Abstract
Endosomes and lysosomes are intracellular vesicular organelles with important roles in cell functions such as protein homeostasis, clearance of extracellular material, and autophagy. Endolysosomes are characterized by an acidic luminal pH that is critical for proper function. Five members of the gene family of voltage-gated ChLoride Channels (CLC proteins) are localized to endolysosomal membranes, carrying out anion/proton exchange activity and thereby regulating pH and chloride concentration. Mutations in these vesicular CLCs cause global developmental delay, intellectual disability, various psychiatric conditions, lysosomal storage diseases, and neurodegeneration, resulting in severe pathologies or even death. Currently, there is no cure for any of these diseases. Here, we review the various diseases in which these proteins are involved and discuss the peculiar biophysical properties of the WT transporter and how these properties are altered in specific neurodegenerative and neurodevelopmental disorders.
1. Introduction—The CLC Family
Physiologically, the most abundant anion is chloride. It is an important substrate of many transport proteins, being carried across the membrane as a single anion or coupled with other ions, and is important, for example, for the regulation of the membrane potential, intracellular vesicles acidification and cell volume regulation [1].
In humans, the CLC family is formed by nine members, which had initially been supposed to be all chloride channels, because of their sequence homology with the founding member, the Torpedo electroplax channel ClC-0 [2]. The discovery that the bacterial Escherichia coli ecClC-1 homologue is not a passive chloride channel but a stoichiometrically coupled secondary active 2 Cl−/1 H+ antiporter has dramatically changed the point of view of the entire CLC group [3]. Based on sequence homology, three branches of human CLCs have been distinguished. The first one includes the plasma membrane-localized chloride channels ClC-1, ClC-2 and the two isoforms ClC-Ka and ClC-Kb. The second branch is formed by ClC-3, ClC-4 and ClC-5, while the third branch contains ClC-6 and ClC-7. ClC-3 to -7 are all Cl−/H+ exchangers and are localized to the intracellular membranes of endosomes and/or lysosomes [1].
All CLC family members share the same dimeric architecture that is unique to this protein family. Except for ClC-6 and ClC-7 [4], the other CLC proteins can form homo- or hetero-dimers with members of the same branch [1]. Biochemical studies and single-channel analysis on the first cloned Torpedo ClC-0, mutants [2,5,6] and biochemical and low-resolution structural analysis of ecClC-1 [7,8] suggested a homodimeric “double-barreled” architecture, with physically separated anion transport pathways in each protomer. This architecture has been fully confirmed by the determination of ecClC-1 and Salmonella typhimurium stClC crystal structures [9,10]. The structures revealed the presence of distinct anion binding sites, formed by residues that are also highly conserved in human CLCs. The sites are denominated Sext, Scen and Sint, with Sext being occupied by the presumably negatively charged side chain of the “gating glutamate” E148 [9,10]. Each monomer presents 18 α-helices (from A to R) of which 17 are partially embedded in the membrane. The two subunits interact in a tight manner and the architecture follows an inverted and parallel orientation [1]. Two C-terminal tandem cystathionine-β-synthase (CBS) domains are present in most eukaryotic CLC proteins [11,12], but are absent in ecClC-1. The two CBS domains may have a role in the so-called common gating process (that will be discussed in more detail below) and confer unique features to the CLC members [1]. Dutzler and colleagues determined the crystal structures of isolated CBS domains of Torpedo ClC-0 [13], human ClC-5 [14] and human ClC-Ka [15]. CBS domains are present in many different protein families, where they are often implicated in the sensing of adenine nucleotides [11,16]. Structurally, so far, ATP has been found to be bound in the isolated domains of ClC-5 and in the full-length structure of ClC-7 [17], but not in isolated domains of ClC-0 and ClC-Ka and not in full-length structures of bovine ClC-K or human ClC-1 [18,19,20].
Single-channel recordings of the Torpedo ClC-0 channel displayed two kinds of gating mechanisms that regulate the open probability (Po) of the channel: a “fast” or “protopore” gate that acts independently on single pores determines the closing or opening state of each pore of the double-barreled structure [1]. The fast gate is mainly determined by the gating glutamate (E166 in ClC-0), in that its neutralization renders CLC-0 channels voltage independent. Protonation of the gating glutamate and its competition with permeant ions underlie the anion and pH dependent protopore gating of most CLC channels [10,21,22,23]. Conversely, a second mechanism, termed a slow or common gate, operates on both pores simultaneously and is still not well understood [1].
Some CLC proteins require association with a small ancillary subunit for proper function or membrane expression. In particular, the kidney ClC-Ka and ClC-Kb channels require association with the barttin subunit [24]. In glia cells, ClC-2 associates with GlialCAM, a protein with the typical architecture of a cell adhesion molecule, which is mutated in megalencephalic leukoencephalopathy with subcortical cysts (MLC) [25]. It leads to clustering of ClC-2 at glial cell–cell contacts and alters biophysical functions of the ClC-2 channel [25,26]. The complex of ClC-7 with its subunit Ostm1 is mandatory for mutual stabilization [4,27].
The plasma membrane localized chloride channels belonging to the first branch of the CLC family are expressed in a tissue-dependent manner that is different for each member according to their physiological role. All channel CLCs are involved in various human genetic diseases, as reviewed in detail elsewhere [1,28,29].
ClC-3 through ClC-7, which are the focus of this review, function as Cl−/H+ exchangers and are localized to intracellular endosomes and/or lysosomes (Figure 1, Table 1). Initially, when the transporter function of the intracellular CLCs was not yet known, it was proposed that they act as charge-shunting chloride channels to assist the luminal acidification of endosomes and lysosomes intracellular organelles [30,31,32,33,34]. Indeed, the maintenance of an acidic pH of the lumen of endo-/lysosomes is required for their proper physiological function. The proton pumping V-ATPase is electrogenic and thus generates an electrical potential difference that would impede acidification if not neutralized by anionic cotransport and/or cationic counter-transport. Somewhat surprisingly and counter-intuitively, model calculations show that a 2 Cl−/H+ exchange activity, contributing to a more inside-negative voltage, allows a more acidic steady-state luminal pH compared to a shunting Cl− channel [35,36].
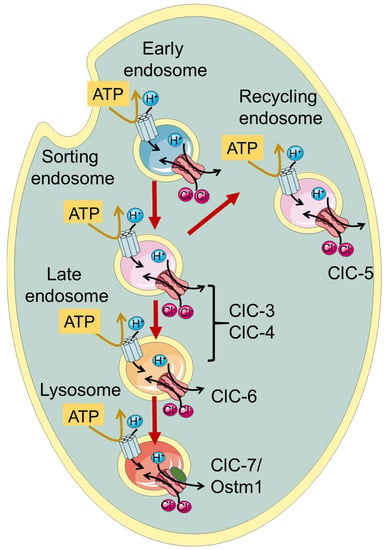
Figure 1.
