1. Introduction
The misuse of antibiotics has led to the development of drug resistance in bacteria, rendering infections challenging to treat and potentially life-threatening [
1]. Antimicrobial peptides (AMPs) present a potential alternative to antibiotics [
2]. They constitute a class of small protein molecules or peptides with antimicrobial activity, widely found in the flora and fauna of the natural world. AMPs possess the capability to combat various microorganisms, including bacteria and fungi. These peptides exert their antimicrobial effects through diverse mechanisms such as disrupting cell membranes, interfering with protein synthesis, and inducing self-destruction in microorganisms. The unique mechanism of action displayed by AMPs sets them apart from antibiotics, making it challenging for bacteria to develop resistance against them [
3].
However, the wet lab experiments required for identifying and characterizing AMPs are complex and time-consuming, necessitating the development of efficient predictive models through modern computational science. Currently, several computation-based AMP predictors have already been developed, ClassAMP utilizes a combination of features, including charge, hydrophobicity, BLOSUM-50 matrix scores, conformational similarity, normalized van der Waals volume, polarity, and polarizability [
4]. Following feature selection, these characteristics are employed as input for a multiclass classification model composed of random forests and support vector machines. This integrated approach enables the accurate prediction and classification of AMPs. IAMPpred is specifically designed for variable-length AMP sequences. It employs a 1D-CNN to process the AMP sequences, extracting feature vectors from the hidden layers to serve as representations of the peptides’ features. Subsequently, these feature vectors are input into a support vector machine (SVM) to accomplish the classification task. This integrated methodology effectively addresses the challenge posed by variable-length sequences and enables accurate classification of AMPs [
5]. IAMPE [
6] categorizes amino acids into different groups using their 13CNM resonance spectra and analyzes the composition and distribution of members within these groups to construct feature vectors for antimicrobial peptide sequences. Then, these vectors were input into SVM and random forests for AMP classification prediction. AMPfun [
7] employs a comprehensive feature set, including n-gram features, amino acid composition (AAC) features, pseudo amino acid composition (PseAAC) features, and physicochemical features. Following feature selection, the model utilizes SVM and Random Forest (RF) algorithms for the classification and prediction of AMP functionalities. iAMPCN [
8] employed diverse encoding methods for input sequences, utilizing distinct CNN to extract features; subsequently, these extracted vectors are input into a multilayer perceptron for classification, culminating in the successful prediction of AMP functionalities. This approach showcases a nuanced strategy, leveraging different encoding techniques and specialized CNN architectures to enhance the accuracy of feature extraction and subsequent functional predictions for AMPs. sAMPpred-GAT [
9] used graph attention mechanisms and incorporated structural features into deep learning networks, further improving the predictive accuracy of AMPs. iAMP-Attenpred [
10] uses the BERT feature extraction method and CNN-BiLSTM-Attention combination model to achieve binary classification prediction of antimicrobial peptides. Despite the success of the aforementioned classifiers on their respective datasets, according to the research conducted by XU, these classifiers did not perform well on a comprehensive dataset [
11]. We analyze that the classifiers were unable to fully capture the diversity of data distributions. Hence, there is a need to develop a novel AMP classifier that enhances overall accuracy and possesses improved generalization capabilities by ensuring a more comprehensive extraction of AMP features.
Protein language models are a specific type of neural network that possess the ability to predict the subsequent character or vocabulary based on preceding text and have found applications in the field of biochemistry as a transfer learning tool [
12]. By inputting protein sequences and learning the inherent biochemical properties, structural information, and other intrinsic patterns, protein language models generate feature vectors that can be applied to various downstream protein tasks. Existing research has demonstrated favorable results in multiple downstream prediction tasks employing protein language models [
13]. However, different protein language models are developed using different training datasets, leading to differences in the emphasis placed on protein representation by each model. Consequently, a single protein language model may suffer from incomplete protein representation.
In order to apply protein language models to the task of antimicrobial peptide prediction and overcome the issue of incomplete protein representation inherent in a single protein language model, we merged three different protein language models: ESM-2, ProtBert, and UniRep. They are based on Transformer, BERT, and RNN, respectively. Therefore, the emphasis on representing proteins also varies, leading to differences in the emphasis on protein feature vectors. We extracted and fused protein features from these three different protein language models. The merged vectors elevate the comprehensiveness of protein representation to a new level.
Peptide sequences can exhibit symmetry through repetitive or mirrored patterns of amino acids, which can be crucial for the peptide’s stability, folding, and interaction with other molecules. Further, 1D-CNN can learn to identify and extract symmetrical features from peptide sequences by adjusting the weights of its convolutional filters. This bidirectional processing ability makes the Bi-LSTM particularly well suited to capturing symmetrical features within sequences, as symmetry often involves the interrelationship between the two ends of the sequence. Therefore, the merged feature vectors are then input into a hybrid deep learning network composed of multi-layered bidirectional LSTM networks, 1D-CNN, and an attention mechanism. The performance score of this model is validated through relevant verification methods, ultimately achieving superior results compared to state-of-the-art research.
