
Cyclodextrins in Asymmetric and Stereospecific Synthesis

Abstract
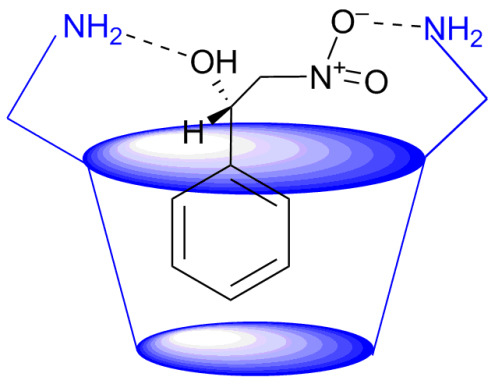
:
1. Introduction
- (1)
- to significantly increase the rate and selectivity of reactions catalyzed by water-soluble organometallic complexes;
- (2)
- to design new water-soluble ligands for aqueous organometallic catalysis;
- (3)
- to stabilize catalytically-active noble metal nanoparticles in water;
- (4)
- to facilitate reactions catalyzed by supported metals or metallic powder in water.
- 1)
- cyclodextrins’ complexes with transition metals as asymmetric and stereospecific catalysts;
- 2)
- cyclodextrins’ non-metallic derivatives as asymmetric and stereospecific catalysts;
- 3)
- cyclodextrins promoting asymmetric and stereospecific catalysis in water.
2. Results and Discussion
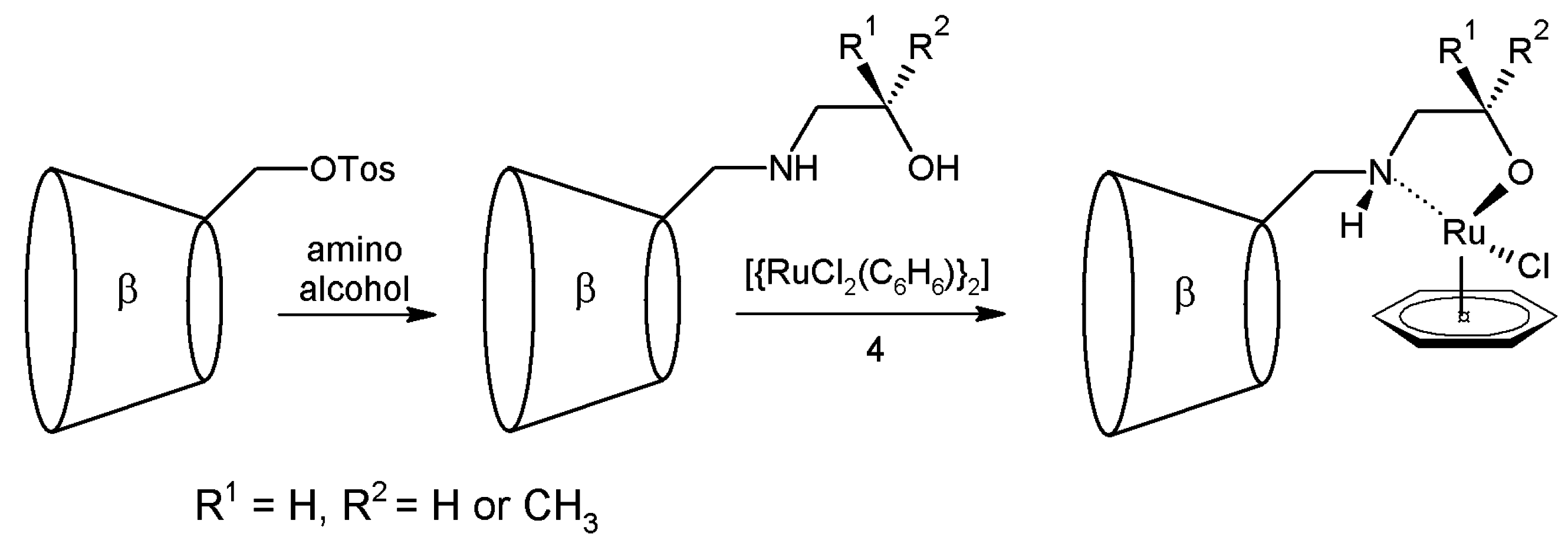
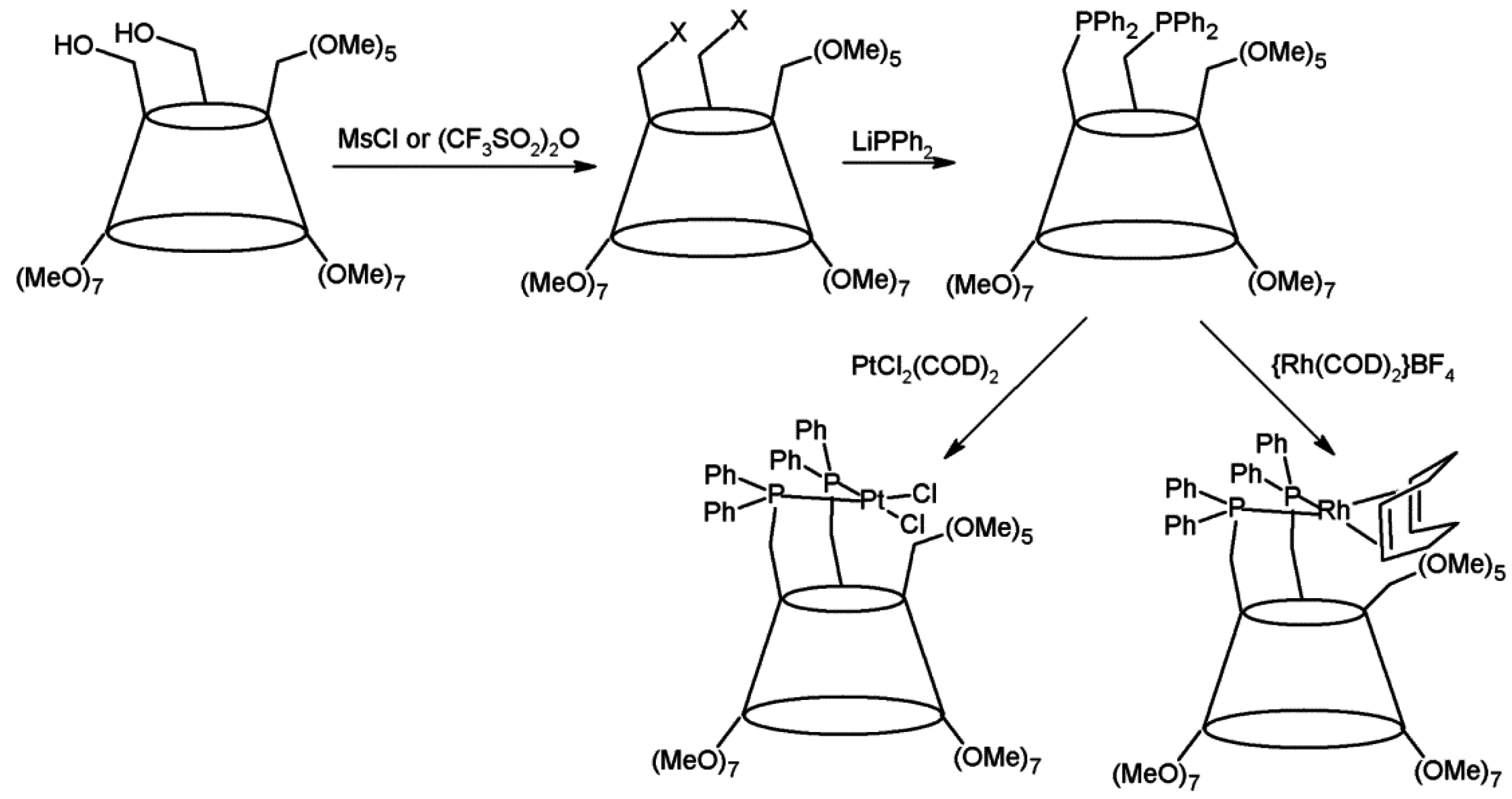
2.1. Cyclodextrins’ Complexes with Transition Metals as Asymmetric and Stereospecific Catalysts
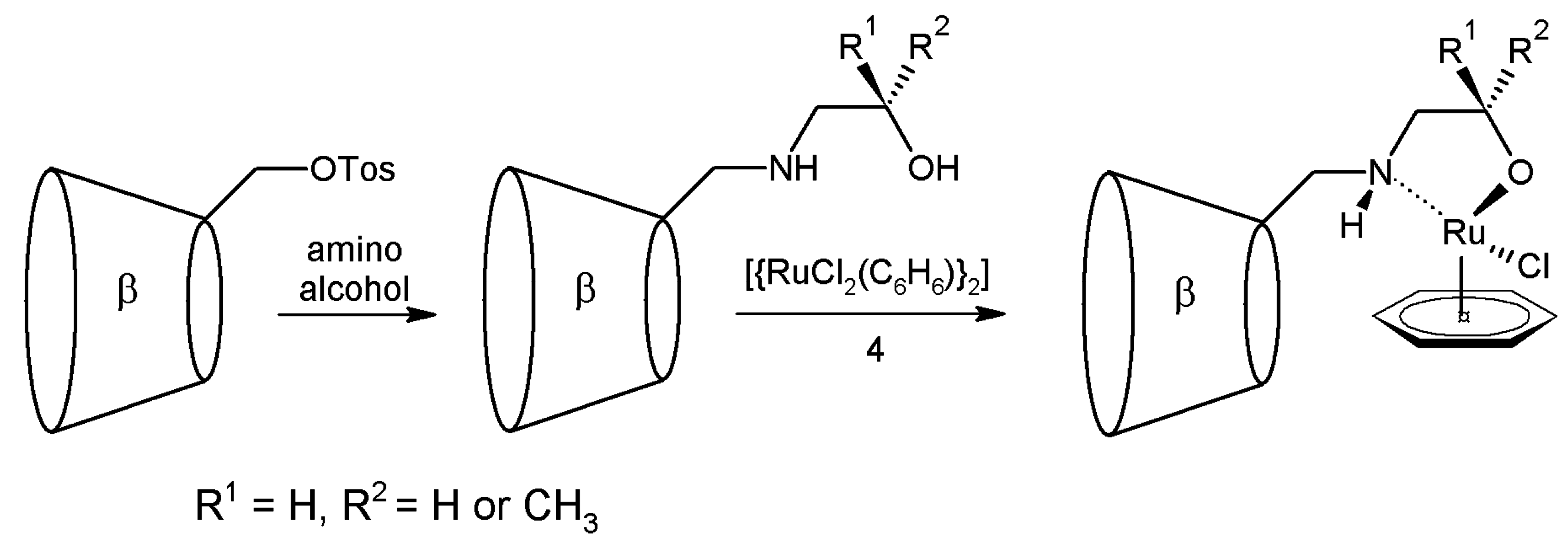




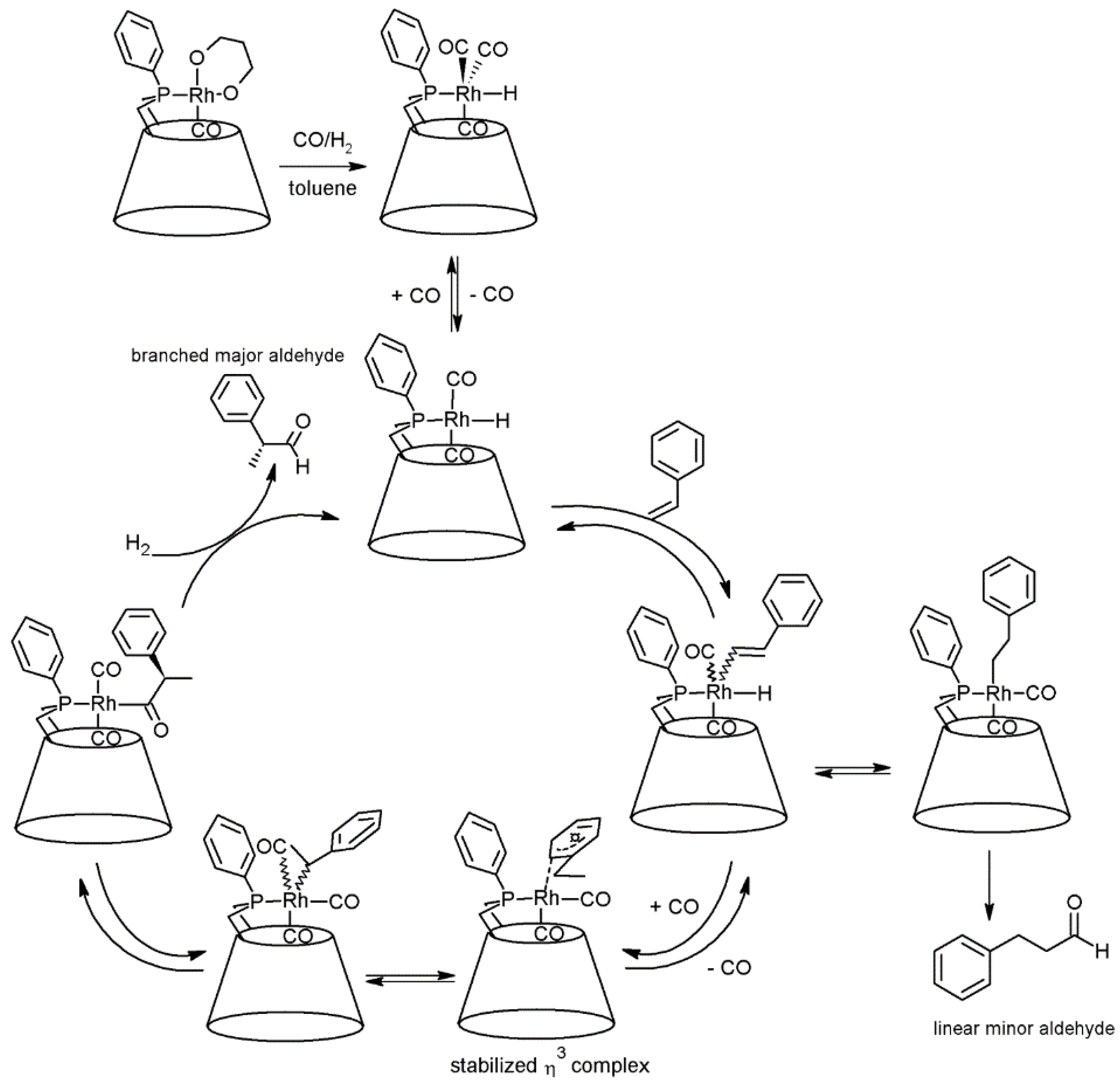
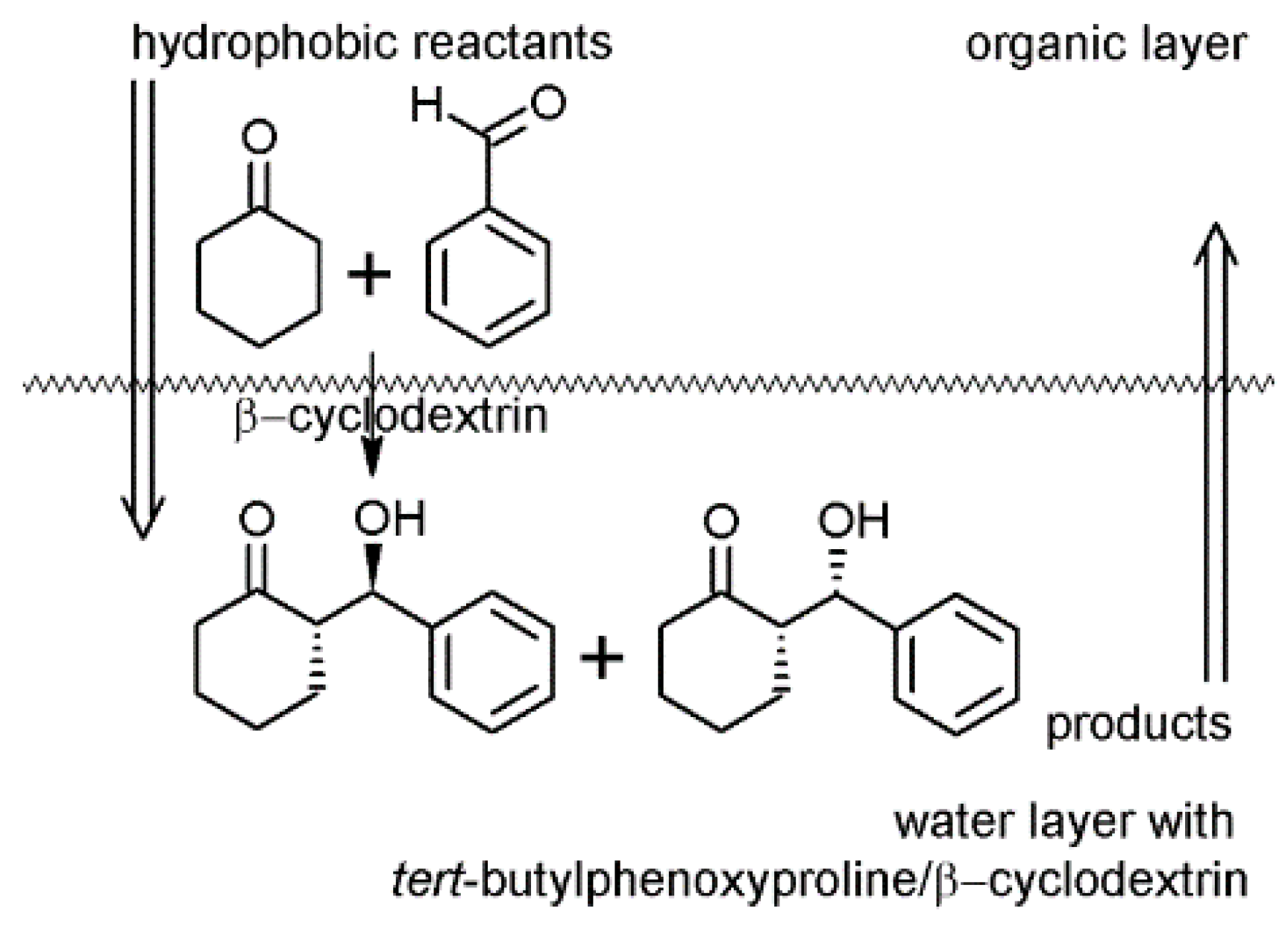
2.2. Cyclodextrins’ Non-Metallic Derivatives as Asymmetric and Stereospecific Catalysts




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
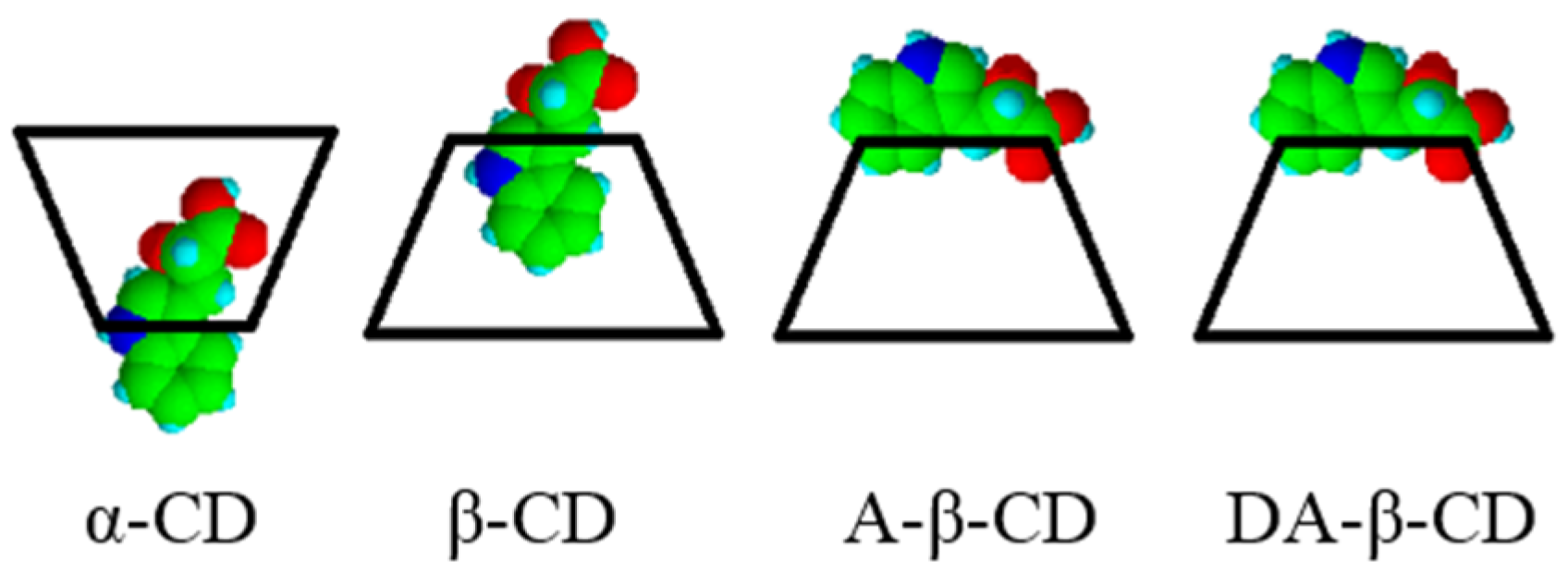
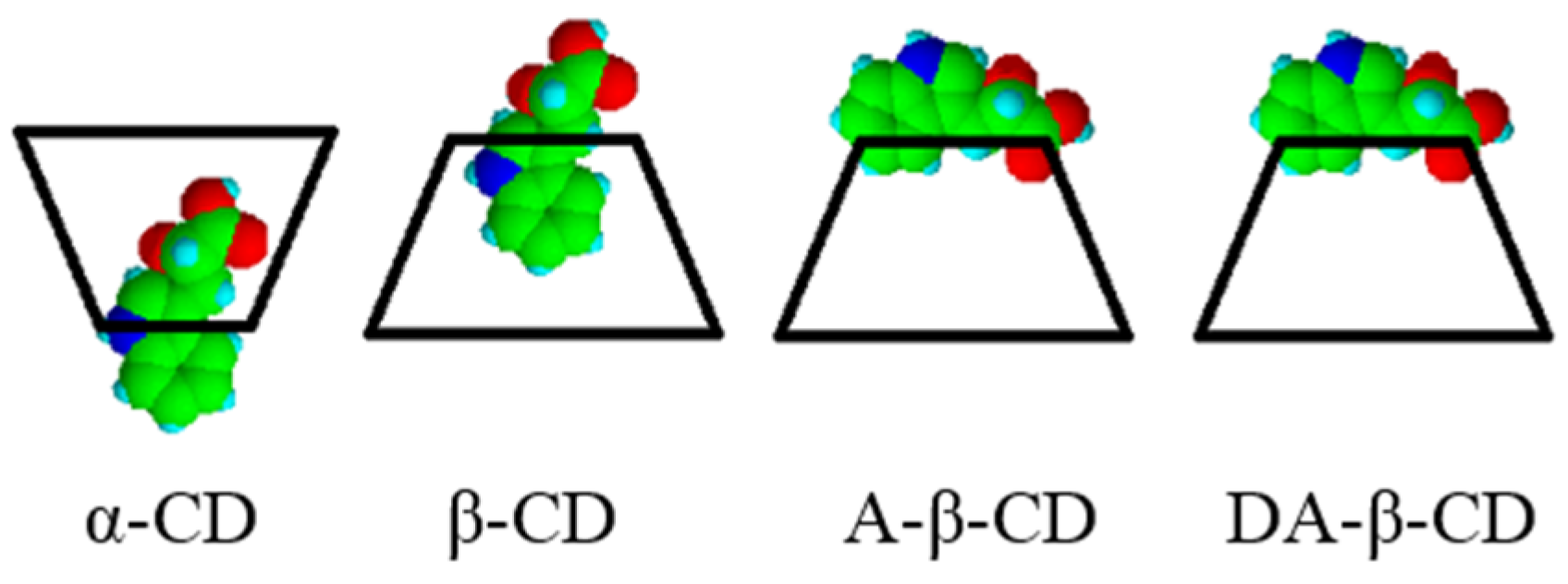
| Substrate | Cyclodextrin | Optical yield%/ee | Configuration |
|---|---|---|---|
| IPA | none | 3 | S |
| α-CD | 1 | S | |
| β-CD | 6 | S | |
| γ-CD | 0 | – | |
| A-β-CD | 17 | R | |
| DA-β-CD | 33 | R |
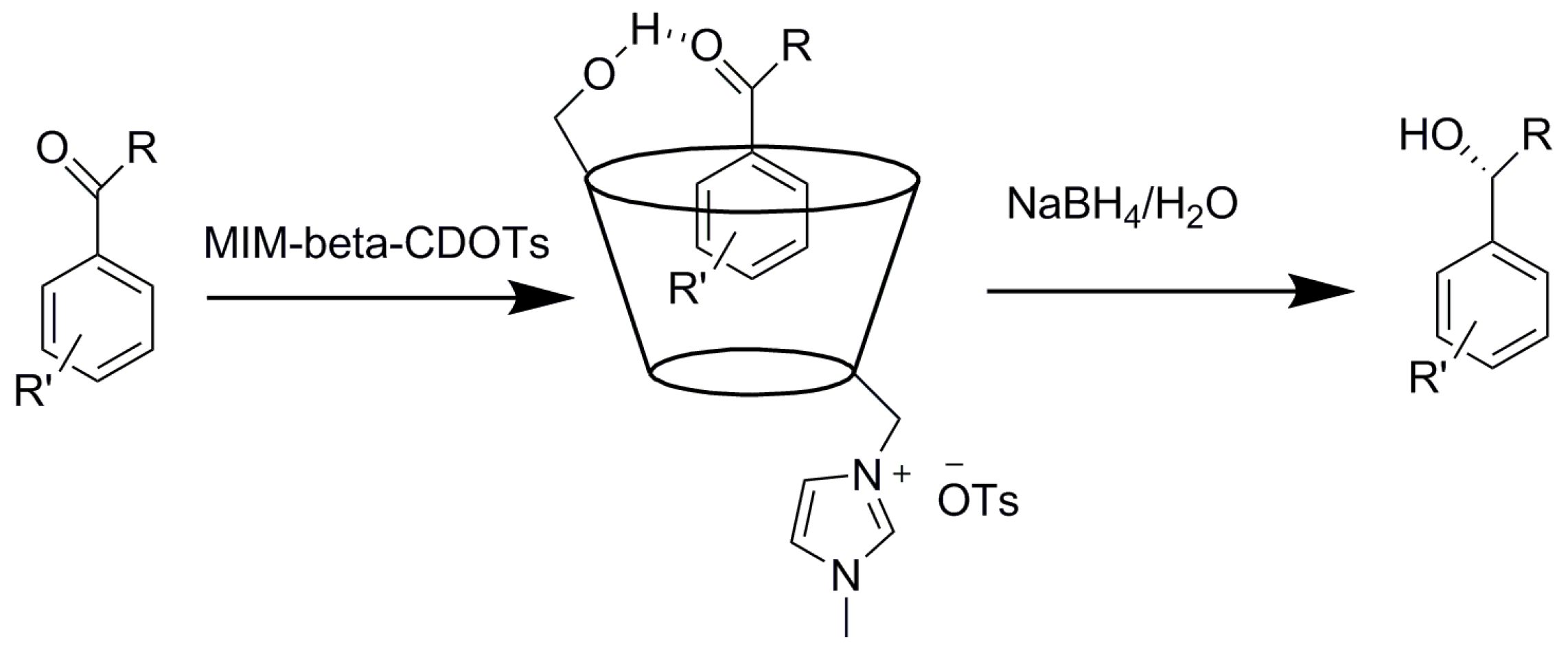

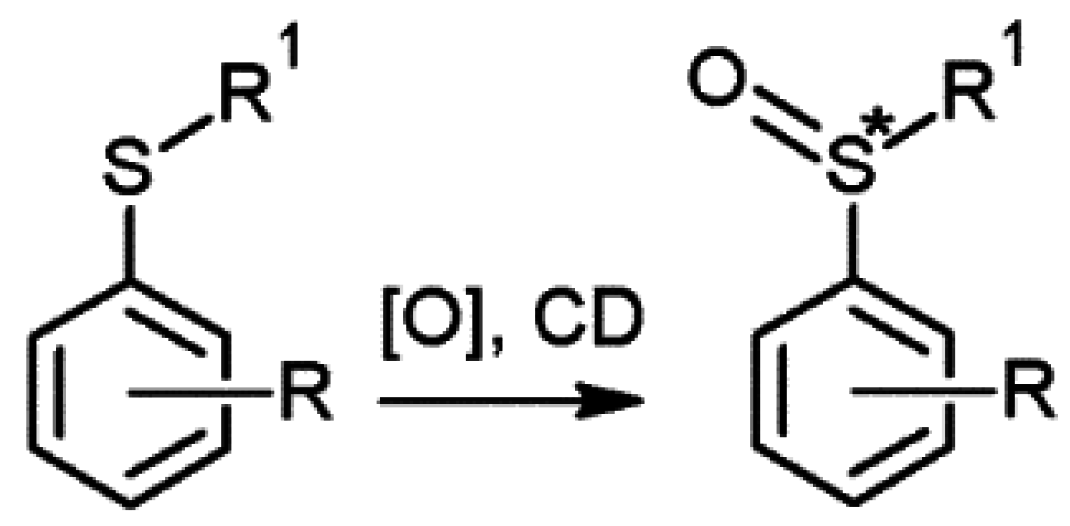


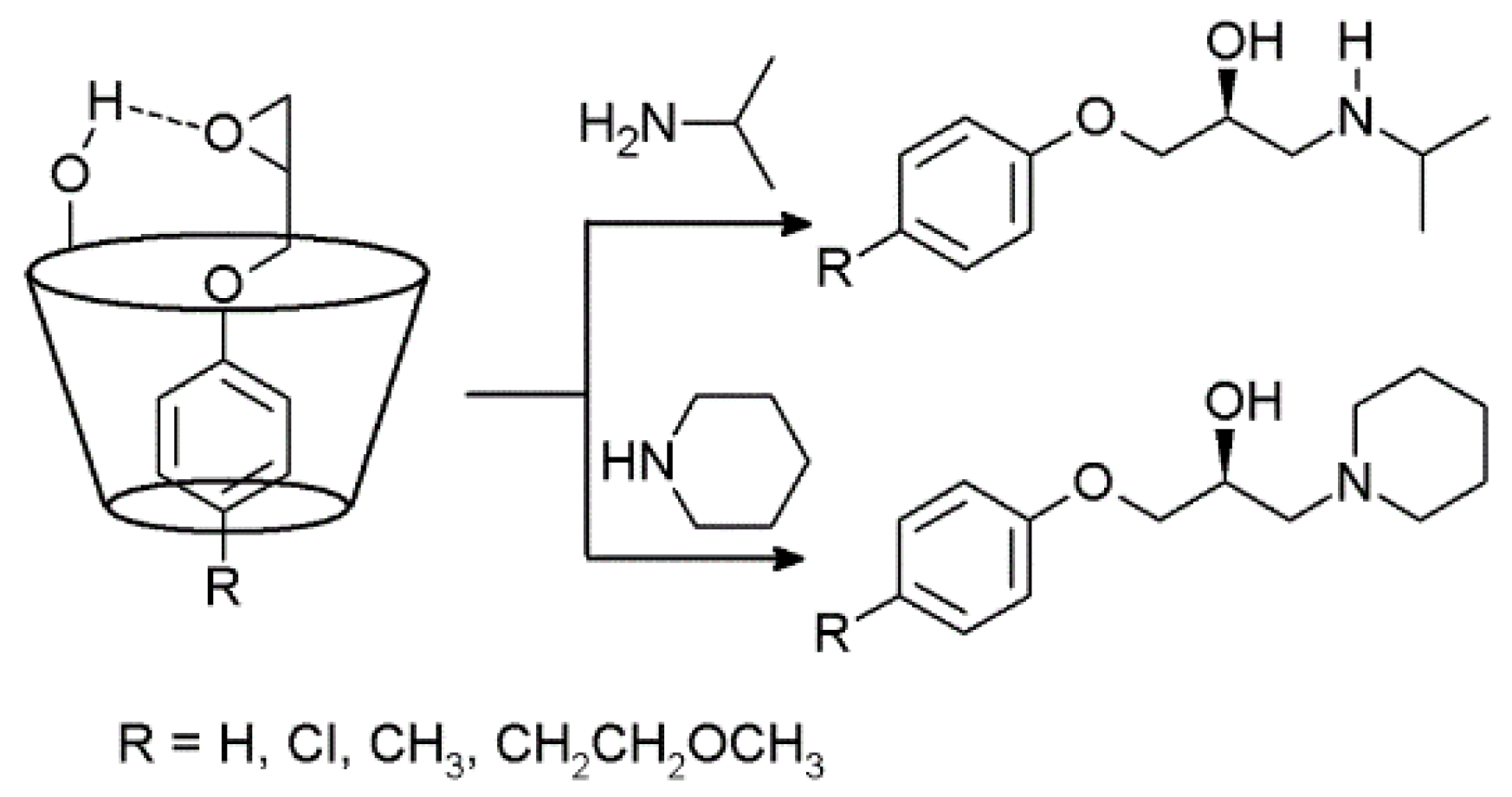


2.3. Cyclodextrins Promoting Asymmetric and Stereospecific Catalysis in Water

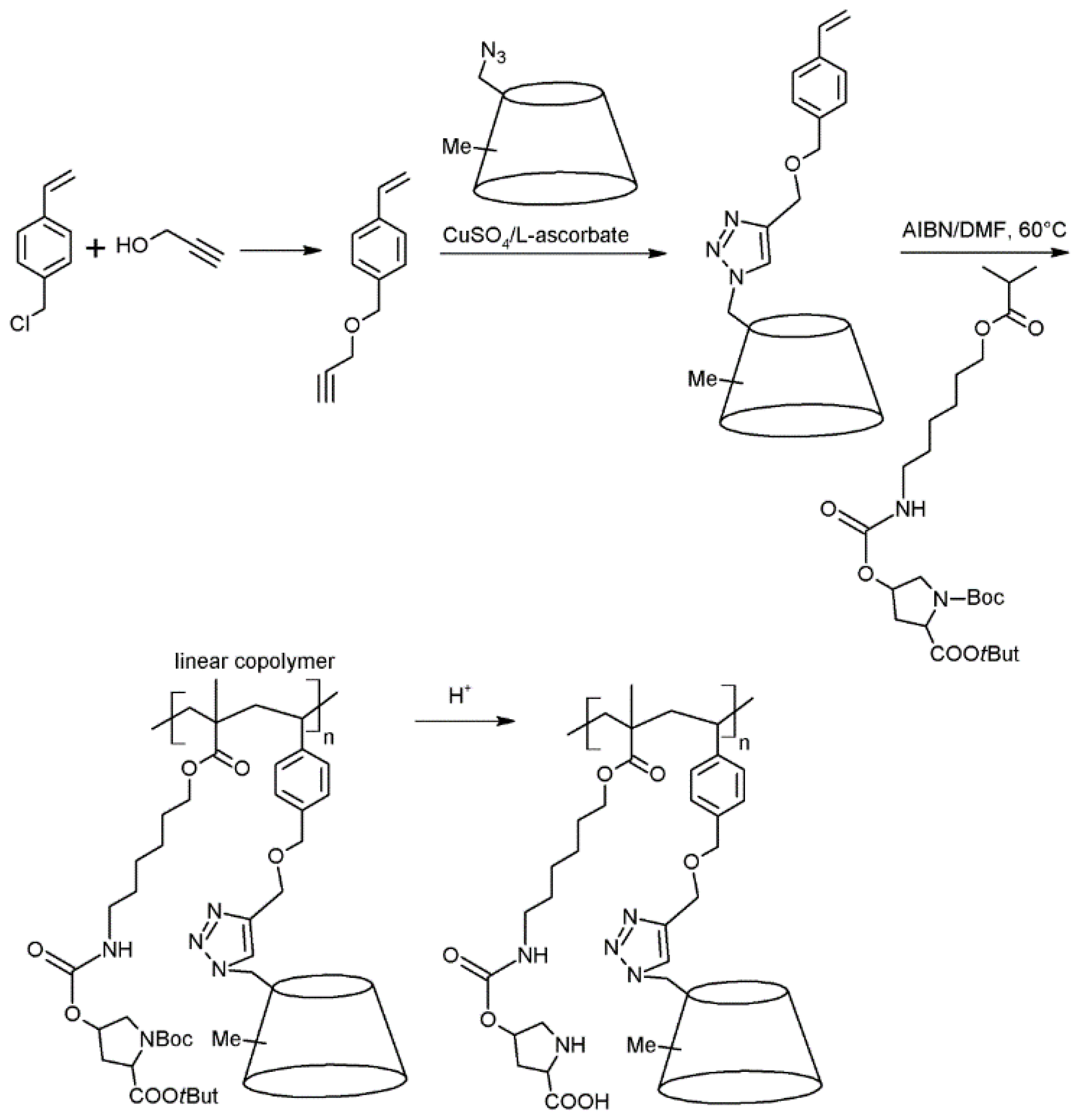
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hapiot, F.; Ponchel, A.; Tilloy, S.; Monflier, E. Cyclodextrins and their applications in aqueous-phase metal-catalyzed reactions. C. R. Chim. 2011, 14, 149–166. [Google Scholar] [CrossRef]
- Takahashi, K. Organic reactions mediated by cyclodextrins. Chem. Rev. 1998, 98, 2013–2034. [Google Scholar] [CrossRef] [PubMed]
- Breslow, R.; Dong, S.D. Biomimetic reactions catalyzed by cyclodextrins and their derivatives. Chem. Rev. 1998, 98, 1997–2012. [Google Scholar] [CrossRef] [PubMed]
