
Oligodendroglial Heterogeneity in Neuropsychiatric Disease


{kind=link}
Abstract
:1. Introduction
2. White Matter Damage and Oligodendroglial Dysfunction in Neuropsychiatric Conditions
2.1. The Role of Oligodendroglia in Huntington’s Disease
2.2. The Role of Oligodendroglia in Other Neuropsychiatric Diseases
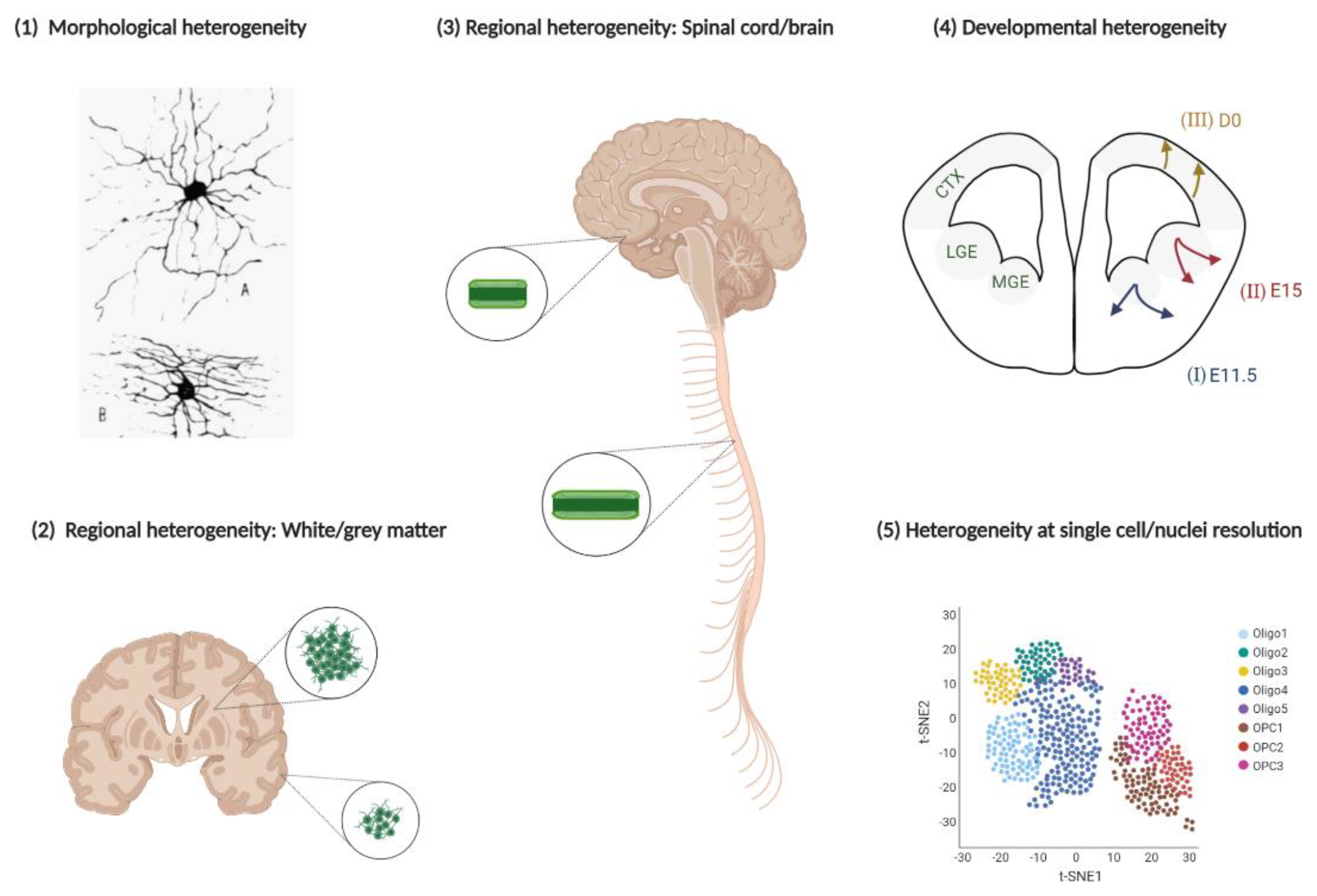
3. Heterogeneity of the Oligodendroglial Lineage
3.1. Developmental Heterogeneity
3.2. Regional Heterogeneity
4. Oligodendroglial Heterogeneity at Single-Cell Resolution
4.1. Major Depressive Disorder
4.2. Alzheimer’s Disease
4.3. Parkinson’s Disease
5. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbrevations
| CNS | Central nervous system |
| OPCs | Oligodendrocyte precursor cells |
| MS | Multiple Sclerosis |
| WM | White matter |
| MRI | Magnetic resonance imaging |
| DTI | Diffusion tensor imaging |
| PM | Post mortem |
| HD | Huntington’s disease |
| AD | Alzheimer’s disease |
| PD | Parkinson’s disease |
| Sz | Schizophrenia |
| MDD | Major depressive disorder |
| AUD | Alcohol use disorder |
| mHTT | Mutant huntingtin |
| MAG | Myelin-associated glycoprotein |
| CNPase | 2′, 3′-Cyclic-nucleotide 3′-phosphodiesterase |
| PFC | Prefrontal cortex |
| E | Embryonic day |
| iPSC | Induced pluripotent stem cell |
| GM | Grey matter |
| SC | Spinal cord |
| scRNAseq | Single-cell RNA sequencing |
| snRNAseq | Single-nucleus RNA sequencing |
| COPs | Committed OPCs |
| NFOLs | Newly formed oligodendrocytes |
| MFOLs | Myelin-forming oligodendrocytes |
| MOLs | Mature oligodendrocytes |
| GWAS | Genome-wide association study |
| SN | Substantia nigra |
References
- Zoupi, L.; Booker, S.A.; Eigel, D.; Werner, C.; Kind, P.C.; Spires-Jones, T.L.; Newland, B.; Williams, A.C. Selective vulnerability of inhibitory networks in multiple sclerosis. Acta Neuropathol. 2021, 1–15. [Google Scholar] [CrossRef]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Philips, T.; Mironova, Y.A.; Jouroukhin, Y.; Chew, J.; Vidensky, S.; Farah, M.H.; Pletnikov, M.V.; Bergles, D.E.; Morrison, B.M.; Rothstein, J.D. MCT1 Deletion in Oligodendrocyte Lineage Cells Causes Late-Onset Hypomyelination and Axonal Degeneration. Cell Rep. 2021, 34, 108610. [Google Scholar] [CrossRef]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef]
- Casella, C.; Lipp, I.; Rosser, A.; Jones, D.K.; Metzler-Baddeley, C. A Critical Review of White Matter Changes in Huntington’s Disease. Mov. Disord. 2020, 1, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, D.; Sandor, C.; Volpato, V.; Caffrey, T.M.; Monzón-Sandoval, J.; Bowden, R.; Alegre-Abarrategui, J.; Wade-Martins, R.; Webber, C. A single-cell atlas of the human substantia nigra reveals cell-specific pathways associated with neurological disorders. Nat. Commun. 2020, 11, 4183. [Google Scholar] [CrossRef]