2. Materials and Methods
2.1. Dataset and Data Preprocessing
To date, plenty of AMP databases exist. The Antimicrobial Peptide Database (APD) [
14] is an early-established repository that aggregates extensively sourced AMP sequences and related information. It encompasses AMP data from various biological domains, including bacteria, fungi, and animals, along with classification, structure, and activity information for these peptides. Linking Antimicrobial Peptide [
15] (LAMP) provides sequences of AMPs from various organisms, both internal and external, accompanied by relevant literature citations and additional annotation data. The Collection of Antimicrobial Peptides [
16] (CAMP) consolidates AMP information from different species, including various structural classification details. The Database of Antimicrobial Activity and Structure of Peptides [
17] (DBAASP) serves as a database for storing and providing information on AMPs, encompassing sequences, structures, antimicrobial activity, and relevant literature citations. The Data Repository of Antimicrobial Peptides [
18] (DRAMP) is a comprehensive AMP database containing structural data and annotation entries. The Structurally Annotated Therapeutic Peptides Database [
19] (SATPdb) offers a wealth of AMP structural data, primarily predicted through computational tools. These database establishments facilitate researchers, aiding in the profound exploration and scientific advancement of the field of AMPs.
To mitigate the impact of varying data distributions across different databases, we utilized a comprehensive benchmark evaluation dataset for training and validation. This benchmark dataset comprises AMP and non-AMP peptide data, the positive data are from six different databases: APD, LAMP, CAMP, DBAASP, DRAMP, and SATPdb, the negative data was randomly extracted from UniProt. As new databases may reference data from earlier databases, leading to potential data overlap between different databases, the CD-HIT tool [
20] was employed to eliminate redundant AMP and non-AMP peptides, and sequences with a similarity exceeding 90% between peptide sequences from different databases were removed. Based on the research by Yue Zhang [
21] and Ke Yan [
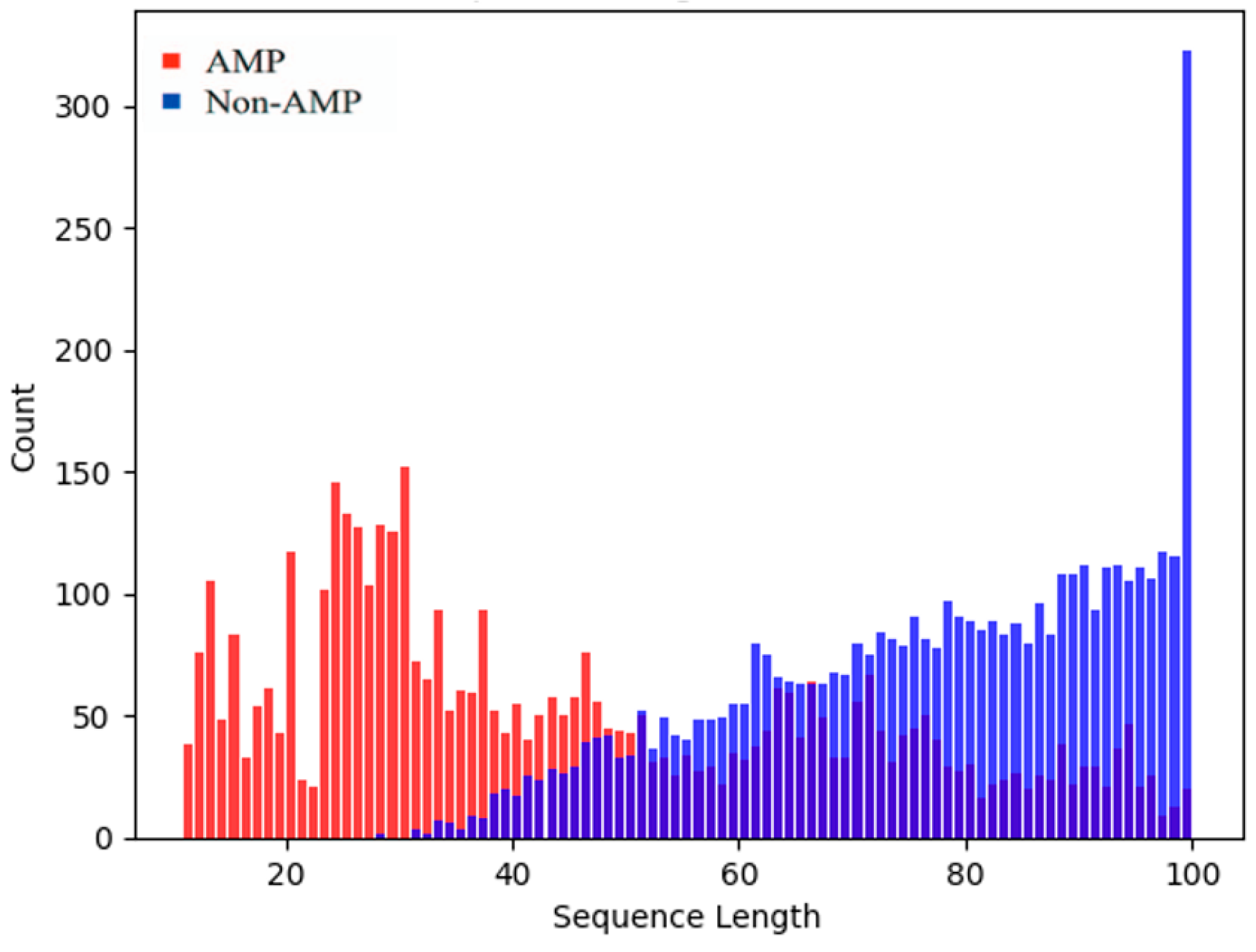
9], excessively long peptide chains may result in more complex structures, making protein synthesis challenging; conversely, overly short AMPs may lack sufficient functional sites or structural domains and are prone to degradation in the environment. Therefore, sequences with lengths ranging from 10 to 100 amino acids were selected, and excluded sequences that contained non-standard amino acids (B, J, O, U). This process resulted in a benchmark evaluation dataset comprising 4550 AMPs and 4550 non-AMPs. The lengths of AMPs typically lean towards the shorter side, predominantly falling within the range of 20–40 amino acids, the antimicrobial activity of AMPs tends to weaken with increasing length, and, as the number of amino acids increases, the quantity of AMPs gradually decreases. In contrast, non-AMPs generally exhibit greater length [
7] from the sequence distribution plot of our constructed dataset, which is shown in
Figure 1. This observation underscores the conformity of our dataset to the aforementioned pattern, implying that the dataset effectively captures the diverse length distribution of AMP. It is important to note that the non-AMP sequences in this dataset are longer on average than the AMP sequences, which may introduce some bias in the experiments. However, since most existing works have evaluated their models based on this dataset [
11], we have also utilized it for our study.