- Noël, S.; Léger, B.; Ponchel, A.; Philippot, K.; Denicourt-Nowicki, A.; Roucoux, A.; Monflier, E. Cyclodextrin-based systems for the stabilization of metallic (0) nanoparticles and their versatile applications in catalysis. Catal. Today 2014, 235, 20–32. [Google Scholar] [CrossRef]
- Hapiot, F.; Bricout, H.; Menuel, S.; Tilloy, S.; Monflier, E. Recent breakthroughs in aqueous cyclodextrin-assisted supramolecular catalysis. Catal. Sci. Technol. 2014, 4, 1899–1908. [Google Scholar] [CrossRef]
- Fillet, M.; Hubert, P.; Crommen, J. Enantiomeric separations of drugs using mixtures of charged and neutral cyclodextrins. J. Chromatogr. A. 2000, 875, 123–134. [Google Scholar] [CrossRef]
- Bicchi, C.; D’Amato, A.; Rubiolo, P. Cyclodextrin derivatives as chiral selectors for direct gas chromatographic separation of enantiomers in the essential oil, aroma and flavour fields. J. Chromatogr. A 1999, 843, 99–121. [Google Scholar] [CrossRef]
- Xiao, Y.; Ng, S.C.; Tan, T.T.Y.; Wang, Y. Recent development of cyclodextrin chiral stationary phases and their applications in chromatography. J. Chromatogr. A. 2012, 1269, 52–68. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Ng, S.C.; Sun, D. Modified Cyclodextrins for Chiral Separation; Springer: New York, NY, USA, 2013. [Google Scholar]
- Schlatter, A.; Kundu, M.K.; Woggon, W.D. Enantioselective reduction of aromatic and aliphatic ketones catalysed by ruthenium complexes attached to β-cyclodextrin. Angew. Chem. 2004, 116, 6899–6902. [Google Scholar] [CrossRef]
- Shen, H.M.; Ji, H.B. Cyclodextrin–[RuCl2(Arene)]2 conjugates: Another way to enhance the enantioselectivity of aromatic ketones reduction by aromatic ligands’ volume. Tetrahedron 2013, 69, 8360–8367. [Google Scholar] [CrossRef]
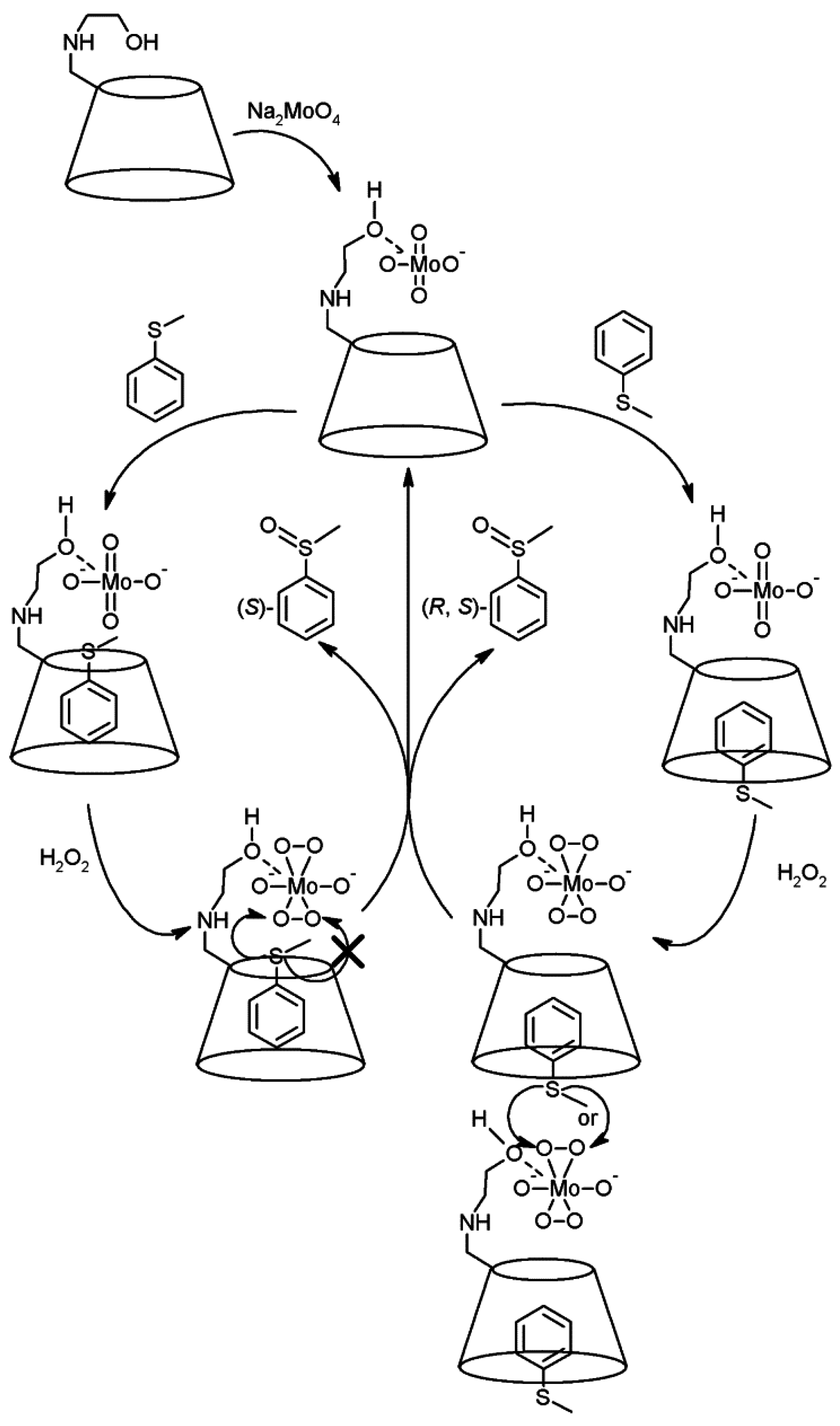
- Sakuraba, H.; Maekawa, H. Enantioselective oxidation of sulfides catalyzed by chiral Mo V and Cu II complexes of catechol-appended β-cyclodextrin derivatives in water. J. Incl. Phenom. Macrocycl. Chem. 2006, 54, 41–45. [Google Scholar] [CrossRef]
- Wong, Y.T.; Yang, C.; Ying, K.C.; Jia, G. Synthesis of a novel β-cyclodextrin-functionalized diphosphine ligand and its catalytic properties for asymmetric hydrogenation. Organometallics 2002, 21, 1782–1787. [Google Scholar] [CrossRef]
