005, 1052, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Bhavnani, B.R. Pharmacokinetics and Pharmacodynamics of Conjugated Equine Estrogens: Chemistry and Metabolism. Exp. Biol. Med. 1998, 217, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Bhavnani, B.R.; Tam, S.-P.; Lu, X. Structure Activity Relationships and Differential Interactions and Functional Activity of Various Equine Estrogens Mediated via Estrogen Receptors (ERs) ERα and ERβ. Endocrinology 2008, 149, 4857–4870. [Google Scholar] [CrossRef][Green Version]
- Jeyakumar, M.; Carlson, K.E.; Gunther, J.R.; Katzenellenbogen, J.A. Exploration of Dimensions of Estrogen Potency. J. Biol. Chem. 2011, 286, 12971–12982. [Google Scholar] [CrossRef]
- Wang, Z.; Chandrasena, E.R.; Yuan, Y.; Peng, K.; van Breemen, R.B.; Thatcher, G.R.J.; Bolton, J.L. Redox Cycling of Catechol Estrogens Generating Apurinic/Apyrimidinic Sites and 8-oxo-Deoxyguanosine via Reactive Oxygen Species Differentiates Equine and Human Estrogens. Chem. Res. Toxicol. 2010, 23, 1365–1373. [Google Scholar] [CrossRef]
- Li, J.J.; Li, S.A.; Oberley, T.D.; Parsons, J.A. Carcinogenic activities of various steroidal and nonsteroidal estrogens in the hamster kidney: Relation to hormonal activity and cell proliferation. Cancer Res. 1995, 55, 4347–4351. [Google Scholar]
- Henderson, B.E.; Feigelson, H.S. Hormonal carcinogenesis. Carcinogenesis 2000, 21, 427–433. [Google Scholar] [CrossRef]
- Revankar, C.M. A Transmembrane Intracellular Estrogen Receptor Mediates Rapid Cell Signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Song, R.X.-D.; Fan, P.; Yue, W.; Chen, Y.; Santen, R.J. Role of receptor complexes in the extranuclear actions of estrogen receptor α in breast cancer. Endocr. Relat. Cancer 2006, 13, S3–S13. [Google Scholar] [CrossRef]
- Rogan, E.G. Relative imbalances in estrogen metabolism and conjugation in breast tissue of women with carcinoma: Potential biomarkers of susceptibility to cancer. Carcinogenesis 2003, 24, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Castagnetta, L.A.M.; Granata, O.M.; Traina, A.; Ravazzolo, B.; Amoroso, M.; Miele, M.; Bellavia, V.; Agostara, B.; Carruba, G. Tissue content of hydroxyestrogens in relation to survival of breast cancer patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 3146–3155. [Google Scholar]
- Chandrasena, R.E.P.; Edirisinghe, P.D.; Bolton, J.L.; Thatcher, G.R.J. Problematic detoxification of estrogen quinones by NAD(P)H-dependent quinone oxidoreductase and glutathione-S-transferase. Chem. Res. Toxicol. 2008, 21, 1324–1329. [Google Scholar] [CrossRef] [PubMed]
- Bolton, J.L.; Thatcher, G.R.J. Potential Mechanisms of Estrogen Quinone Carcinogenesis. Chem. Res. Toxicol. 2008, 21, 93–101. [Google Scholar] [CrossRef]
- Stack, D.E.; Li, G.; Hill, A.; Hoffman, N. Mechanistic Insights into the Michael Addition of Deoxyguanosine to Catechol Estrogen-3,4-quinones. Chem. Res. Toxicol. 2008, 21, 1415–1425. [Google Scholar] [CrossRef]
- Stack, D. Identifying the Tautomeric Form of a Deoxyguanosine-Estrogen Quinone Intermediate. Metabolites 2015, 5, 475–488. [Google Scholar] [CrossRef]
- Cavalieri, E.L.; Rogan, E.G. Depurinating estrogen-DNA adducts, generators of cancer initiation: Their minimization leads to cancer prevention. Clin. Transl. Med. 2016, 5. [Google Scholar] [CrossRef]
- Cavalieri, E.; Chakravarti, D.; Guttenplan, J.; Hart, E.; Ingle, J.; Jankowiak, R.; Muti, P.; Rogan, E.; Russo, J.; Santen, R.; et al. Catechol estrogen quinones as initiators of breast and other human cancers: Implications for biomarkers of susceptibility and cancer prevention. Biochim. Biophys. Acta–Rev. Cancer 2006, 1766, 63–78. [Google Scholar] [CrossRef]