- Duncan, G.W.; Firbank, M.J.; Yarnall, A.J.; Khoo, T.K.; Brooks, D.J.; Barker, R.A.; Burn, D.J.; O’Brien, J.T. Gray and white matter imaging: A biomarker for cognitive impairment in early Parkinson’s disease? Mov. Disord. 2016, 31, 103–110. [Google Scholar] [CrossRef]
- Raabe, F.J.; Slapakova, L.; Rossner, M.J.; Cantuti-Castelvetri, L.; Simons, M.; Falkai, P.G.; Schmitt, A. Oligodendrocytes as A New Therapeutic Target in Schizophrenia: From Histopathological Findings to Neuron-Oligodendrocyte Interaction. Cells 2019, 8, 1496. [Google Scholar] [CrossRef] [Green Version]
- Nagy, C.; Maitra, M.; Tanti, A.; Suderman, M.; Théroux, J.F.; Davoli, M.A.; Perlman, K.; Yerko, V.; Wang, Y.C.; Tripathy, S.J.; et al. Single nucleus transcriptomics of the prefrontal cortex in major depressive disorder implicates oligodendrocyte precursor cells and excitatory neurons. Nat. Neurosci. 2020, 23, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Brenner, E.; Tiwari, G.R.; Kapoor, M.; Liu, Y.; Brock, A.; Mayfield, R.D. Single cell transcriptome profiling of the human alcohol-dependent brain. Hum. Mol. Genet. 2020, 29, 1144–1153. [Google Scholar] [CrossRef]
- Zhang, F.F.; Peng, W.; Sweeney, J.A.; Jia, Z.Y.; Gong, Q.Y. Brain structure alterations in depression: Psychoradiological evidence. CNS Neurosci. Ther. 2018, 24, 994–1003. [Google Scholar] [CrossRef] [Green Version]
- Chang, E.H.; Argyelan, M.; Aggarwal, M.; Chandon, T.S.S.; Karlsgodt, K.H.; Mori, S.; Malhotra, A.K. The role of myelination in measures of white matter integrity: Combination of diffusion tensor imaging and two-photon microscopy of CLARITY intact brains. NeuroImage 2017, 147, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Skene, N.G.; Bryois, J.; Bakken, T.E.; Breen, G.; Crowley, J.J.; Gaspar, H.A.; Giusti-Rodriguez, P.; Hodge, R.D.; Miller, J.A.; Muñoz-Manchado, A.B.; et al. Genetic identification of brain cell types underlying schizophrenia. Nat. Genet. 2018, 50, 825–833. [Google Scholar] [CrossRef]
- Gregory, S.; Crawford, H.; Seunarine, K.; Leavitt, B.; Durr, A.; Roos, R.A.; Scahill, R.I.; Tabrizi, S.J.; Rees, G.; Langbehn, D.; et al. Natural biological variation of white matter microstructure is accentuated in Huntington’s disease. Hum. Brain Mapp. 2018, 39, 3516–3527. [Google Scholar] [CrossRef] [PubMed]
- Phillips, O.R.; Joshi, S.H.; Squitieri, F.; Sanchez-Castaneda, C.; Narr, K.; Shattuck, D.W.; Caltagirone, C.; Sabatini, U.; Di Paola, M. Major superficial white matter abnormalities in Huntington’s disease. Front. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [Green Version]