Schematic illustration of localization of vesicular CLCs in the endo-/lysosomal pathway.
Among the endo-/lysosomal CLCs, ClC-5 is rather specifically expressed in the kidney with a predominant presence in epithelial cells of the proximal tubule, where it is involved in endocytic uptake [30,31,33]. Indeed, mutations causing impaired ClC-5 transport activity are associated with Dent’s disease, a kidney disorder characterized by the primary symptom of low molecular weight proteinuria, and a series of secondary symptoms including kidney stones and renal failure, caused by defective endocytosis in the proximal tubule [33,37]. ClC-7, together with its subunit Ostm1 [27], is rather ubiquitously expressed in the body and is localized to lysosomes and in the ruffled border of osteoclasts functioning as a 2Cl−/H+ antiporter [1]. Accordingly, impaired bone resorption in osteoclast, caused by a functionally defective ClC-7/Ostm1 complex, causes osteopetrosis, a disease characterized by stiff and fragile bones [38].

Table 1.
List of the CLC genes, subcellular localization and CLC related neurological diseases.
Table 1.
List of the CLC genes, subcellular localization and CLC related neurological diseases.
| Protein | Gene | Cellular Localization | Neurological Disorder | Symptoms | References |
|---|---|---|---|---|---|
| ClC-3 | CLCN3 | Sorting and late endosomes | Global developmental delay | Intellectual disability, agenesis of the corpus callosum, epilepsy, visual impairment, hypotonia, anxiety, dysmorphic facial features | [39] |
| ClC-4 | CLCN4 | Sorting and late endosomes | CLCN4-related X linked intellectual disability syndrome | Intellectual disability, epilepsy, autism, growth and feeding difficulties, epilepsy, movement disorders, gastrointestinal conditions, dysmorphic facial features | [40,41,42] |
| ClC-6 | CLCN6 | Late endosomes | |||
| Early Onset Neurodegeneration | Severe neurodegeneration, severe generalized hypotonia and respiratory insufficiency, brain atrophy | [43] | |||
| Kufs’ disease | Adult-onset neuronal ceroid Lipofuscinosis, movement and cognitive function impairment | [44,45] | |||
| West syndrome | Severe developmental delay, autism, movement disorder, microcephaly, facial dysmorphism, visual impairment | [46] | |||
| ClC-7/ Ostm1 | CLCN7/ OSTM1 | Lysosomes | |||
| Autosomal Recessive Osteopetrosis | Osteopetrosis, lysosomal storage disease, neurodegeneration, visual impairment | [38,47,48] | |||
| Gain of function CLCN7 related disease | Delayed myelination and development, organomegaly, and hypopigmentation | [49] |
A large phenotypic spectrum of neuronal diseases is associated with mutations in the genes encoding ClC-3/-4/-6 and ClC-7, as will be described in detail in the following paragraphs (see Table 1).
For all vesicular CLCs, an unsolved question pertains to the direction of exchanger transport. Despite being physiologically localized to endo-/lysosomes, ClC-3 to -6 can reach the plasma membrane when heterologously expressed in HEK293 cells, allowing the investigation of their biophysical properties using the patch clamp technique [45,50,51,52,53]. For ClC-7, the elimination of N-terminal lysosomal targeting motifs leads to plasma membrane expression [4,54]. ClC-3 to -5 all exhibit extreme outward rectification of currents with very little or nonresolvable activation kinetics [50,51,52,53,55]. This current direction corresponds to the transport of luminal Cl− out of lysosomes with a parallel influx of cytosolic H+. However, it remains unclear whether the direction of transport is physiologically relevant and whether CLC exchangers work synergistically with V-ATPase, contributing to luminal acidification.
Additionally, ClC-6 and ClC-7 exhibit strongly outwardly rectifying currents, which are, however, characterized by slow activation kinetics and measurable inward “tail” currents [4,45].
2. ClC-3 and ClC-4
The second branch of the CLC family comprises ClC-3, -4 and 5. These three endosomal transporters share high sequence similarity and have similar functional properties [1,29]. Among the human CLCs, they are the most similar to the Escherichia coli ecClC-1 homologue. The renal-specific ClC-5 is found mostly in recycling endosomes and its physiological role will not be discussed in detail [1]. ClC-3 and ClC-4 are localized to sorting endosomes, and ClC-3 is probably localized in late endosomes as well [1,29]. ClC-3 has also been proposed to play a role in synaptic vesicles. This is, however, still controversial and will not be discussed in detail here (see [29] for a discussion).
Functionally, these transporters are characterized by an extremely outwardly rectifying current–voltage relationship with almost instantaneous activation at positive voltages [50,51,52,55,56,57,58] (Figure 2A). The extreme outward rectification precluded the determination of transport stoichiometry by reversal potentials measurements [59]. Using a fluorescent assay, a 2 Cl−/1 H+ stoichiometry was determined for ClC-5, which is probably similar for ClC-3 and ClC-4 [55]. The outward rectification at least partially reflects a gating process, as evidenced by a single point mutation (D76H) that conferred detectable inward tail currents to ClC-5 [60]. The mutant also allowed us to measure reversal potentials for ClC-5 for the first time, confirming the 2:1 Cl−/H+ transport stoichiometry [60].
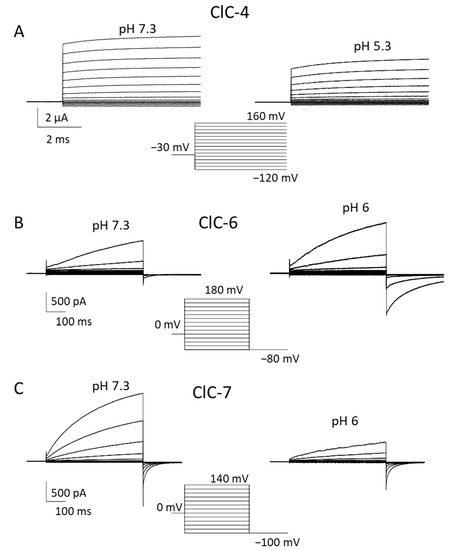
Figure 2.
Biophysical properties of ClC-4, ClC-6 and ClC-7. Typical voltage-clamp current traces of ClC-4 ((A), expressed in Xenopus oocytes), ClC-6 ((B), expressed in HEK cells), and ClC-7PM ((C), expressed in HEK cells), elicited by the indicated voltage-clamp protocols, measured using the two-electrode voltage clamp method (A) or the whole-cell recording mode of the patch clamp technique (B,C) at neutral (left panels) and acidic (right panels) pH. Extracellular solutions were 100 mM NaCl, 10 mM Hepes or MES, 5 mM MgSO4 (A) or 150 mM NaCl, 10 mM Hepes or MES, 4 mM MgSO4 (B,C). In B and C the pipette solution contained 130 mM NaCl, 10 mM Hepes, 2 mM EGTA, 2 mM MgSO4. ClC-3 is similar to ClC-4.