To assess the model’s generalization ability, an independent test set named XUAMP, created by Xu et al. [
11], was utilized. This dataset includes 1536 AMPs and 1536 non-AMPs. The CD-HIT tool was applied to the independent test set and the benchmark evaluation dataset to remove sequences with a similarity exceeding 90%, ensuring the independence of the data and providing a more objective evaluation of the model’s generalization ability.
To better compare our model with the latest model iAMP-Attenpred, we also utilized the datasets from Xing et al.’s [
10] research: Xingdataset1 and Xingdataset2. The AMPs in Xingdataset1 were collected from the AMPer [
22], APD3 [
14], and ADAM [
23] databases, and only sequences containing standard amino acids were chosen, with sequences having a similarity higher than 90% removed, and non-AMPs peptides were collected from the UniProt database, resulting in a total of 3594 AMPs and 3925 non-AMPs. For Xingdataset2, AMPs were collected from APD [
14], and only sequences containing standard amino acids were chosen, and, with sequences having a similarity higher than 40% removed, non-AMPs were collected from the UniProt database, resulting in a total of 879 AMPs and 2405 non-AMPs.
2.2. The Framework of UniproLcad
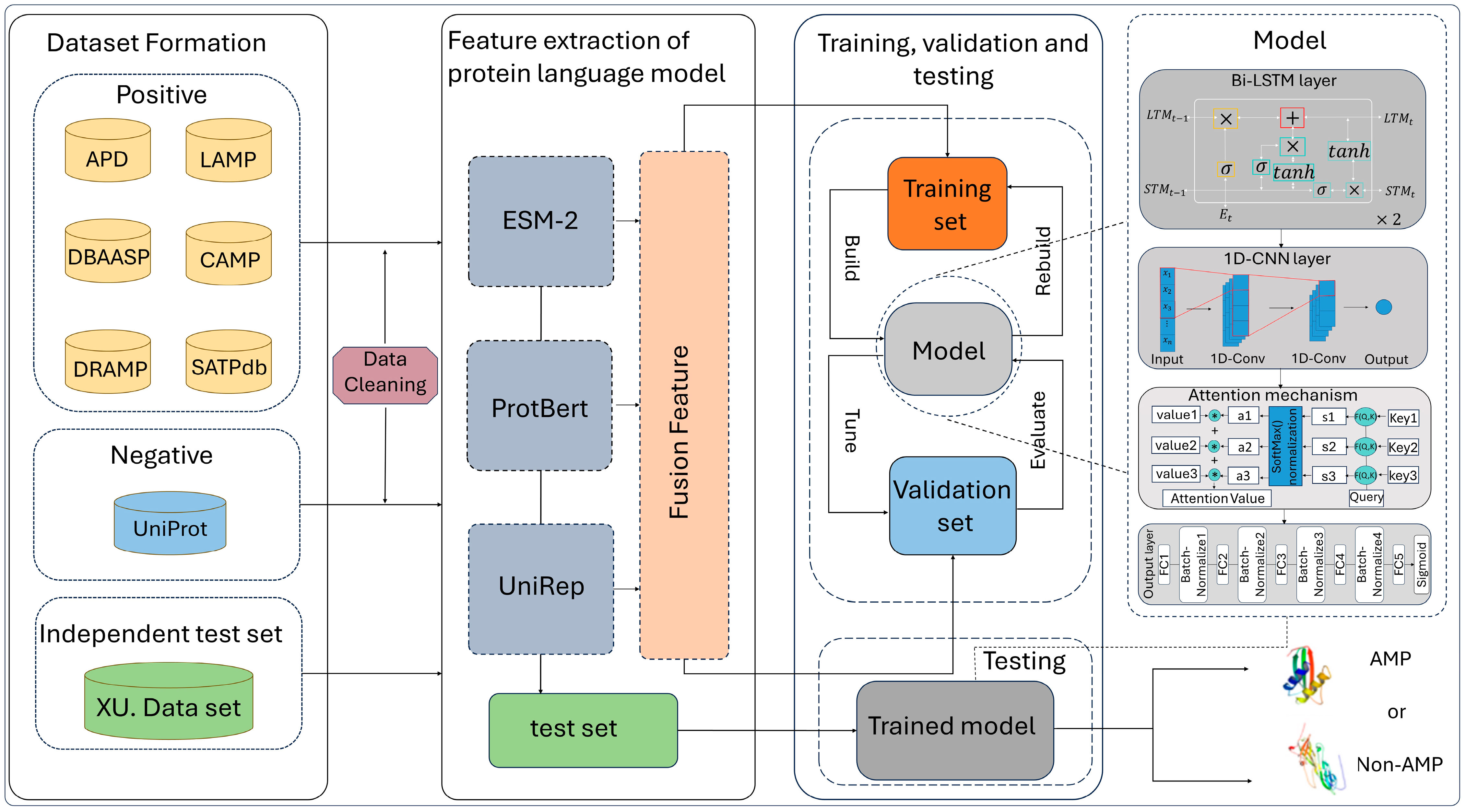
In this study, we have created a predictor for AMPs using a deep learning approach. Our predictor incorporates multi-perspective features extracted from various protein language models, enhancing its predictive capabilities. As shown in
Figure 2, the predictor consists of the following key components.