- Reading, S.A.; Yassa, M.A.; Bakker, A.; Dziorny, A.C.; Gourley, L.M.; Yallapragada, V.; Rosenblatt, A.; Margolis, R.L.; Aylward, E.H.; Brandt, J.; et al. Regional white matter change in pre-symptomatic Huntington’s disease: A diffusion tensor imaging study. Psychiatry Res. Neuroimaging 2005, 140, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Bohanna, I.; Georgiou-Karistianis, N.; Sritharan, A.; Asadi, H.; Johnston, L.; Churchyard, A.; Egan, G. Diffusion Tensor Imaging in Huntington’s disease reveals distinct patterns of white matter degeneration associated with motor and cognitive deficits. Brain Imaging Behav. 2011, 5, 171–180. [Google Scholar] [CrossRef]
- Rosas, H.D.; Wilkens, P.; Salat, D.H.; Mercaldo, N.D.; Vangel, M. NeuroImage: Clinical Complex spatial and temporally de fi ned myelin and axonal degeneration in Huntington disease. NeuroImage Clin. 2020, 20, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.; Scahill, R.I.; Seunarine, K.K.; Stopford, C.; Zhang, H.; Zhang, J.; Orth, M.; Durr, A.; Roos, R.A.; Langbehn, D.R.; et al. Neuropsychiatry and White Matter Microstructure in Huntington’s Disease. J. Huntington’s Dis. 2015, 4, 239–249. [Google Scholar] [CrossRef] [Green Version]
- Stoffers, D.; Sheldon, S.; Kuperman, J.M.; Goldstein, J.; Corey-Bloom, J.; Aron, A.R. Contrasting gray and white matter changes in preclinical Huntington disease: An MRI study. Neurology 2010, 74, 1208–1216. [Google Scholar] [CrossRef] [Green Version]
- Faria, A.V.; Ratnanather, J.T.; Tward, D.J.; Lee, D.S.; Van Den Noort, F.; Wu, D.; Brown, T.; Johnson, H.; Paulsen, J.S.; Ross, C.A.; et al. Linking white matter and deep gray matter alterations in premanifest Huntington disease. NeuroImage Clin. 2016, 11, 450–460. [Google Scholar] [CrossRef] [Green Version]
- McColgan, P.; Seunarine, K.K.; Gregory, S.; Razi, A.; Papoutsi, M.; Long, J.D.; Mills, J.A.; Johnson, E.; Durr, A.; Roos, R.A.; et al. Topological length of white matter connections predicts their rate of atrophy in premanifest Huntington’s disease. JCI Insight 2017, 2, e92641. [Google Scholar] [CrossRef]
- McColgan, P.; Gregory, S.; Razi, A.; Seunarine, K.K.; Gargouri, F.; Durr, A.; Roos, R.A.; Leavitt, B.R.; Scahill, R.I.; Clark, C.A.; et al. White matter predicts functional connectivity in premanifest Huntington’s disease. Ann. Clin. Transl. Neurol. 2017, 4, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Teo, R.T.Y.; Hong, X.; Yu-Taeger, L.; Huang, Y.; Tan, L.J.; Xie, Y.; To, X.V.; Guo, L.; Rajendran, R.; Novati, A.; et al. Structural andmolecularmyelination deficits occur prior to neuronal loss in the YAC128 and BACHD models of Huntington disease. Hum. Mol. Genet. 2016, 25, 2621–2632. [Google Scholar] [CrossRef]