- Jouffroy, M.; Sémeril, D.; Armspach, D.; Matt, D. Phosphane-phosphite chelators built on a α-cyclodextrin scaffold: Application in Rh-catalysed asymmetric hydrogenation and hydroformylation. Eur. J. Org. Chem. 2013, 2013, 6069–6077. [Google Scholar] [CrossRef]
- Yang, J.; Gabriele, B.; Belvedere, S.; Huang, Y.; Breslow, R. Catalytic oxidations of steroid substrates by artificial cytochrome P-450 enzymes. J. Org. Chem. 2002, 67, 5057–5067. [Google Scholar] [CrossRef] [PubMed]
- Engeldinger, É.; Armspach, D.; Matt, D.; Toupet, L.; Wesolek, M. Synthesis of large chelate rings with diphosphites built on a cyclodextrin scaffold. Unexpected formation of 1, 2-phenylene-capped α-cyclodextrins. C. R. Chim. 2002, 5, 359–372. [Google Scholar]
- Cassez, A.; Kania, N.; Hapiot, F.; Fourmentin, S.; Monflier, E.; Ponchel, A. Chemically modified cyclodextrins adsorbed on Pd/C particles: New opportunities to generate highly chemo- and stereoselective catalysts for Heck reaction. Catal. Commun. 2008, 9, 1346–1351. [Google Scholar] [CrossRef]
- Senra, J.D.; Malta, L.F.B.; Souza, A.L.F.; Aguiar, L.; Antunes, O.A.C. Palladium on calcium carbonate combined to 2-hydroxypropyl-α/β-cyclodextrins: A selective catalytic system for aqueous heck coupling and hydroarylation. Adv. Synth. Catal. 2008, 350, 2551–2558. [Google Scholar] [CrossRef]
- Shen, H.M.; Ji, H.B. Biomimetic asymmetric aldol reactions catalyzed by proline derivatives attached to β-cyclodextrin in water. Tetrahedron Lett. 2012, 53, 3541–3545. [Google Scholar] [CrossRef]
- Jouffroy, M.; Gramage-Doria, R.; Sémeril, D.; Armspach, D.; Matt, D.; Oberhauser, W.; Toupet, L. Phosphinocyclodextrins as confining units for catalytic metal centres. Applications to carbon-carbon bond forming reactions. Beilstein J. Org. Chem. 2014, 10, 2388–2405. [Google Scholar] [CrossRef] [PubMed]