Most evidence on the physiological roles of ClC-3 and ClC-4 was obtained from mouse models and their involvement in human genetic diseases. ClC-3 KO mice display severe postnatal neurodegeneration with almost total loss of the hippocampus after 3 months [34,61,62]. Yet, ClC-3 KO mice have a normal life span. Neurodegeneration is unlikely to be caused by impaired synaptic function [1,29]. Another set of mouse models showed that ClC-4 protein stability relies on the presence of ClC-3 via heterodimerization [63,64]. While ClC-4 KO mice have no overt phenotype, the double KO of ClC-3 and ClC-4 has a more severe phenotype than CLC-3 KO mice [64]. Notably, differently from humans, Clcn4 is not X-linked in laboratory mice. For this reason, a rat model might be more useful for the investigation of mechanisms underlying CLCN4-related disease, because in rats, as in humans, Clcn4 is X-linked.
While ClC-4 KO mice have no overt phenotype, in 2013 and 2016, patients (mostly pediatric) with a range of neurodevelopmental and psychiatric complications have been described with X-linked CLCN4 variants [40,42,65] (see Table 1). In heterologous expression, these and some novel variants [66] showed variable loss of function effects. It is important to note that complete loss of ClC-4 protein leads to non-syndromic intellectual disability in males and no disease in heterozygous females. In contrast, de novo and inherited missense variants can lead to severe syndromic neurological disease in males as well as in females, suggesting a dominant effect. In a recent study, a large number of CLCN4 families was investigated, describing a large spectrum of clinical phenotypes and studying > 50 missense variants in heterologous expression [41]. Novel biophysical mechanisms were discovered for new and already described variants. These included a toxic gain of function characterized by the presence of negative currents at acidic extracellular (luminal) pH, and a shift in the voltage dependence of gating to more positive voltages [41]. Both effects can be expected to exert dominant negative effects in ClC-3/ClC-4 heterodimers.
Almost simultaneously came the discovery of the first variants in CLCN3 that cause global developmental delay, intellectual disability and neurodevelopmental disorders [39] (Table 1). Detailed functional analysis revealed a toxic gain of function for two missense variants, similar to the above-described effects in some CLCN4 variants [39].
3. ClC-6
The third branch of the CLC family comprises ClC-6 and ClC-7, which both function as Cl−/H+ exchangers [1,45]. Even though the expression of ClC-6 mRNA appears to be ubiquitous in many tissues [67], biochemical analysis detected native ClC-6 protein predominantly in neurons, where it localizes to late endosomes and partially lysosomes [44]. For a long time, the biophysical profile of ClC-6 remained completely unknown. In the first attempts at heterologous expression, no currents attributable to ClC-6 could be detected [67], possibly caused by the intracellular localization of most of the overexpressed protein [68].
A subtype of lysosomal storage disease, referred to as neuronal ceroid lipofuscinosis (NCL), was observed in ClC-6 knockout mice presenting a mild phenotype with features of reduced pain sensitivity, probably due to strong accumulation of materials in axon initial segments, mild cognitive abnormalities and no impact on their span life [44]. This evidence suggested that CLCN6 variants could be involved in human NCL [44]. Indeed, in a sample of 75 adult-onset variants, including late-onset forms of NLC and Kufs’ disease, two individuals were found to be heterozygous for CLCN6 missense variants (V580M and T628R) [44]. However, no functional analysis had been performed at the time of that study because the transporter had not been successfully functionally characterized.
Preliminary electrophysiological characterization was obtained when the N-terminus of ClC-6 tagged with GFP (GFP-ClC-6) was reported to enhance its cell surface localization [69]. However, the reported currents were small and barely above background levels.
In 2020, the same de novo variant in CLCN6 leading to the amino acid change Y553C was reported in three pediatric patients with no parental correlation [43]. The patients exhibited a severe syndrome characterized by early-onset neurodegeneration. The mutated tyrosine, located in the extracellular P-Q loop, is highly conserved among CLC antiporters. Electrophysiological measurements of ClC-6Y553C revealed large currents activated at positive voltages (≥60 mV), representing a clear gain of function effect [43]. HEK cells overexpressing the mutant showed a dramatic vacuolization [43]. The luminal pH of these organelles did not reach low values, suggesting that the mutant impairs endolysosomal ionic homeostasis in patients.
In 2022, Zifarelli et al. were able to obtain robust recordings of WT ClC-6 currents by applying very large positive voltages beyond 140 mV [45,70]. Interestingly, the magnitude and properties of currents were independent of the N-terminal GFP tag. Similar to ClC-7, ClC-6-mediated currents showed slow activation at positive voltages; however, they required voltages of at least 140 mV to measure appreciable amplitudes [45] (Figure 2B). The necessity of the large voltages explained why these currents had escaped detection so far. Currents represent the coupled Cl−/H+ antiport, as assayed by reversal potential measurements [45]. Similar to ClC-7 [71], ClC-6 also exhibits so called “transient” or “capacitive” currents, whose possible physiological role is, however, obscure [45]. Interestingly, neutralizing the so-called proton glutamate did not completely abolish transport currents [45]. In contrast to most other CLC proteins, ClC-6-mediated currents were enhanced at acidic pH (6.3) compared to neutral pH [45] (Figure 2B), a finding that is of likely physiological relevance. In light of these novel findings on the functional properties of ClC-6, Zifarelli et al. performed a reexamination of the disease-causing variant ClC-6Y553C, concluding that the mutation causes a gain of function by “shifting” the voltage dependence of ClC-6 gating to less positive voltages [45]. Possibly, Y553, being localized at the subunit interface of the homodimer, is involved in the common gating mechanism that acts on both ion-transporting units of the dimer.
The discovery of suitable recording protocols allowed Zifarelli et al. to study the functional impact of the two above-mentioned variants found in Kufs’ disease patients. While T628R was indistinguishable from WT ClC-6, precluding firm conclusions regarding its causative nature for the disease, variant V580M showed a clearly reduced function, suggesting a causal relationship [45]. Since the variant was found in heterozygosity, it might exert a dominant negative effect in WT/mutant heterodimers.
Patients with the completely unrelated West syndrome, characterized by epilepsy, among other symptoms, have been reported to carry the ClC-6 E200A variant [46]. E200 is the critical gating glutamate of the exchanger and it is known that its neutralization in all studied vCLCs, including ClC-6, eliminates H+ transport and transforms it into an uncoupled ohmic chloride channel [69]. In a heterologous expression system, ClC-6E200A caused an impairment of the autophagosome-mediated degradation system, likely because the fusion with lysosomes was compromised [46].
The three classes of disease related CLCN6 mutations, i.e., gain of function (Y553C), reduction of function (V580M), and uncoupling (E200A), have different effects on the functional properties of the ClC-6 antiporter, leading to clinical phenotypes with different degrees of severity. In particular, ClC-6Y553C causing a gain of function is associated with drastic neurodegeneration, whereas ClC-6E200A could be defined as a loss of function in the respect of the uncoupling transport generated and related to a mild phenotype.