(1) In the Bi-LSTM layer, the model utilizes two stacked Bi-LSTM layers to process input features. This approach enables the learning of hidden representations and captures dependencies among contextual information. Theoretically speaking, increasing the number of Bi-LSTM layers can improve the fitting effect of the model; however, experiments have shown that using two Bi-LSTM layers can achieve higher model effects than more layers. The first Bi-LSTM hidden layer has a size of 128, and the size of the second Bi-LSTM hidden layer is 2.
(2) In the 1D-CNN Layer, we utilize a 1D-CNN to extract protein information from the hidden layers of a Bi-LSTM network, obtaining higher-dimensional protein feature vectors to better adapt to our model. Experimental results indicate that the model performs optimally using a single convolution layer with convolutional units having a kernel size of 2000, input channels of 4, and output channels of 2.
(3) Attention mechanism: Following the 1D-CNN layer, an attention mechanism is employed, which can effectively assign attention weights to high-dimensional sequence features from the output of the 1D-CNN layer. Specifically, it refers to the attention of the output
at a certain moment on various parts of the input
, which is the redistribution of weights. In other words, it involves the reassignment of weights for each part of
at each moment with respect to its contribution to
. The calculation formulas for the attention mechanism are as follows.
Firstly, obtain the hidden representation of the hidden state through a fully connected layer . Here, and represent the parameter matrix and bias of a single-layer perceptron. Then, calculate the importance values of the elements as the similarity between the element-wise context vector and . The variable represents a high-level representation of a fixed query. During the training process, the values of are initialized randomly and then optimized collectively to learn the most effective representation. Normalize the importance weights using a SoftMax function. Finally, the output is the product of and .
(4) Output layer: the role of the output layer is to reshape and process the feature vector produced by the neural network, ultimately yielding classification results. This involves operations such as flattening, batch normalization, and the Sigmoid activation function. The output of the attention mechanism is fed into Multilayer Perceptron (MLP). The first fully connected layer comprises 1024 nodes, followed by layers with 512, 256, 128, and 8 nodes, respectively. A linear layer with high discriminative power is necessary for this purpose, and the linear layer is defined as follows:
where
and
are the output and input vectors, respectively,
is the initially flattened vector,
is the weight matrix, and
is the bias for the linear layer.
Batch normalization is applied after each fully connected layer to maintain reasonable data distributions. Batch normalization operates on the principle of normalizing data within each training batch to ensure a stable distribution of input data. The specific procedure involves calculating the mean and standard deviation for each batch of data and then normalizing the data to have a mean of 0 and a standard deviation of 1. The computation is expressed as follows:
where
represents the data after batch normalization,
denotes the mean of the current batch,
represents the standard deviation of the samples, and
and
are used for scaling and shifting the data samples, respectively.
The Sigmoid function is applied to the output of the final layer. The Sigmoid function transforms the output to a range between 0 and 1, treating outputs greater than or equal to 0.5 as AMPs and those less than 0.5 as non-AMPs. The specific formulation is expressed as follows:
where
is the output of the final linear layer.
2.3. Protein Language Model Feature Extraction
Traditional manual feature extraction methods have limitations as they tend to overlook the differences between protein sequences. Protein language models have been successfully applied to various downstream protein prediction tasks. Therefore, this study utilized the UniRep protein language model, the ProtBert protein language model, and the ESM-2 protein language for feature extraction from AMP sequences. The extracted feature vectors were then fused to comprehensively obtain the feature representation of AMPs.
2.3.1. UniRep Protein Language Model
The UniRep protein language model employs LSTM neural networks as its foundational architecture. It continuously optimizes the LSTM neural network by predicting whether the next amino acid value in the sequence is the same as the true amino acid value. Ultimately, the model uses the average of the hidden layer units of multiple LSTM networks as the feature representation of the sequence. UniRep utilizes multiple GPUs and undergoes three weeks of training on approximately 24 million protein sequences from the UniRef50 database. The model can map protein sequences of different lengths to a unified length of 1900-dimensional feature vectors. UniRep effectively categorizes protein sequences with lower uniformity on the sequence into categories with higher structural similarity. In this model, UniRep was used to extract features from AMPs, mapping them to 1900-dimensional feature vectors to better represent the global features of AMPs.
2.3.2. ProtBert Protein Language Model
The ProtBert protein language model employs Transformer/BERT architecture and is trained extensively using over two billion protein sequences from the BDF protein database and UniRep protein database. The model successfully maps protein sequences of varying lengths to a unified length of 1024-dimensional feature vectors. ProtBert has demonstrated successful applications across various downstream tasks, producing favorable outcomes. It leverages the multi-head attention mechanism derived from the Transformer architecture, enabling it to highlight the local characteristics of sequences while retaining a strong representation of global features [
24,
25]. In this model, ProtBert was used to extract features from AMPs, mapping them to 1024-dimensional feature vectors. This ensures a high level of global features while highlighting the local features of AMPs.