- Jouffroy, M.; Gramage-Doria, R.; Armspach, D.; Sémeril, D.; Oberhauser, W.; Matt, D.; Toupet, L. Confining phosphanes derived from cyclodextrins for efficient regio- and enantioselective hydroformylation. Angew. Chem. Int. Ed. 2014, 53, 3937–3940. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Sakuraba, H.; Nakanishi, H. Asymmetric halogenation and hydrohalogenation of olefins in crystalline cyclodextrin complexes. J. Chem. Soc. Chem. Commun. 1983, 17, 947–948. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sakuraba, H.; Oka, Y.; Nakanishi, H. Asymmetric halogenation and hydrohalogenation of ethyl trans-cinnamate in crystalline cyclodextrin complexes. J. Incl. Phenom. 1984, 2, 841–850. [Google Scholar] [CrossRef]
- Sakuraba, H.; Nakai, T.; Tanaka, Y. Asymmetric halogenation and hydrohalogenation of trans-cinnamic acid in crystalline cyclodextrin complexes. J. Incl. Phenom. 1984, 2, 829–839. [Google Scholar] [CrossRef]
- Sakuraba, H.; Ishizaki, H.; Tanaka, Y.; Shimizu, T. Asymmetric halogenation and hydrohalogenation of styrene in crystalline cyclodextrin complexes. J. Incl. Phenom. 1987, 5, 449–458. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sakuraba, H.; Nakanishi, H. Asymmetric halogenation and hydrohalogenation of trans-2-butenoic acid in a crystalline α-cyclodextrin complex. J. Org. Chem. 1990, 55, 564–567. [Google Scholar] [CrossRef]
- Pitchumani, K.; Velusamy, P.; Sabithamala, S.; Srinivasan, C. Modification of chemical reactivity upon cyclodextrin encapsulation: Asymmetric bromination of chalcone and benzylideneacetone. Tetrahedron 1994, 50, 7903–7912. [Google Scholar] [CrossRef]
- Kanagaraj, K.; Pitchumani, K. Per-6-amino-β-cyclodextrin as a chiral base catalyst promoting one-pot asymmetric synthesis of 2-aryl-2,3-dihydro-4-quinolones. J. Org. Chem. 2012, 78, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Kanagaraj, K.; Suresh, P.; Pitchumani, K. Per-6-amino-β-cyclodextrin as a reusable promoter and chiral host for enantioselective Henry reaction. Org. Lett. 2010, 12, 4070–4073. [Google Scholar] [CrossRef] [PubMed]
- Suresh, P.; Pitchumani, K. Per-6-amino-β-cyclodextrin catalysed asymmetric Michael addition of nitromethane and thiols to chalcones in water. Tetrahedron Asymmetry 2008, 19, 2037–2044. [Google Scholar] [CrossRef]
- Baba, N.; Matsumura, Y.; Sugimoto, T. Asymmetric reduction of aryl trifluoromethyl ketones with an achiral NADH model compound in a chiral hydrophobic binding site of sodiumcholate micelle, β-cyclodextrin and bovine serum albumin. Tetrahedron Lett. 1978, 19, 4281–4284. [Google Scholar] [CrossRef]