- Zhang, F.; Chen, Y.; Pisha, E.; Shen, L.; Xiong, Y.; van Breemen, R.B.; Bolton, J.L. The Major Metabolite of Equilin, 4-Hydroxyequilin, Autoxidizes to an o -Quinone Which Isomerizes to the Potent Cytotoxin 4-Hydroxyequilenin- o -quinone. Chem. Res. Toxicol. 1999, 12, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Shen, L. Bioreductive activation of catechol estrogen-ortho-quinones: Aromatization of the B ring in 4-hydroxyequilenin markedly alters quinoid formation and reactivity. Carcinogenesis 1997, 18, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wijewickrama, G.T.; Peng, K.; Dietz, B.M.; Yuan, L.; van Breemen, R.B.; Bolton, J.L.; Thatcher, G.R.J. Estrogen Receptor α Enhances the Rate of Oxidative DNA Damage by Targeting an Equine Estrogen Catechol Metabolite to the Nucleus. J. Biol. Chem. 2009, 284, 8633–8642. [Google Scholar] [CrossRef]
- Cavalieri, E.; Saeed, M.; Zahid, M.; Cassada, D.; Snow, D.; Miljkovic, M.; Rogan, E. Mechanism of DNA depurination by carcinogens in relation to cancer initiation. IUBMB Life 2012, 64, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Benedek, Z.; Papp, M.; Oláh, J.; Szilvási, T. Demonstrating the Direct Relationship between Hydrogen Evolution Reaction and Catalyst Deactivation in Synthetic Fe Nitrogenases. ACS Catal. 2020, 10, 12555–12568. [Google Scholar] [CrossRef]
- Soyemi, A.; Szilvási, T. Trends in computational molecular catalyst design. Dalt. Trans. 2021, 50, 10325–10339. [Google Scholar] [CrossRef]
- Dewyer, A.L.; Argüelles, A.J.; Zimmerman, P.M. Methods for exploring reaction space in molecular systems. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1354. [Google Scholar] [CrossRef]
- Vogiatzis, K.D.; Polynski, M.V.; Kirkland, J.K.; Townsend, J.; Hashemi, A.; Liu, C.; Pidko, E.A. Computational Approach to Molecular Catalysis by 3d Transition Metals: Challenges and Opportunities. Chem. Rev. 2019, 119, 2453–2523. [Google Scholar] [CrossRef]
- Blomberg, M.R.A.; Borowski, T.; Himo, F.; Liao, R.-Z.; Siegbahn, P.E.M. Quantum Chemical Studies of Mechanisms for Metalloenzymes. Chem. Rev. 2014, 114, 3601–3658. [Google Scholar] [CrossRef]
- Sheng, X.; Kazemi, M.; Planas, F.; Himo, F. Modeling Enzymatic Enantioselectivity using Quantum Chemical Methodology. ACS Catal. 2020, 10, 6430–6449. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61. [Google Scholar] [CrossRef]
- Eyring, H. The Activated Complex in Chemical Reactions. J. Chem. Phys. 1935, 3, 107–115. [Google Scholar] [CrossRef]
- An, R.; Jia, Y.; Wan, B.; Zhang, Y.; Dong, P.; Li, J.; Liang, X. Non-Enzymatic Depurination of Nucleic Acids: Factors and Mechanisms. PLoS ONE 2014, 9, e115950. [Google Scholar] [CrossRef]
- Saeed, M.; Zahid, M.; Gunselman, S.J.; Rogan, E.; Cavalieri, E. Slow loss of deoxyribose from the N7deoxyguanosine adducts of estradiol-3,4-quinone and hexestrol-3′,4′-quinone. Steroids 2005, 70, 29–35. [Google Scholar] [CrossRef]
- Zahid, M.; Kohli, E.; Saeed, M.; Rogan, E.; Cavalieri, E. The Greater Reactivity of Estradiol-3,4-quinone vs Estradiol-2,3-quinone with DNA in the Formation of Depurinating Adducts: Implications for Tumor-Initiating Activity. Chem. Res. Toxicol. 2006, 19, 164–172. [Google Scholar] [CrossRef]
- Harvey, J.N.; Himo, F.; Maseras, F.; Perrin, L. Scope and Challenge of Computational Methods for Studying Mechanism and Reactivity in Homogeneous Catalysis. ACS Catal. 2019, 9, 6803–6813. [Google Scholar] [CrossRef]
- Tamura, A.; Privalov, P.L. The entropy cost of protein association. J. Mol. Biol. 1997, 273, 1048–1060. [Google Scholar] [CrossRef] [PubMed]
- Lábas, A.; Krámos, B.; Oláh, J. Combined Docking and Quantum Chemical Study on CYP-Mediated Metabolism of Estrogens in Man. Chem. Res. Toxicol. 2017, 30, 583–594. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benedek, Z.; Girnt, P.; Olah, J. The Reactivity of Human and Equine Estrogen Quinones towards Purine Nucleosides. Symmetry 2021, 13, 1641. https://doi.org/10.3390/sym13091641
Benedek Z, Girnt P, Olah J. The Reactivity of Human and Equine Estrogen Quinones towards Purine Nucleosides. Symmetry. 2021; 13(9):1641. https://doi.org/10.3390/sym13091641
Chicago/Turabian StyleBenedek, Zsolt, Peter Girnt, and Julianna Olah. 2021. "The Reactivity of Human and Equine Estrogen Quinones towards Purine Nucleosides" Symmetry 13, no. 9: 1641. https://doi.org/10.3390/sym13091641
APA StyleBenedek, Z., Girnt, P., & Olah, J. (2021). The Reactivity of Human and Equine Estrogen Quinones towards Purine Nucleosides. Symmetry, 13(9), 1641. https://doi.org/10.3390/sym13091641