2.3.3. ESM-2 Protein Language Model
The ESM-2 protein language model is an unsupervised protein language model that operates without the need for annotated data pertaining to protein structure and function. Instead, it relies solely on vast amounts of protein sequence data. This characteristic empowers ESM-2 to harness the extensive information present in protein databases, free from the constraints of experimental data. Another distinctive feature of ESM-2 is its ability to achieve zero-shot or few-shot predictions. In other words, it does not require additional training or fine-tuning for each specific task. Utilizing the feature representation generated by ESM-2 as input enables its direct application to various protein-related tasks. Based on the aforementioned characteristics, we believe that using the ESM-2 language model to encode AMPs can yield strong feature representations, the variable-length AMP sequences were uniformly encoded into 1024-dimensional eigenvectors.
In this study, we adopted a comprehensive approach by integrating three distinct protein language models—UniRep, ProtBert, and ESM-2—to comprehensively extract features from AMP sequences. The rationale behind combining these models lies in their unique strengths and complementary capabilities. The integration aims to fully leverage their respective advantages. UniRep focuses on capturing sequence dependencies, ProtBert simultaneously emphasizes local and global features, and ESM-2 provides insightful perspectives from an unsupervised standpoint. The amalgamation of these models seeks to overcome individual model limitations, providing a more robust and nuanced representation of AMP sequences. This integrated approach is anticipated to yield a more comprehensive feature set, better capturing the multifaceted characteristics of AMPs and ultimately enhancing the accuracy and generalization of downstream prediction tasks.
2.4. Deep Learning Network Model
2.4.1. Bi-LSTM Networks
RNN is a recurrent neural network structure that possesses memory capabilities when processing sequential data, allowing it to capture contextual information within the sequence [
26]. However, traditional RNNs have limitations. In longer text sequences, the traditional RNN structure faces issues like vanishing or exploding gradients, making it challenging to effectively capture long-term dependencies.
LSTM is designed to address the issues encountered by traditional RNNs. Introducing three gates (input gate, forget gate, and output gate) and an internal cell state, LSTM enables better control over the flow of information [
27]. Through carefully designed gate mechanisms, LSTM can selectively remember or forget information, thereby capturing long-term dependencies more effectively and mitigating the gradient-related challenges.
Traditional RNNs and LSTM networks transmit information in a unidirectional manner, lacking the ability to gather information about future states. However, protein sequences can be seen as a form of biological language, akin to sentences where peptide segments represent sentences and individual amino acid residues function as words. To accurately predict outcomes, it is essential to consider the contextual relationships among these residues [
28].
To address the limitations of LSTMs, this study employs bidirectional LSTM (Bi-LSTM) networks. By processing input from both directions, Bi-LSTM learns long-distance dependencies in peptide sequences in a bidirectional manner, capturing information from both the front and back ends. This enhances the neural network’s expressive power and enables it to better understand the complex relationships within protein sequences [
29,
30]. With this architectural design, the output layer incorporates both historical and prospective information.
2.4.2. Convolutional Neural Networks
Next, 1D-CNN is a variant of convolutional neural networks specifically designed to process one-dimensional sequential data [
31], such as time series data or text sequences in natural language processing. Unlike traditional two-dimensional CNNs used in image processing, 1D-CNN primarily focuses on feature extraction along a single direction, making it suitable for data with sequential structures. It has demonstrated excellent performance in various applications, including processing time-series data, analyzing speech signals, and handling text sequences in NLP.
In the field of bioinformatics, 1D-CNN finds extensive applications, especially in tasks involving the processing of protein sequences [
32]. This network architecture proves effective in capturing local features and patterns within sequential data, yielding favorable results across diverse tasks. In this study, we utilize a 1D-CNN to extract protein information from the hidden layers of a Bi-LSTM network, obtaining higher-dimensional protein feature vectors to better adapt to our model.
2.4.3. Attention Mechanisms
Attention mechanisms are a prevalent technique in deep learning that emulates the selective focus observed in the human visual system or cognitive processes [
33,
34]. Attention mechanisms have found widespread applications in the fields of natural language processing and computer vision. The fundamental idea behind attention mechanisms is to assign different attention weights to different parts of information when processing sequential data, emphasizing more on crucial information, which allows models to concentrate on the most relevant information for a given task. This capability proves beneficial for enhancing the handling of long sequences or complex data, ultimately improving model performance. Furthermore, attention mechanisms are frequently employed in bioinformatics in conjunction with recurrent neural networks, showcasing competitive performance across a broad spectrum of biological sequence analysis problems [
35,
36]. In this research, attention mechanisms are utilized to identify crucial information influencing AMP prediction by feeding the high-dimensional protein feature vectors through a deep learning network structure, and an attention mechanism is applied to assign attention weights to these vectors.