- Fornasier, R.; Reniero, F.; Scrimin, P.; Tonellato, U. Asymmetric reductions by sodium borhydride of ketone-β-cyclodextrin complexes. J. Org. Chem. 1985, 50, 3209–3211. [Google Scholar] [CrossRef]
- Sakuraba, H.; Inomata, N.; Tanaka, Y. Asymmetric reduction of ketones with crystalline cyclodextrin complexes of amine-boranes. J. Org. Chem. 1989, 54, 3482–3484. [Google Scholar] [CrossRef]
- Kawajiri, Y.; Motohashi, N. Strong asymmetric induction without covalent bond connection. J. Chem. Soc. Chem. Commun. 1989, 1336–1337. [Google Scholar] [CrossRef]
- Hattori, K.; Takahashi, K.; Uematsu, M.; Sakai, N. Multiple interactions between host cyclodextrin and guest compound assisting asymmetrically selective reduction with NaBH4 in aqueous media. Chem. Lett. 1990, 8, 1463–1466. [Google Scholar] [CrossRef]
- Hattori, K.; Takahashi, K.; Sakai, N. Enantioface differentiating reduction of keto acid in the presence of 6-deoxy-6-amino-β-cyclodextrin with NaBH4 in aqueous media. Bull. Chem. Soc. Jpn. 1992, 65, 2690–2696. [Google Scholar] [CrossRef]
- Takahashi, K.; Yokomizo, H.; Ishiyama, K.; Kitsuta, M.; Ohashi, M. New aspects of cyclodextrin chemistry induced by outside complex formation; asymmetric reduction of indol-3-pyruvic acid with NaBH4 in aqueous solution. J. Incl. Phenom. Macrocycl. Chem. 2006, 56, 95–99. [Google Scholar] [CrossRef]
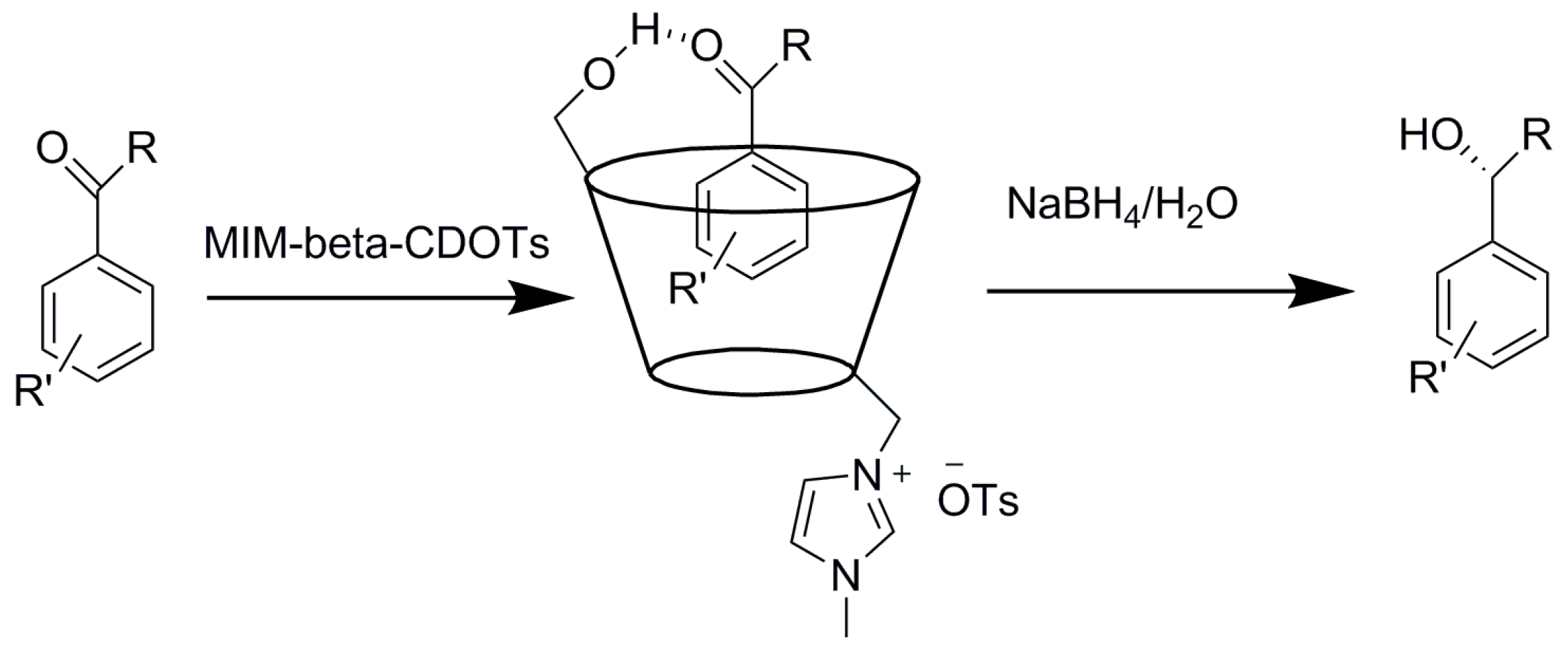
- Tang, W.; Tang, J.; Ng, S.C.; Chan, H.S.O. Asymmetric reduction of acetophenones with NaBH4 in the presence of mono-6-(1-methyl-3-imidazolium)-6-deoxy-β-cyclodextrin tosylate. J. Incl. Phenom. Macrocycl. Chem. 2006, 56, 287–290. [Google Scholar] [CrossRef]
- Wan, Y.; Wang, X.; Liu, N. Interaction of β-cyclodextrin as catalyst with acetophenone in asymmetric reaction: A theoretical survey. J. Mol. Model. 2014, 20, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.A.; Bhanumathi, N.; Rao, K.R. Asymmetric synthesis of 2-azido-1-arylethanols from azido aryl ketone-beta-cyclodextrin complexes and sodium borohydride in water. Chem. Commun. Camb. 2001, 19, 1974–1975. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, R. Stereoselective hydrogenation of (2S, 5R)-(−)-menthone in presence of β-cyclodextrin. J. Mol. Catal. A Chem. 2006, 256, 216–218. [Google Scholar] [CrossRef]