- Ferrari Bardile, C.; Garcia-Miralles, M.; Caron, N.S.; Rayan, N.A.; Langley, S.R.; Harmston, N.; Rondelli, A.M.; Teo, R.T.Y.; Waltl, S.; Anderson, L.M.; et al. Intrinsic mutant HTT-mediated defects in oligodendroglia cause myelination deficits and behavioral abnormalities in Huntington disease. Proc. Natl. Acad. Sci. USA 2019, 116, 201818042. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Wei, W.J.; Wang, G.; Gaertig, M.A.; Feng, Y.; Wang, W.; Li, X.J.; Li, S. Mutant huntingtin downregulates myelin regulatory factor-mediated myelin gene expression and affects mature oligodendrocytes. Neuron 2015, 85, 1212–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakak, Y.; Walker, J.R.; Li, C.; Wong, W.H.; Davis, K.L.; Buxbaum, J.D.; Haroutunian, V.; Fienberg, A.A. Genome-wide expression analysis reveals dysregulation of myelination related genes in chronic schizophrenia. Proc. Natl. Acad. Sci. USA 2001, 98, 4746–4751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Rio-Hortega, P. Tercera Aportacion al Conocimiento Morfologico e Interpretacion Funcional de la Oligodendroglia. Mem. Real Soc. Esp. Hist. Nat. 1928, 14, 5–122. [Google Scholar]
- Foerster, S.; Hill, M.F.; Franklin, R.J. Diversity in the oligodendrocyte lineage: Plasticity or heterogeneity? Glia 2019, 67, 1797–1805. [Google Scholar] [CrossRef]
- Viganò, F.; Möbius, W.; Götz, M.; Dimou, L. Transplantation reveals regional differences in oligodendrocyte differentiation in the adult brain. Nat. Neurosci. 2013, 16, 1370–1372. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.A.; Patel, K.D.; Medved, J.; Reiss, A.M.; Nishiyama, A. NG2 cells in white matter but not gray matter proliferate in response to PDGF. J. Neurosci. 2013, 33, 14558–14566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, S.Y.; Rosenberg, S.S.; Fancy, S.P.; Zhao, C.; Shen, Y.A.A.; Hahn, A.T.; McGee, A.W.; Xu, X.; Zheng, B.; Zhang, L.I.; et al. Neurite outgrowth inhibitor Nogo-A establishes spatial segregation and extent of oligodendrocyte myelination. Proc. Natl. Acad. Sci. USA 2012, 109, 1299–1304. [Google Scholar] [CrossRef] [Green Version]
- Bechler, M.E.; Byrne, L.; Ffrench-Constant, C. CNS Myelin Sheath Lengths Are an Intrinsic Property of Oligodendrocytes. Curr. Biol. 2015, 25, 2411–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, A.H.; Tripathi, R.B.; Richardson, W.D.; Franklin, R.J. Developmental Origin of Oligodendrocyte Lineage Cells Determines Response to Demyelination and Susceptibility to Age-Associated Functional Decline. Cell Rep. 2016, 15, 761–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessaris, N.; Fogarty, M.; Iannarelli, P.; Grist, M.; Wegner, M.; Richardson, W.D. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat. Neurosci. 2006, 9, 173–179. [Google Scholar] [CrossRef]
- Marques, S.; Zeisel, A.; Codeluppi, S.; Van Bruggen, D.; Falcão, A.M.; Xiao, L.; Li, H.; Häring, M.; Hochgerner, H.; Romanov, R.A.; et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 2016, 352, 1326–1329. [Google Scholar] [CrossRef] [Green Version]