For effective training, this study employs a dynamic learning rate algorithm, ReduceLROnPlateau. This algorithm is one of the learning rate schedulers available in PyTorch, designed to dynamically adjust the learning rate during training based on performance metrics from the validation set. The primary objective of this scheduler is to reduce the learning rate when the model’s performance on the validation set ceases to improve, thereby facilitating more effective convergence. The pseudocode for this scheduler is presented in Algorithm 1. Specifically, when the accuracy (
ACC) on the test set remains unchanged for two consecutive epochs, the learning rate is adjusted to 70% of its original value. The utilization of such a scheduler aims to enhance the model’s convergence capabilities and adaptability to varying complexities in the training process.
| Algorithm 1. ReduceLROnPlateau algorithm. |
| ReduceLROnPlateau: dynamic learning rate algorithm |
| | input: ACC values for each test set |
| | Output: Updated learning rate |
| 1 | L Current learning rate |
| 2 | if the ACC value does not change do |
| 3 | L L*0.7 |
| 4 | else |
| 5 | L L |
| 6 | return L |
In order to accomplish the classification task, the proposed model in this study employs a dynamic learning rate algorithm: in this study, a binary cross-entropy loss function was employed during the model training process, and the computation of the loss is as follows.
where
represents the model’s output, and
denotes the true label.
The model is evaluated using ten-fold cross-validation, and parameter settings, based on ESM-2, ProtBert, and UniRep in the train dataset, including a batch size of 64, a learning rate of 0.001, and an Adam [
37] optimizer for model optimization. The training epoch is set to 20.
2.5. Model Performance Evaluation
By evaluating the performance of the model, one can choose the most suitable combination of parameters among numerous possibilities for the prediction task, thereby effectively predicting AMPs. In this study, five metrics are employed to assess the proposed method, and their calculation formulas are presented in Equation (9):
where
TP,
FP, and
FN represent true positives, false positives, true negatives, and false negatives, respectively.
ACC (accuracy) denotes the model’s accuracy;
MCC (Matthew’s correlation coefficient) represents the Pearson correlation coefficient;
Sn (sensitivity) indicates the model’s sensitivity; and
Sp (specificity) represents the model’s specificity. These four metrics are commonly used for statistical predictions of model performance.
When ACC = 1, it indicates that all AMP predictions are correct, and when ACC = 0, it implies that all AMP predictions are incorrect. Sensitivity (Sn) and specificity (Sp), respectively, represent the model’s ability to predict AMPs and non-AMPs. The closer MCC is to 1, the model is considered to be more perfect; the closer it is to 0, the closer the model’s performance is to a random classifier; and the closer it is to -1, the more opposite the model’s predictions are to reality.
AUC (Area Under the Curve) is a widely used metric for evaluating the performance of machine learning models in binary classification problems. It assesses the quality of a model’s predictions by measuring the area under the Receiver Operating Characteristic (ROC) curve. The ROC curve represents the trade-off between the true positive rate (sensitivity or recall) and the false positive rate, with the true positive rate plotted on the y-axis and the false positive rate on the x-axis. The AUC is calculated as the area under this curve. One key advantage of AUC is its robustness in handling class imbalance, meaning that it remains reliable even when there is a substantial difference in the number of positive and negative instances in the dataset. This makes AUC a valuable performance evaluation metric in practical applications. It allows for the comparison of different models and facilitates the selection of the model with superior performance.
3. Results and Discussion
To validate the effectiveness of the structures we employed, we conducted ablation experiments aimed at demonstrating the correctness of the selected architecture. Furthermore, in order to better align the model with our dataset, we performed parameter tuning optimization. These endeavors were undertaken to ensure the model’s adherence to our data and to affirm the suitability of the chosen structures and parameter configurations for our task. Through these systematic steps, we enhance our confidence in the selected architecture and parameter settings, thereby improving the model’s adaptability to our specific requirements.
3.1. Selecting Model Architecture of UniproLcad
Based on the training set, using ten-fold cross-validation, we evaluated the performance of protein language models including ESM-2, ProtBert, and UniRep features individually, as well as their combinations: ESM-2 features, ProtBert features, UniRep features, ESM-2 features combined with ProtBert features, ESM-2 features combined with UniRep features, ProtBert features combined with UniRep features, and ESM-2 features combined with ProtBert and UniRep features, using the Bi-LSTM architecture and 1D-CNN architecture. This comprehensive assessment allowed us to analyze the predictive capabilities of these protein language models. The results are depicted in
Table 1.
For individual protein language models, the feature representation of ESM-2 exhibited the highest performance, surpassing ProtBert and UniRep by 1.4% and 1.9%, respectively. In the case of two protein language models, the combination of ESM-2 and ProtBert features outperformed ESM-2 + UniRep and ProtBert + UniRep by 0.6% and 0.4%, respectively. Ultimately, the feature representation of ESM-2 + ProtBert + UniRep achieved the highest overall performance, surpassing ESM-2 and ESM-2 + ProtBert by 2.1% and 1.9%. This highlights the superiority of combining multiple language models in capturing diverse aspects of protein sequences.
3.2. Ablation Experiment
In order to ensure the effectiveness of the selected network model, we conducted ablation experiments on three network modules based on the training set, namely removing Bi-LSTM, 1D-CNN, and the attention mechanism. The experimental results are depicted in
Table 2.
The removal of Bi-LSTM resulted in a decrease in model accuracy by 2.8%, the removal of 1D-CNN led to a decrease of 1.9% in accuracy, and the removal of the attention mechanism caused a reduction of 1.2% in model accuracy.
3.3. Model Parameter Selection
To explore the optimal parameter combination for the predictor, we selected parameters on the training set based on the average results of the model’s ten-fold cross-validation. Initially, considering the impact of the convolutional kernel size on model performance, we conducted experiments with different convolutional kernel sizes.