The recent remarkable progress that has been made regarding the functional analysis of ClC-6 activity will allow us to decode further mechanisms underlying disease caused by defective ClC-6 proteins.
4. ClC-7
Belonging to the third mammalian CLC branch, ClC-7 shares 45% of sequence homology with ClC-6. It was cloned in parallel with ClC-6 in 1995 [67], but could not be functionally analyzed for a long time. Intriguingly, ClC-7 is the only subcellular CLC member to be present almost exclusively in lysosomes [44]. Moreover, it has also been found in the ruffled border of osteoclasts, where it participates in bone resorption [38]. Unlike the other CLC transporters, ClC-7 requires association with a type I transmembrane protein, called Ostm1, for proper function and stability [4,27].
Similarly to ClC-6, no information about electrophysiological ClC-7 characterization has been available for a long time, due to its intracellular localization upon heterologous expression [1]. Ion flux studies with isolated mouse lysosomes showed that ClC-7 is the dominant anion permeation pathway of lysosomal membranes and that it performs 2 Cl−/1 H+ antiport activity [72]. A breakthrough was achieved by Stauber and Jentsch, who discovered the sorting motifs that mediate lysosomal targeting [54]. In particular, they found that when four leucine residues localized in the N-terminal portion are changed to alanine, the transporter is at least partially targeted to the plasma membrane [54]. Notably, Ostm1 follows ClC-7 in its expression location. ClC-7PM, the ClC-7 variant in which the two dileucine motifs are mutated to alanine, elicited robust transmembrane, outwardly rectifying voltage-activated currents [4] (Figure 2C). Even though some electrophysiological properties of ClC-7 are similar to that of other vesicular CLCs, including the inhibitory effect of acidic pH, ClC-7 differs substantially from ClC-3 to -5. Most importantly, ClC-7PM exhibits very slow activation kinetics in the seconds time range [4] (Figure 2C). This slow “gating” phenomenon is strictly linked to conformational changes in the proteins, where the interactions between transmembrane as well as cytoplasmic domains play a key role [73]. In addition to the transport currents, Pusch and Zifarelli discovered that the transporter also exhibits rather large “transient” or “capacitive” currents that reflect charge rearrangements within the protein. These are most likely mediated by movements of the gating glutamate and chloride binding/unbinding events [71]. Similar currents have been observed in ClC-5 and ClC-3 [52,74,75]. The transient currents probably have no physiological role, but represent a biophysical feature that can be useful in deciphering molecular mechanisms of gating and transport. Interestingly, while in ClC-5, neutralization of the so-called proton glutamate completely abolished transport currents, leaving only transient currents [74,75], in ClC-7, residual transport currents were observed in the corresponding E312A mutant [71].
The physiological role of ClC-7 remained unclear for a long time. The first insights were obtained with a mouse KO model that was characterized by severe osteopetrosis [38]. The involvement of ClC-7 in bone resorption was confirmed by the presence of CLCN7 mutations in a human patient with malignant osteopetrosis [38]. Further evidence came from the identification of a spontaneous Ostm1 mutation to be associated with the onset of a severe osteopetrosis in gray lethal mice presenting a fur color defect [76]. In Clcn7−/− mice, even though the number of osteoclasts was normal, their ability to reabsorb calcified bone was impaired [38]. Interestingly, however, no impact on lysosomal acidification was observed, suggesting that the osteoclasts’ ability to acidify intracellular vesicles was preserved in Clcn7−/− mice [38]. The life span of the KO mice was limited to 6–7 weeks.
Importantly, in addition to osteopetrosis, ClC-7−/− mice also presented severe lysosomal storage associated with central nervous system and retinal degeneration [47]. Using a lacZ fusion protein, the expression profile of ClC-7 was determined in the nervous tissue of WT e KO mice revealing the hippocampus CA3 region, the cortex and the cerebellum as the main regions experiencing neuronal loss in KO mice [47]. Electron microscopy analysis revealed the presence of autofluorescent lipopigment in the regions affected by neurodegeneration and deficient of CLC-7 [47]. This factor, together with the detection of microglial activation and astrogliosis, represents three important hallmarks of neuronal ceroid lipofuscinosis (NCL) [47]. Importantly, no significant difference in pH values in lysosomes of cultured neurons and fibroblasts was found in Clnc7−/− mice, but rather a reduction in lysosomal Cl concentration was observed [47]. Several pieces of evidence suggest that osteopetrosis and neurodegeneration are independent outcomes. First, an osteopetrotic mouse model with a mutation in the a3 subunit of V-type H+-ATPase (oc/oc mice) does not show retinal or neurodegeneration [47]. Moreover, the osteoporotic phenotype in Clnc7−/− mice could be rescued by transgenically expressing ClC-7 in osteoclasts and macrophages under the control of tartrate-resistant acid phosphatase (TRAP) promoter [47]. This treatment achieved a lifespan increase, but it was not enough to ensure their survival due to the enduring neurological problems [47]. Surprisingly, the same approach failed when TRAP promoter-mediated Ostm1 expression was applied to rescue osteopetrosis in gray lethal (gl) mice which also displayed neuronal loss [77]. The severity of the phenotype and the short life span of the mice were serious problems, preventing a better understanding of the mechanisms underlying the progression of neurodegeneration in lysosomal pathologies [1]. The first information was collected when Wartosch et al. designed a floxed Clcn7 mouse model allowing tissue-specific ClC-7 depletion [78]. No difference in lifespan between neuron-specific Clcn7 KO and WT mice was observed. Moreover, neuron-specific Clcn7 KO mice had no osteopetrotic phenotype; thus, the quality of life of these mice was improved compared to Clcn7−/− [78]. Importantly, it was observed that neuronal loss occurs in regions, where ClC-7 had been disrupted and neurodegeneration started in the CA3 region of the hippocampus as in constitutive Clcn7−/−. Accordingly, in previous studies, astrogliosis and microglia activation were observed in the regions lacking ClC-7. Impaired lysosomal protein degradation was suggested after the detection of increased levels of LC3-II, a marker of autophagy [78].
A somewhat surprising finding was that several, mostly dominantly inherited, CLCN7 variants causing osteopetrosis (but not neurodegeneration) produce a significant acceleration of gating kinetics [4,79,80]. It is unclear how this biophysical defect is related to ClC-7 malfunction.