- Lake, B.B.; Chen, S.; Sos, B.C.; Fan, J.; Kaeser, G.E.; Yung, Y.C.; Duong, T.E.; Gao, D.; Chun, J.; Kharchenko, P.V.; et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 2018, 36, 70–80. [Google Scholar] [CrossRef]
- Kim, H.; Xu, R.; Padmashri, R.; Dunaevsky, A.; Liu, Y.; Dreyfus, C.F.; Jiang, P. Pluripotent Stem Cell-Derived Cerebral Organoids Reveal Human Oligodendrogenesis with Dorsal and Ventral Origins. Stem Cell Rep. 2019, 12, 890–905. [Google Scholar] [CrossRef] [Green Version]
- Hildebrand, C.; Remahl, S.; Persson, H.; Bjartmar, C. Myelinated nerve fibres in the CNS. Prog. Neurobiol. 1993, 40, 319–384. [Google Scholar] [CrossRef]
- Jäkel, S.; Agirre, E.; Mendanha Falcão, A.; van Bruggen, D.; Lee, K.W.; Knuesel, I.; Malhotra, D.; Ffrench-Constant, C.; Williams, A.; Castelo-Branco, G. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 2019, 566, 543–547. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Regev, A.; Mccarroll, S.A.; Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [Green Version]
- Van Bruggen, D.; Agirre, E.; Castelo-Branco, G. Single-cell transcriptomic analysis of oligodendrocyte lineage cells. Curr. Opin. Neurobiol. 2017, 47, 168–175. [Google Scholar] [CrossRef]
- Zeisel, A.; Moz-Manchado, A.B.; Codeluppi, S.; Lönnerberg, P.; Manno, G.L.; Juréus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C.; et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef]
- Schirmer, L.; Velmeshev, D.; Holmqvist, S.; Kaufmann, M.; Werneburg, S.; Jung, D.; Vistnes, S.; Stockley, J.H.; Young, A.; Steindel, M.; et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 2019, 573, 75–82. [Google Scholar] [CrossRef]
- Bakken, T.E.; Jorstad, N.L.; Hu, Q.; Lake, B.B.; Tian, W.; Kalmbach, B.E.; Hodge, R.D.; Krienen, F.M.; Sorensen, S.A.; Eggermont, J.; et al. Evolution of cellular diversity in primary motor cortex of human, marmoset monkey, and mouse. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Wray, N.R.; Ripke, S.; Mattheisen, M.; Trzaskowski, M.; Byrne, E.M.; Abdellaoui, A.; Adams, M.J.; Agerbo, E.; Air, T.M.; Andlauer, T.M.; et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 2018, 50, 668–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bøstrand, S.M.K.; Williams, A. Oligodendroglial Heterogeneity in Neuropsychiatric Disease. Life 2021, 11, 125. https://doi.org/10.3390/life11020125
Bøstrand SMK, Williams A. Oligodendroglial Heterogeneity in Neuropsychiatric Disease. Life. 2021; 11(2):125. https://doi.org/10.3390/life11020125
Chicago/Turabian StyleBøstrand, Sunniva M. K., and Anna Williams. 2021. "Oligodendroglial Heterogeneity in Neuropsychiatric Disease" Life 11, no. 2: 125. https://doi.org/10.3390/life11020125
APA StyleBøstrand, S. M. K., & Williams, A. (2021). Oligodendroglial Heterogeneity in Neuropsychiatric Disease. Life, 11(2), 125. https://doi.org/10.3390/life11020125