Table 3 shows a performance comparison of various 1D-CNN parameters on the training set. When the convolutional kernel is set to 2000, the
ACC value is maximized, surpassing 1900 and 2100 by 2.2% and 1.9%, respectively. Additionally, the
MCC value increased by 0.32% and 0.27%, compared to 1900 and 2100.
For the LSTM model, we compared the model performance under different LSTM structures on the training set using ten-fold cross-validation, including 1-layer unidirectional and bidirectional LSTM, 2-layer unidirectional and bidirectional LSTM, and 3-layer unidirectional and bidirectional LSTM. The results are presented in
Table 4.
As indicated in
Table 4, a one-layer bidirectional LSTM model structure surpasses its unidirectional counterpart. Our analysis reveals that the unidirectional LSTM state transmission, occurring only from front to back, imposes a directional constraint on information propagation. In contrast, bidirectional LSTM can assimilate information in both forward and backward directions, thereby amplifying the neural network’s expressive capacity. Concerning the layers of the Bi-LSTM model, the two-layer configuration attains superior performance. In comparison to the three-layer model, it achieves elevated values in
ACC,
MCC,
Sn, and
Sp, with improvements of 0.8%, 0.7%, 0.6%, and 0.7%, respectively.
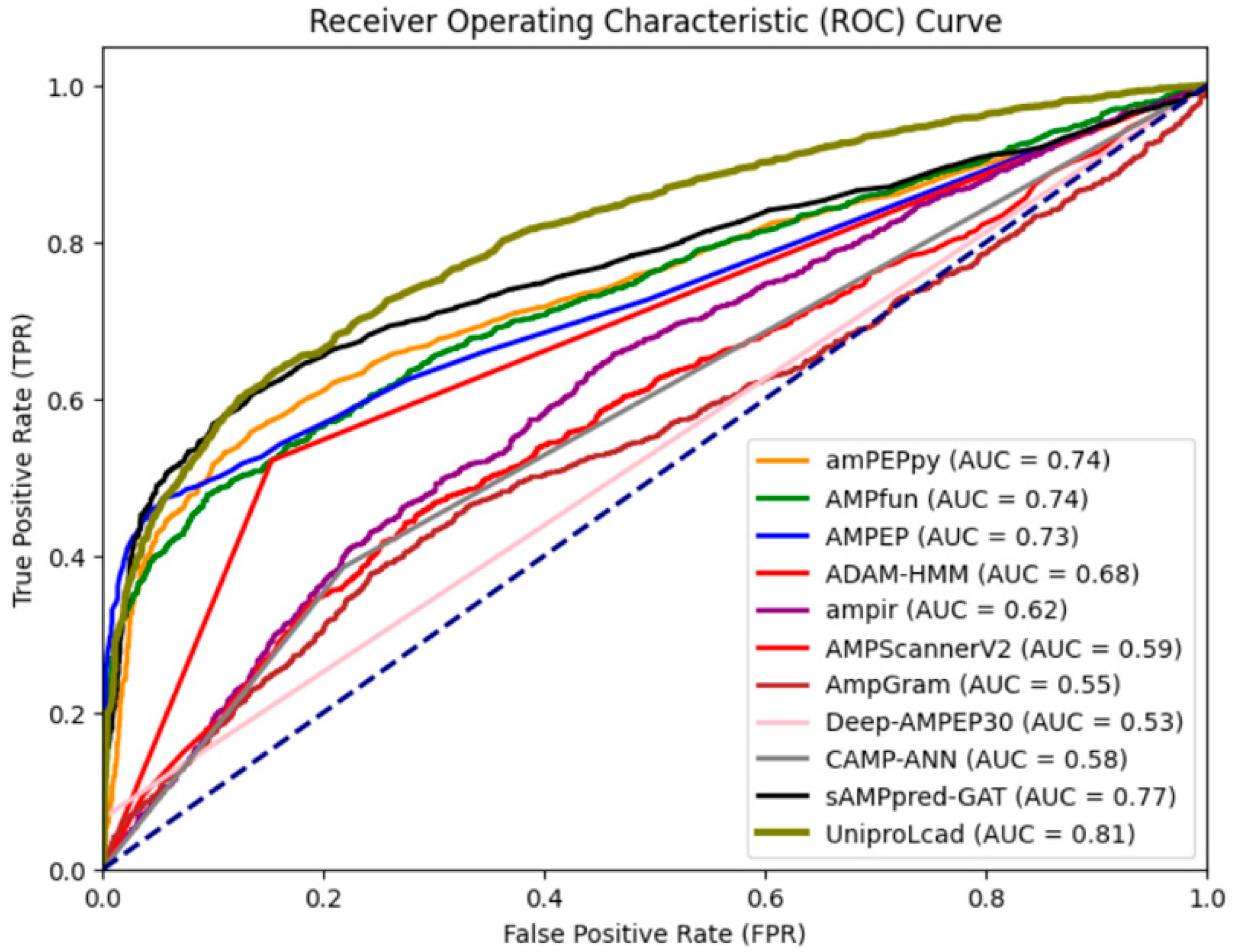
In order to ensure the advancement of our model, we compare the proposed method with nine state-of-the-art approaches on the independent XUAMP test set, and the AUC values for each model were computed on the test set. The evaluated methods include amPEPpy [
38], AMPfun [
7], AMPEP [
39], ADAM-HMM [
40], AMPIR [
41], AMPScannerV2 [
42], AMPGram [
43], Deep-AMPEP30 [
44], CAMP-ANN [
4], and sAMPpred-GAT [
9]. The results are detailed in
Table 5, and the AUC values are visually represented in
Figure 3.