- Drabowicz, J.; Mikołajczyk, M. Asymmetric oxidation of sulfides to sulfoxides catalyzed by β-cyclodextrin. Phosphorus Sulfur Relat. Elem. 1984, 21, 245–248. [Google Scholar] [CrossRef]
- Czarnik, A.W. Cyclodextrin-mediated chiral sulfoxidations. J. Org. Chem. 1984, 49, 924–927. [Google Scholar] [CrossRef]
- Rao, K.R.; Sattur, P.B. Asymmetric synthesis of α,α-dichlorosulphoxides from substituted thioacetates and sodium hypochlorite in β-cyclodextrin complexes. J. Chem. Soc. Chem. Commun. 1989, 342–343. [Google Scholar] [CrossRef]
- Sakuraba, H.; Natori, K.; Tanaka, Y. Asymmetric oxidation of alkyl aryl sulfides in crystalline cyclodextrin complexes. J. Org. Chem. 1991, 56, 4124–4129. [Google Scholar] [CrossRef]
- Fornasier, R.; Scrimin, P.; Tecilla, P.; Tonellato, U. Asymmetric oxidation of sulfides in the presence of cyclodextrins: Effect of the precomplexation of the reactants. Phosphorus Sulfur Relat. Elem. 1988, 35, 211–213. [Google Scholar] [CrossRef]
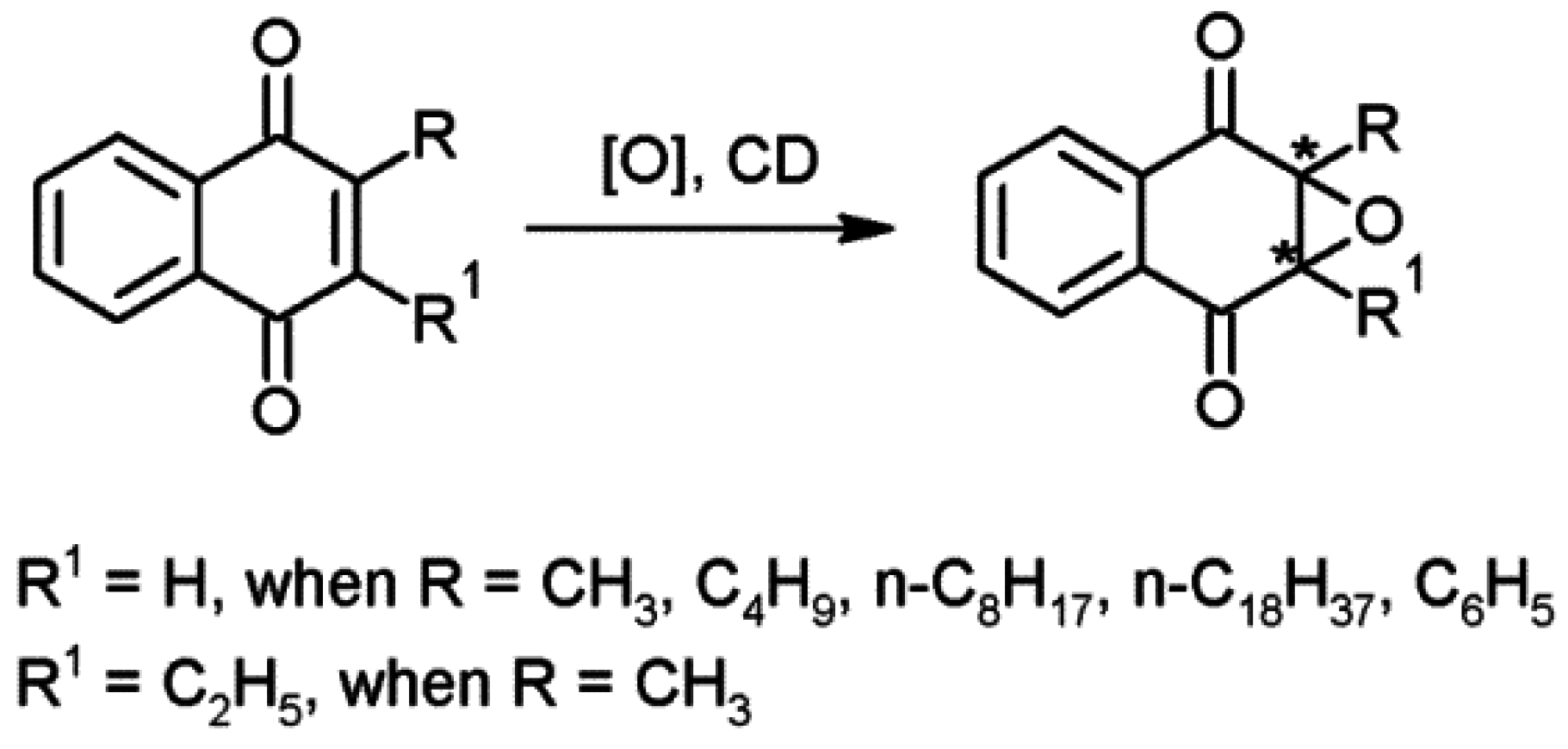
- Deary, M.E.; Davies, D.M. Evidence for cyclodextrin dioxiranes. Part 2. Catalytic and enantioselective properties of cyclodextrin dioxiranes formed from keto-derivatised hydroxypropyl-cyclodextrins. Carbohydr. Res. 1999, 317, 10–18. [Google Scholar] [CrossRef]
- Mojr, V.; Herzig, V.; Buděšínský, M.; Cibulka, R.; Kraus, T. Flavin–cyclodextrin conjugates as catalysts of enantioselective sulfoxidations with hydrogen peroxide in aqueous media. Chem. Commun. 2010, 46, 7599–7601. [Google Scholar] [CrossRef] [PubMed]
- Mojr, V.; Buděšínský, M.; Cibulka, R.; Kraus, T. Alloxazine–cyclodextrin conjugates for organocatalytic enantioselective sulfoxidations. Org. Biomol. Chem. 2011, 9, 7318–7326. [Google Scholar] [CrossRef] [PubMed]
- Hartman, T.; Herzig, V.; Buděšínský, M.; Jindřich, J.; Cibulka, R.; Kraus, T. Flavin–cyclodextrin conjugates: Effect of the structure on the catalytic activity in enantioselective sulfoxidations. Tetrahedron Asymmetry 2012, 23, 1571–1583. [Google Scholar] [CrossRef]
- Colonna, S.; Manfredi, A.; Annunziata, R.; Gaggero, N.; Casella, L. Biomimetic asymmetric synthesis. Enantioselective Weitz-Scheffer epoxidation of vitamin K3 and analogs in the presence of cyclodextrins. J. Org. Chem. 1990, 55, 5862–5866. [Google Scholar] [CrossRef]