More recently, a completely different disease characterized by delayed myelination and development, organomegaly and hypopigmentation was found in two children who both carried the de novo Y715C variant [49]. Surprisingly, none of the patients showed osteopetrosis (see Table 1). The variant, located in the C-terminus, was associated with larger currents when directed to the plasma membrane, representing a clear gain of function effect. Interestingly, the lysosomes of patient fibroblasts were enlarged and had a lower pH (0.2 pH units) than control lysosomes [49]. Even more excitingly, in the ClC-7 structure, Y715 is relatively close to the bound PI(3)P molecule (see below) and Leray et al. recently reported that intracellular phosphatidylinositol-3,5-bisphosphate (PI(3,5)P2) appears to tonically inhibit ClC-7 function, and that the Y715C variant was insensitive to PI(3,5)P2 [81]. The regulation of CLC-transporters by these signaling molecules is clearly an exciting aspect that needs to be understood in more detail.
The structure of the ClC-7/Ostm1 complex has been determined by two independent groups [17,82]. The structures revealed that the heavily N-glycosylated luminal region of Ostm1 forms a sort of cap on the luminal portion of ClC-7, preventing its degradation by lysosomal proteases [17,82]. Importantly, the mutual protein stabilization between ClC-7 and Ostm1 is suggested by the observation that the gray lethal mouse line, which lacks Ostm1, showed very weak ClC-7 staining; similarly, in Clcn7−/− mice, there was only weak Ostm1 staining [27,76]. Both cryo-EM structures revealed strong intramolecular interactions between the cytosolic N-terminal portion and the CBS domains [17,82]. This important feature is also conserved in ClC-6 (Hite, personal communication), but the role of these interactions in other CLCs remains unclear because no information about the cytosolic structure of other CLC members is available. Similarly to ClC-5, the ClC-7 CBS domains bind ATP and additionally, a Mg2+ ion was found to be bound [17,82]. However, ATP had no effect on transport activity and its role remains to be understood [4]. Moreover, in the structure of ClC-7 an endolysosomal phosphatidylinositol 3-phosphate (PI3P) lipid was found to be bound at the interface between the CBS and the membrane domains [17].
5. Conclusions
While significant progress has been made in elucidating the functional properties of vesicular CLC transporters and their involvement in various neurological diseases, highlighting their importance in nervous system development and homeostasis, their precise physiological role is still largely unknown. Even for the most studied ClC-7, it is still disputed whether it is primarily necessary for proper luminal acidification or the regulation of the luminal chloride concentration. Most recent evidence favors the idea that ClC-7 is responsible for achieving a high luminal chloride concentration, which is important for phagosomal clearance [83]. Less clear are the roles of ClC-3 and ClC-4 in endosomes and the possible involvement of ClC-3/ClC-4 dimers in human genetic diseases. For all vesicular CLCs, and in particular for ClC-6, the significance of the activation at highly positive voltages remains enigmatic, since similar voltages are not expected to be achieved in endosomes. However, it is clear that ClC-3 and ClC-4 need to be inactive at negative voltages, since even a small amount of activity at these voltages, caused by gate disrupting mutations, leads to severe disease for both transporters [39,41]. In general, it appears that gain of function mutations lead to more severe phenotypes than loss-of function mutations for all vesicular CLCs [39,41,43,49]. In this respect, specific pharmacological inhibitors of vesicular CLCs are highly desirable. Unfortunately, thus far, no useful pharmacological tools are available for any of them.
Author Contributions
M.P. and M.A.C. conceived the idea and prepared the figures. M.A.C., A.T.-M., P.I., P.G., A.L. and M.P. contributed to writing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was partially funded by the European Union—NextGenerationEU (Missione 4 Componente 2, “Dalla ricerca all’impresa”, Innovation Ecosystem RAISE “Robotics and AI for Socio-economic Empowerment”, ECS00000035). However, the views and opinions expressed are those of the authors alone and do not necessarily reflect those of the European Union or the European Commission. Neither the European Union nor the European Commission can be held responsible for them. In addition, this research was partially funded by the Fondazione AIRC per la Ricerca sul Cancro (grant # IG 21558), PRIN-MIUR 2017 Prot. 20174TB8KW, Fondazione Telethon (grant # GMR22T102) granted to M.P. and Fondazione Telethon/Cariplo (grant # GJC22008) to M.P.
Data Availability Statement
Data of Figure 2 are available upon reasonable request.
Acknowledgments
We would like to thank Francesca Quartino and Alessandro Barbin for their excellent technical assistance. This work was partially carried out within the framework of the project “RAISE—Robotics and AI for Socio-economic Empowerment” and has been supported by the European Union—NextGenerationEU.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jentsch, T.J.; Pusch, M. CLC chloride channels and transporters: Structure, function, physiology, and disease. Physiol. Rev. 2018, 98, 1493–1590. [Google Scholar] [CrossRef]
- Jentsch, T.J.; Steinmeyer, K.; Schwarz, G. Primary structure of Torpedo marmorata chloride channel isolated by expression cloning in Xenopus oocytes. Nature 1990, 348, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Accardi, A.; Miller, C. Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature 2004, 427, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Leisle, L.; Ludwig, C.F.; Wagner, F.A.; Jentsch, T.J.; Stauber, T. ClC-7 is a slowly voltage-gated 2Cl−/1H+-exchanger and requires Ostm1 for transport activity. EMBO J. 2011, 30, 2140–2152. [Google Scholar] [CrossRef] [PubMed]