From
Table 5, it can be noted that the model proposed in this study achieves the best performance in terms of
ACC,
MCC, and
Sn. On the other hand, Deep-AMPEP30 attains the highest
Sp but with a very small
Sn, indicating that the model is heavily biased towards predicting positive samples. This suggests that the model has a weak generalization ability.
We also presented the AUC scores of all the mentioned models based on the XU independent test set in
Figure 3. From
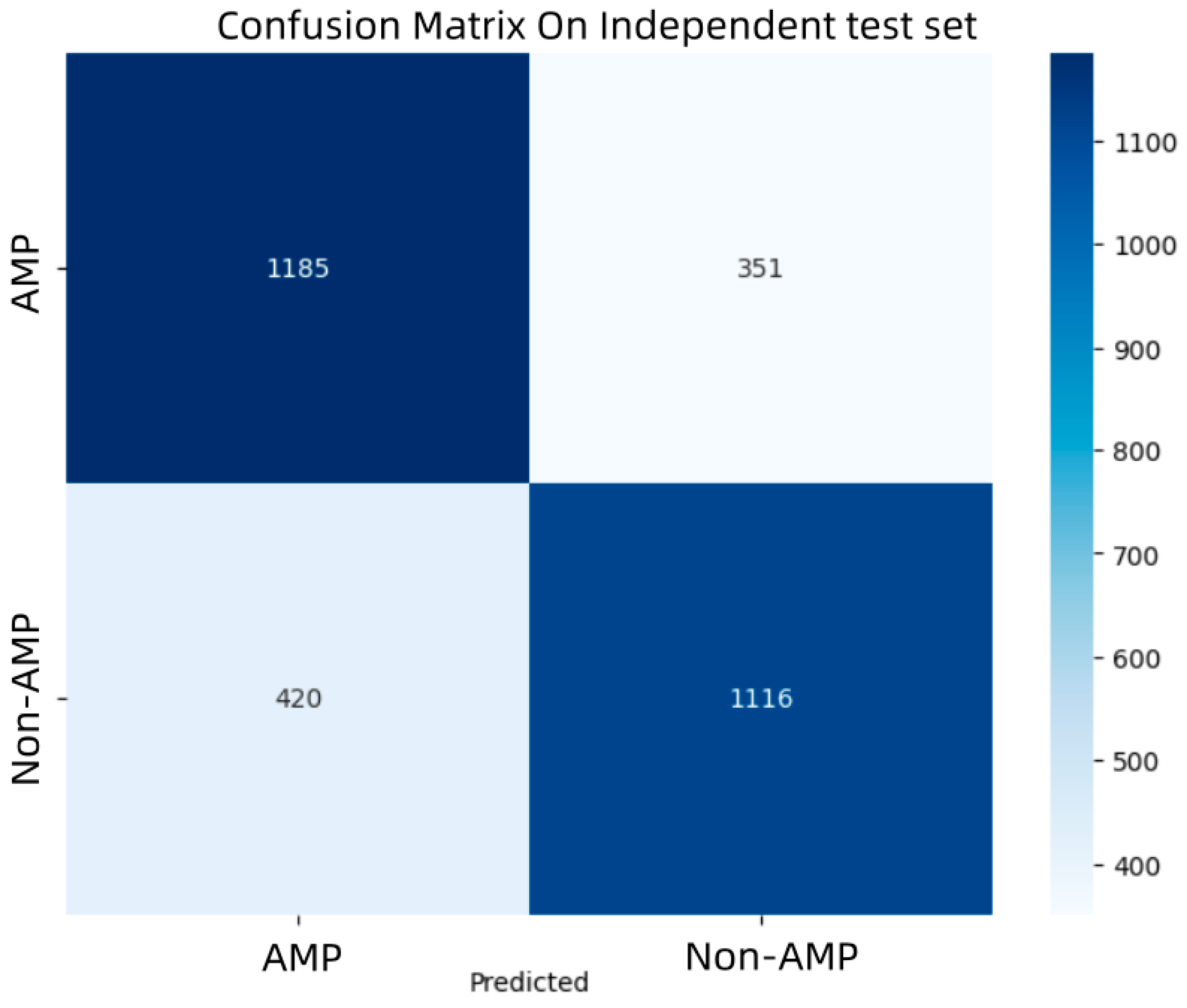
Figure 3, it can be seen that the model proposed in this study achieves the highest AUC, indicating that the model possesses the best predictive capability and has a relatively low false positive rate. And confusion matrix of the model is shown in
Figure 4.
To further evaluate the performance of our proposed model, we compared it with the latest method iAMP-Attenpred [
10] using ten-fold cross-validation on the training sets Xingdataset1 and Xingdataset2. The experimental results are presented in
Table 6 and
Table 7, respectively.
Table 6 and
Table 7 reveal that our proposed method UniproLcad and iAMP-Attenpred have achieved comparable performance results. Specifically, on the Xingdataset1 dataset, UniproLcad slightly outperforms iAMP-Attenpred in terms of
ACC and
Sp, while its performance is marginally lower than UniproLcad in
MCC and
Sn. On the Xingdataset2 dataset, UniproLcad is superior to iAMP-Attenpred across all metrics. The similar performance results achieved by the two methods may be attributed to their use of similar network architectures and protein language models.









{kind=link}
{kind=link}
{kind=link}
{kind=link}