- Chan, W.K.; Yu, W.Y.; Che, C.M.; Wong, M.K. A cyclodextrin-modified ketoester for stereoselective epoxidation of alkenes. J. Org. Chem. 2003, 68, 6576–6582. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, C.; Christensen, B.; Petersen, T.E.; Bols, M. Cyclodextrins containing an acetone bridge. Synthesis and study as epoxidation catalysts. Org. Biomol. Chem. 2004, 2, 3476–3482. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, C.; Christensen, B.; Bols, M. Artificial epoxidase II. Synthesis of cyclodextrin ketoesters and epoxidation of alkenes. Eur. J. Org. Chem. 2005, 13, 2734–2739. [Google Scholar] [CrossRef]
- Shen, Z.; Ma, J.; Liu, Y.; Jiao, C.; Li, M.; Zhang, Y. β-cyclodextrin-immobilized (4S)-phenoxy-(S)-proline as a catalyst for direct asymmetric aldol reactions. Chirality 2005, 17, 556–558. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.M.; Ji, H.B. Amino alcohol-modified β-cyclodextrin inducing biomimetic asymmetric oxidation of thioanisole in water. Carbohydr. Res. 2012, 354, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Zhang, G. Direct asymmetric aldol reactions in aqueous media catalyzed by a β-cyclodextrin–proline conjugate with a urea linker. Tetrahedron Lett. 2015, 56, 243–246. [Google Scholar] [CrossRef]
- Bonetto, L.; Fornasier, R.; Tonellato, U. Enantioselective addition of diethylzinc to aldehydes promoted by alkylated-cyclodextrin derivatives. Gazz. Chim. Ital. 1995, 125, 63–63. [Google Scholar]
- Fornasier, R.; Marcuzzi, F.; Piva, M.; Tonellato, U. Enantioselective allylation and tert-butylation of 2-cyclohexanone in aqueous media in the presence of β-cyclodextrins as chiral auxiliaries. Gazz. Chim. Ital. 1996, 126, 633–634. [Google Scholar]
- Reddy, L.R.; Bhanumathi, N.; Rama Rao, K. Dynamic kinetic asymmetric synthesis of β-aminoalcohols from racemic epoxides in cyclodextrin complexes under solid state conditions. Chem. Commun. 2000, 23, 2321–2322. [Google Scholar] [CrossRef]
- Asahara, H.; Kida, T.; Iwamoto, T.; Hinoue, T.; Akashi, M. Kinetic resolution of primary amines via enantioselective N-acylation with acyl chlorides in the presence of supramolecular cyclodextrin nanocapsules. Tetrahedron 2014, 70, 197–203. [Google Scholar] [CrossRef]