- Middleton, R.E.; Pheasant, D.J.; Miller, C. Homodimeric architecture of a ClC-type chloride ion channel. Nature 1996, 383, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Ludewig, U.; Pusch, M.; Jentsch, T.J. Two physically distinct pores in the dimeric ClC-0 chloride channel. Nature 1996, 383, 340–343. [Google Scholar] [CrossRef]
- Maduke, M.; Pheasant, D.J.; Miller, C. High-level expression, functional reconstitution, and quaternary structure of a prokaryotic ClC-type chloride channel. J. Gen. Physiol. 1999, 114, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Mindell, J.A.; Maduke, M.; Miller, C.; Grigorieff, N. Projection structure of a ClC-type chloride channel at 6.5 Å resolution. Nature 2001, 409, 219–223. [Google Scholar] [CrossRef]
- Dutzler, R.; Campbell, E.B.; Cadene, M.; Chait, B.T.; MacKinnon, R. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature 2002, 415, 287–294. [Google Scholar] [CrossRef]
- Dutzler, R.; Campbell, E.B.; MacKinnon, R. Gating the selectivity filter in ClC chloride channels. Science 2003, 300, 108–112. [Google Scholar] [CrossRef]
- Ponting, C.P. CBS domains in CIC chloride channels implicated in myotonia and nephrolithiasis (kidney stones). J. Mol. Med. 1997, 75, 160–163. [Google Scholar]
- Estévez, R.; Jentsch, T.J. CLC chloride channels: Correlating structure with function. Curr. Opin. Struct. Biol. 2002, 12, 531–539. [Google Scholar] [CrossRef]
- Meyer, S.; Dutzler, R. Crystal structure of the cytoplasmic domain of the chloride channel ClC-0. Structure 2006, 14, 299–307. [Google Scholar] [CrossRef]
- Meyer, S.; Savaresi, S.; Forster, I.C.; Dutzler, R. Nucleotide recognition by the cytoplasmic domain of the human chloride transporter ClC-5. Nat. Struct. Mol. Biol. 2007, 14, 60–67. [Google Scholar] [CrossRef]
- Markovic, S.; Dutzler, R. The structure of the cytoplasmic domain of the chloride channel ClC-Ka reveals a conserved interaction interface. Structure 2007, 15, 715–725. [Google Scholar] [CrossRef]
- Bateman, A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem. Sci. 1997, 22, 12–13. [Google Scholar] [CrossRef]
- Schrecker, M.; Korobenko, J.; Hite, R.K. Cryo-EM structure of the lysosomal chloride-proton exchanger CLC-7 in complex with OSTM1. eLife 2020, 9, e59555. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Campbell, E.B.; MacKinnon, R. Structure of a CLC chloride ion channel by cryo-electron microscopy. Nature 2017, 541, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; MacKinnon, R. Structure of the CLC-1 chloride channel from Homo sapiens. eLife 2018, 7, e36629. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Preisler, S.S.; Zhang, L.; Cui, Y.; Missel, J.W.; Gronberg, C.; Gotfryd, K.; Lindahl, E.; Andersson, M.; Calloe, K.; et al. Structure of the human ClC-1 chloride channel. PLoS Biol. 2019, 17, e3000218. [Google Scholar] [CrossRef]
- Pusch, M.; Ludewig, U.; Rehfeldt, A.; Jentsch, T.J. Gating of the voltage-dependent chloride channel CIC-0 by the permeant anion. Nature 1995, 373, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Traverso, S.; Elia, L.; Pusch, M. Gating competence of constitutively open CLC-0 mutants revealed by the interaction with a small organic Inhibitor. J. Gen. Physiol. 2003, 122, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Zifarelli, G.; Murgia, A.R.; Soliani, P.; Pusch, M. Intracellular proton regulation of ClC-0. J. Gen. Physiol. 2008, 132, 185–198. [Google Scholar] [CrossRef]
- Estévez, R.; Boettger, T.; Stein, V.; Birkenhäger, R.; Otto, E.; Hildebrandt, F.; Jentsch, T.J. Barttin is a Cl− channel beta-subunit crucial for renal Cl− reabsorption and inner ear K+ secretion. Nature 2001, 414, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Jeworutzki, E.; López-Hernández, T.; Capdevila-Nortes, X.; Sirisi, S.; Bengtsson, L.; Montolio, M.; Zifarelli, G.; Arnedo, T.; Müller, C.S.; Schulte, U.; et al. GlialCAM, a protein defective in a leukodystrophy, serves as a ClC-2 Cl− channel auxiliary subunit. Neuron 2012, 73, 951–961. [Google Scholar] [CrossRef]
- Jeworutzki, E.; Lagostena, L.; Elorza-Vidal, X.; Lopez-Hernandez, T.; Estevez, R.; Pusch, M. GlialCAM, a CLC-2 Cl− channel subunit, activates the slow gate of CLC chloride channels. Biophys. J. 2014, 107, 1105–1116. [Google Scholar] [CrossRef]
- Lange, P.F.; Wartosch, L.; Jentsch, T.J.; Fuhrmann, J.C. ClC-7 requires Ostm1 as a beta-subunit to support bone resorption and lysosomal function. Nature 2006, 440, 220–223. [Google Scholar] [CrossRef]
- Stauber, T.; Weinert, S.; Jentsch, T.J. Cell biology and physiology of CLC chloride channels and transporters. Compr. Physiol. 2012, 2, 1701–1744. [Google Scholar] [CrossRef]
- Bose, S.; He, H.; Stauber, T. Neurodegeneration upon dysfunction of endosomal/lysosomal CLC chloride transporters. Front. Cell Dev. Biol. 2021, 9, 639231. [Google Scholar] [CrossRef]
- Günther, W.; Lüchow, A.; Cluzeaud, F.; Vandewalle, A.; Jentsch, T.J. ClC-5, the chloride channel mutated in Dent’s disease, colocalizes with the proton pump in endocytotically active kidney cells. Proc. Natl. Acad. Sci. USA 1998, 95, 8075–8080. [Google Scholar] [CrossRef]
- Günther, W.; Piwon, N.; Jentsch, T.J. The ClC-5 chloride channel knock-out mouse—An animal model for Dent’s disease. Pflügers Arch. 2003, 445, 456–462. [Google Scholar] [CrossRef]
- Jentsch, T.J.; Günther, W. Chloride channels: An emerging molecular picture. Bioessays 1997, 19, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Piwon, N.; Günther, W.; Schwake, M.; Bösl, M.R.; Jentsch, T.J. ClC-5 Cl−-channel disruption impairs endocytosis in a mouse model for Dent’s disease. Nature 2000, 408, 369–373. [Google Scholar] [CrossRef]
- Stobrawa, S.M.; Breiderhoff, T.; Takamori, S.; Engel, D.; Schweizer, M.; Zdebik, A.A.; Bösl, M.R.; Ruether, K.; Jahn, H.; Draguhn, A.; et al. Disruption of ClC-3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron 2001, 29, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Weinert, S.; Jabs, S.; Supanchart, C.; Schweizer, M.; Gimber, N.; Richter, M.; Rademann, J.; Stauber, T.; Kornak, U.; Jentsch, T.J. Lysosomal pathology and osteopetrosis upon loss of H+-driven lysosomal Cl− accumulation. Science 2010, 328, 1401–1403. [Google Scholar] [CrossRef]