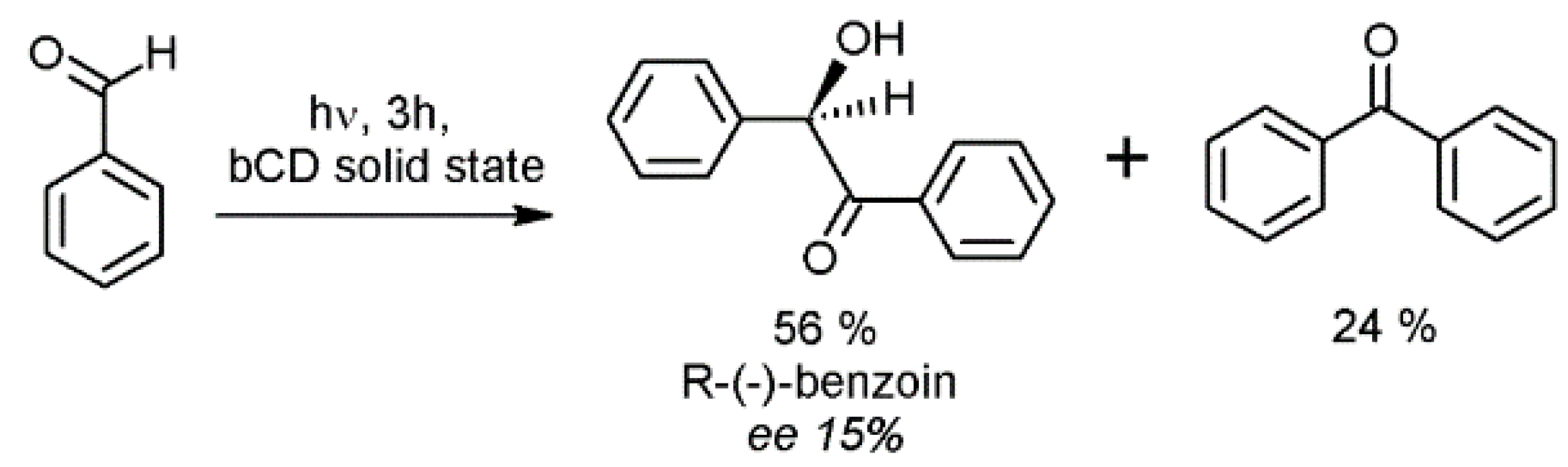
- Rao, V.P.; Turro, N.J. Asymmetric induction in benzoin by photolysis of benzaldehyde adsorbed in cyclodextrin cavities. Tetrahedron Lett. 1989, 30, 4641–4644. [Google Scholar] [CrossRef]
- Trotta, F.; Cravotto, G.; Rossignoli, S. Asymmetric synthesis in the presence of cyclodextrins. J. Incl. Phenom. Macrocycl. Chem. 2002, 44, 293–296. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, X.; Armstrong, D.W. Highly efficient asymmetric direct stoichiometric aldol reactions on/in water. Angew. Chem. Int. Ed. 2007, 46, 9073–9077. [Google Scholar] [CrossRef] [PubMed]
- Doyaguez, E.G.; Rodríguez-Hernández, J.; Corrales, G.; Fernández-Mayoralas, A.; Gallardo, A. Water-soluble pendant copolymers bearing proline and permethylated β-cyclodextrin: pH-dependent catalytic nanoreactors. Macromolecules 2012, 45, 7676–7683. [Google Scholar] [CrossRef]
- Zelinski, T.; Kula, M.R. Asymmetric enzymatic reduction of lipophilic ketones in aqueous solution containing cyclodextrins. Biocatal. Biotransform. 1997, 15, 57–74. [Google Scholar] [CrossRef]
- Zelinski, T.; Liese, A.; Wandrey, C.; Kula, M.R. Asymmetric reductions in aqueous media: Enzymatic synthesis in cyclodextrin containing buffers. Tetrahedron Asymmetry 1999, 10, 1681–1687. [Google Scholar] [CrossRef]
- Zhao, W.; Zhong, Q. Recent advance of cyclodextrins as nanoreactors in various organic reactions: A brief overview. J. Incl. Phenom. Macrocycl. Chem. 2012, 72, 1–14. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macaev, F.; Boldescu, V. Cyclodextrins in Asymmetric and Stereospecific Synthesis. Symmetry 2015, 7, 1699-1720. https://doi.org/10.3390/sym7041699
Macaev F, Boldescu V. Cyclodextrins in Asymmetric and Stereospecific Synthesis. Symmetry. 2015; 7(4):1699-1720. https://doi.org/10.3390/sym7041699
Chicago/Turabian StyleMacaev, Fliur, and Veaceslav Boldescu. 2015. "Cyclodextrins in Asymmetric and Stereospecific Synthesis" Symmetry 7, no. 4: 1699-1720. https://doi.org/10.3390/sym7041699
APA StyleMacaev, F., & Boldescu, V. (2015). Cyclodextrins in Asymmetric and Stereospecific Synthesis. Symmetry, 7(4), 1699-1720. https://doi.org/10.3390/sym7041699