- Ishida, Y.; Nayak, S.; Mindell, J.A.; Grabe, M. A model of lysosomal pH regulation. J. Gen. Physiol. 2013, 141, 705–720. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, S.E.; Pearce, S.H.; Fisher, S.E.; Steinmeyer, K.; Schwappach, B.; Scheinman, S.J.; Harding, B.; Bolino, A.; Devoto, M.; Goodyer, P.; et al. A common molecular basis for three inherited kidney stone diseases. Nature 1996, 379, 445–449. [Google Scholar] [CrossRef]
- Kornak, U.; Kasper, D.; Bösl, M.R.; Kaiser, E.; Schweizer, M.; Schulz, A.; Friedrich, W.; Delling, G.; Jentsch, T.J. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell 2001, 104, 205–215. [Google Scholar] [CrossRef]
- Duncan, A.R.; Polovitskaya, M.M.; Gaitan-Penas, H.; Bertelli, S.; VanNoy, G.E.; Grant, P.E.; O’Donnell-Luria, A.; Valivullah, Z.; Lovgren, A.K.; England, E.M.; et al. Unique variants in CLCN3, encoding an endosomal anion/proton exchanger, underlie a spectrum of neurodevelopmental disorders. Am. J. Hum. Genet. 2021, 108, 1450–1465. [Google Scholar] [CrossRef]
- Hu, H.; Haas, S.A.; Chelly, J.; Van Esch, H.; Raynaud, M.; de Brouwer, A.P.; Weinert, S.; Froyen, G.; Frints, S.G.; Laumonnier, F.; et al. X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol. Psychiatry 2016, 21, 133–148. [Google Scholar] [CrossRef]
- Palmer, E.E.; Pusch, M.; Picollo, A.; Forwood, C.; Nguyen, M.H.; Suckow, V.; Gibbons, J.; Hoff, A.; Sigfrid, L.; Megarbane, A.; et al. Functional and clinical studies reveal pathophysiological complexity of CLCN4-related neurodevelopmental condition. Mol. Psychiatry 2023, 28, 668–697. [Google Scholar] [CrossRef] [PubMed]
- Palmer, E.E.; Stuhlmann, T.; Weinert, S.; Haan, E.; Van Esch, H.; Holvoet, M.; Boyle, J.; Leffler, M.; Raynaud, M.; Moraine, C.; et al. De novo and inherited mutations in the X-linked gene CLCN4 are associated with syndromic intellectual disability and behavior and seizure disorders in males and females. Mol. Psychiatry 2016, 23, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Polovitskaya, M.M.; Barbini, C.; Martinelli, D.; Harms, F.L.; Cole, F.S.; Calligari, P.; Bocchinfuso, G.; Stella, L.; Ciolfi, A.; Niceta, M.; et al. A Recurrent Gain-of-Function Mutation in CLCN6, Encoding the ClC-6 Cl−/H+-Exchanger, Causes Early-Onset Neurodegeneration. Am. J. Hum. Genet. 2020, 107, 1062–1077. [Google Scholar] [CrossRef]
- Poët, M.; Kornak, U.; Schweizer, M.; Zdebik, A.A.; Scheel, O.; Hoelter, S.; Wurst, W.; Schmitt, A.; Fuhrmann, J.C.; Planells-Cases, R.; et al. Lysosomal storage disease upon disruption of the neuronal chloride transport protein ClC-6. Proc. Natl. Acad. Sci. USA 2006, 103, 13854–13859. [Google Scholar] [CrossRef]
- Zifarelli, G.; Pusch, M.; Fong, P. Altered voltage-dependence of slowly activating chloride-proton antiport by late endosomal ClC-6 explains distinct neurological disorders. J. Physiol. 2022, 600, 2147–2164. [Google Scholar] [CrossRef]
- He, H.; Cao, X.; Yin, F.; Wu, T.; Stauber, T.; Peng, J. West syndrome caused by a chloride/proton exchange-uncoupling CLCN6 mutation related to autophagic-lysosomal dysfunction. Mol. Neurobiol. 2021, 58, 2990–2999. [Google Scholar] [CrossRef] [PubMed]
- Kasper, D.; Planells-Cases, R.; Fuhrmann, J.C.; Scheel, O.; Zeitz, O.; Ruether, K.; Schmitt, A.; Poët, M.; Steinfeld, R.; Schweizer, M.; et al. Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. EMBO J. 2005, 24, 1079–1091. [Google Scholar] [CrossRef]
- Pangrazio, A.; Pusch, M.; Caldana, E.; Frattini, A.; Lanino, E.; Tamhankar, P.M.; Phadke, S.; Lopez, A.G.; Orchard, P.; Mihci, E.; et al. Molecular and clinical heterogeneity in CLCN7-dependent osteopetrosis: Report of 20 novel mutations. Hum. Mutat. 2010, 31, E1071–E1080. [Google Scholar] [CrossRef] [PubMed]
- Nicoli, E.R.; Weston, M.R.; Hackbarth, M.; Becerril, A.; Larson, A.; Zein, W.M.; Baker, P.R., 2nd; Burke, J.D.; Dorward, H.; Davids, M.; et al. Lysosomal storage and albinism due to effects of a de novo CLCN7 variant on lysosomal acidification. Am. J. Hum. Genet. 2019, 104, 1127–1138. [Google Scholar] [CrossRef]
- Steinmeyer, K.; Schwappach, B.; Bens, M.; Vandewalle, A.; Jentsch, T.J. Cloning and functional expression of rat CLC-5, a chloride channel related to kidney disease. J. Biol. Chem. 1995, 270, 31172–31177. [Google Scholar] [CrossRef]
- Friedrich, T.; Breiderhoff, T.; Jentsch, T.J. Mutational analysis demonstrates that ClC-4 and ClC-5 directly mediate plasma membrane currents. J. Biol. Chem. 1999, 274, 896–902. [Google Scholar] [CrossRef]
- Guzman, R.E.; Grieschat, M.; Fahlke, C.; Alekov, A.K. ClC-3 is an intracellular chloride/proton exchanger with large voltage-dependent nonlinear capacitance. ACS Chem. Neurosci. 2013, 4, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Guzman, R.E.; Miranda-Laferte, E.; Franzen, A.; Fahlke, C. Neuronal ClC-3 splice variants differ in subcellular localizations, but mediate identical transport functions. J. Biol. Chem. 2015, 290, 25851–25862. [Google Scholar] [CrossRef] [PubMed]
- Stauber, T.; Jentsch, T.J. Sorting motifs of the endosomal/lysosomal CLC chloride transporters. J. Biol. Chem. 2010, 285, 34537–34548. [Google Scholar] [CrossRef] [PubMed]
- Zifarelli, G.; Pusch, M. Conversion of the 2 Cl−/1 H+ antiporter ClC-5 in a NO3−/H+ antiporter by a single point mutation. Embo J. 2009, 28, 175–182. [Google Scholar] [CrossRef]
- Li, X.; Shimada, K.; Showalter, L.A.; Weinman, S.A. Biophysical properties of ClC-3 differentiate it from swelling-activated chloride channels in Chinese hamster ovary-K1 cells. J. Biol. Chem. 2000, 275, 35994–35998. [Google Scholar] [CrossRef]
- Zifarelli, G.; Pusch, M. Intracellular regulation of human ClC-5 by adenine nucleotides. EMBO Rep. 2009, 10, 1111–1116. [Google Scholar] [CrossRef]
- De Stefano, S.; Pusch, M.; Zifarelli, G. Extracellular determinants of anion discrimination of the Cl−/H+ antiporter CLC-5. J. Biol. Chem. 2011, 286, 44134–44144. [Google Scholar] [CrossRef]
- Picollo, A.; Pusch, M. Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature 2005, 436, 420–423. [Google Scholar] [CrossRef]
- De Stefano, S.; Pusch, M.; Zifarelli, G. A single point mutation reveals gating of the human ClC-5 Cl−/H+ antiporter. J. Physiol. 2013, 591, 5879–5893. [Google Scholar] [CrossRef]
- Dickerson, L.W.; Bonthius, D.J.; Schutte, B.C.; Yang, B.; Barna, T.J.; Bailey, M.C.; Nehrke, K.; Williamson, R.A.; Lamb, F.S. Altered GABAergic function accompanies hippocampal degeneration in mice lacking ClC-3 voltage-gated chloride channels. Brain Res. 2002, 958, 227–250. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Uchida, S.; Ezaki, J.; Rai, T.; Hayama, A.; Kobayashi, K.; Kida, Y.; Noda, M.; Koike, M.; Uchiyama, Y.; et al. CLC-3 deficiency leads to phenotypes similar to human neuronal ceroid lipofuscinosis. Genes Cells 2002, 7, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Guzman, R.E.; Bungert-Plumke, S.; Franzen, A.; Fahlke, C. Preferential association with ClC-3 permits sorting of ClC-4 into endosomal compartments. J. Biol. Chem. 2017, 292, 19055–19065. [Google Scholar] [CrossRef] [PubMed]
- Weinert, S.; Gimber, N.; Deuschel, D.; Stuhlmann, T.; Puchkov, D.; Farsi, Z.; Ludwig, C.F.; Novarino, G.; Lopez-Cayuqueo, K.I.; Planells-Cases, R.; et al. Uncoupling endosomal CLC chloride/proton exchange causes severe neurodegeneration. EMBO J. 2020, 39, e103358. [Google Scholar] [CrossRef] [PubMed]
- Veeramah, K.R.; Johnstone, L.; Karafet, T.M.; Wolf, D.; Sprissler, R.; Salogiannis, J.; Barth-Maron, A.; Greenberg, M.E.; Stuhlmann, T.; Weinert, S.; et al. Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia 2013, 54, 1270–1281. [Google Scholar] [CrossRef]
- He, H.; Guzman, R.E.; Cao, D.; Sierra-Marquez, J.; Yin, F.; Fahlke, C.; Peng, J.; Stauber, T. The molecular and phenotypic spectrum of CLCN4-related epilepsy. Epilepsia 2021, 62, 1401–1415. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.; Jentsch, T.J. ClC-6 and ClC-7 are two novel broadly expressed members of the CLC chloride channel family. FEBS Lett. 1995, 377, 15–20. [Google Scholar] [CrossRef]
- Ignoul, S.; Simaels, J.; Hermans, D.; Annaert, W.; Eggermont, J. Human ClC-6 is a late endosomal glycoprotein that associates with detergent-resistant lipid domains. PLoS ONE 2007, 2, e474. [Google Scholar] [CrossRef]
- Neagoe, I.; Stauber, T.; Fidzinski, P.; Bergsdorf, E.Y.; Jentsch, T.J. The late endosomal ClC-6 mediates proton/chloride countertransport in heterologous plasma membrane expression. J. Biol. Chem. 2010, 285, 21689–21697. [Google Scholar] [CrossRef]
- Kobertz, W.R. Want to hear ClC-6 sing? Push your amp to eleven. J. Physiol. 2022, 600, 2019–2020. [Google Scholar] [CrossRef]
- Pusch, M.; Zifarelli, G. Large transient capacitive currents in wild-type lysosomal Cl−/H+ antiporter ClC-7 and residual transport activity in the proton glutamate mutant E312A. J. Gen. Physiol. 2021, 153, e202012583. [Google Scholar] [CrossRef] [PubMed]
- Graves, A.R.; Curran, P.K.; Smith, C.L.; Mindell, J.A. The Cl−/H+ antiporter ClC-7 is the primary chloride permeation pathway in lysosomes. Nature 2008, 453, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, C.F.; Ullrich, F.; Leisle, L.; Stauber, T.; Jentsch, T.J. Common gating of both CLC transporter subunits underlies voltage-dependent activation of the 2Cl−/1H+ exchanger ClC-7/Ostm1. J. Biol. Chem. 2013, 288, 28611–28619. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Lippiat, J.D. Voltage-dependent charge movement associated with activation of the CLC-5 2Cl−/1H+ exchanger. Faseb J. 2010, 24, 3696–3705. [Google Scholar] [CrossRef]
- Zifarelli, G.; De Stefano, S.; Zanardi, I.; Pusch, M. On the mechanism of gating charge movement of ClC-5, a human Cl−/H+ antiporter. Biophys. J. 2012, 102, 2060–2069. [Google Scholar] [CrossRef]
- Chalhoub, N.; Benachenhou, N.; Rajapurohitam, V.; Pata, M.; Ferron, M.; Frattini, A.; Villa, A.; Vacher, J. Grey-lethal mutation induces severe malignant autosomal recessive osteopetrosis in mouse and human. Nat. Med. 2003, 9, 399–406. [Google Scholar] [CrossRef]
- Pata, M.; Héraud, C.; Vacher, J. OSTM1 bone defect reveals an intercellular hematopoietic crosstalk. J. Biol. Chem. 2008, 283, 30522–30530. [Google Scholar] [CrossRef]
- Wartosch, L.; Fuhrmann, J.C.; Schweizer, M.; Stauber, T.; Jentsch, T.J. Lysosomal degradation of endocytosed proteins depends on the chloride transport protein ClC-7. FASEB J. 2009, 23, 4056–4068. [Google Scholar] [CrossRef]
- Sartelet, A.; Stauber, T.; Coppieters, W.; Ludwig, C.F.; Fasquelle, C.; Druet, T.; Zhang, Z.; Ahariz, N.; Cambisano, N.; Jentsch, T.J.; et al. A missense mutation accelerating the gating of the lysosomal Cl−/H+-exchanger ClC-7/Ostm1 causes osteopetrosis with gingival hamartomas in cattle. Dis. Model Mech. 2014, 7, 119–128. [Google Scholar] [CrossRef]
- Di Zanni, E.; Palagano, E.; Lagostena, L.; Strina, D.; Rehman, A.; Abinun, M.; De Somer, L.; Martire, B.; Brown, J.; Kariminejad, A.; et al. Pathobiologic Mechanisms of Neurodegeneration in Osteopetrosis Derived From Structural and Functional Analysis of 14 ClC-7 Mutants. J. Bone Miner. Res. 2021, 36, 531–545. [Google Scholar] [CrossRef]
- Leray, X.; Hilton, J.K.; Nwangwu, K.; Becerril, A.; Mikusevic, V.; Fitzgerald, G.; Amin, A.; Weston, M.R.; Mindell, J.A. Tonic inhibition of the chloride/proton antiporter ClC-7 by PI(3,5)P2 is crucial for lysosomal pH maintenance. eLife 2022, 11, e74136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, Y.; Zhang, B.; Zhou, J.; Li, T.; Liu, Z.; Li, Y.; Yang, M. Molecular insights into the human CLC-7/Ostm1 transporter. Sci. Adv. 2020, 6, eabb4747. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Z.; Zeziulia, M.; Kwon, W.; Jentsch, T.J.; Grinstein, S.; Freeman, S.A. ClC-7 drives intraphagosomal chloride accumulation to support hydrolase activity and phagosome resolution. J. Cell Biol. 2023, 222, e202208155. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).