
Targeting Ion Channels and Purkinje Neuron Intrinsic Membrane Excitability as a Therapeutic Strategy for Cerebellar Ataxia


Abstract
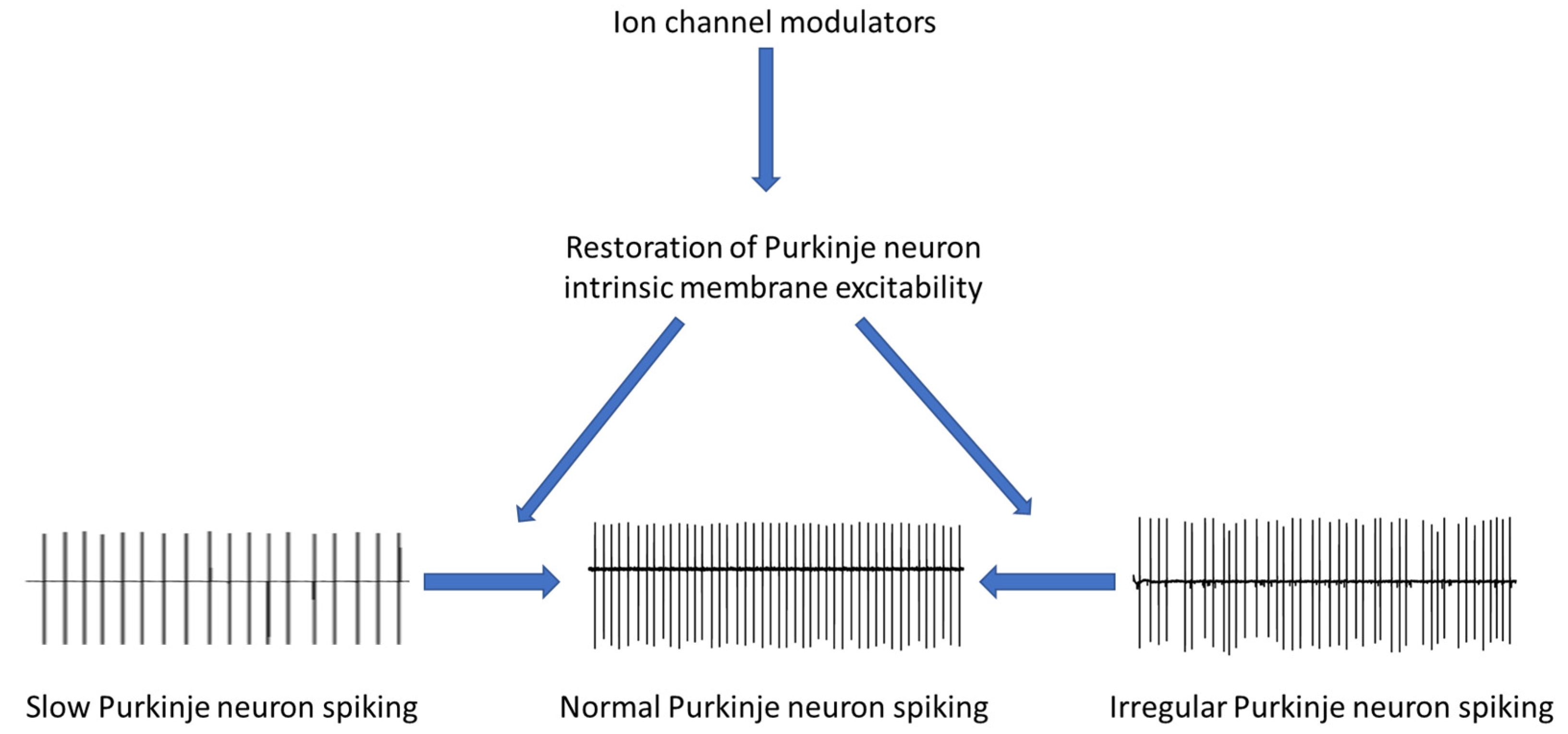
:1. Introduction
2. Ion Channel Gene Mutations That Cause Ataxia
2.1. Voltage-Gated Sodium Channels

{kind=link}
| Types | Ion Channel | Disease | Cerebellar Pathology |
|---|---|---|---|
| Voltage-gated sodium channels | Nav1.1 | Dravet syndrome | Cerebellar atrophy on MRI; Cerebellar atrophy with loss of Purkinje neuron on post-mortem tissue [30]. |
| Nav1.6 | Epileptic encephalopathy | Cerebellar atrophy on MRI in one patient [31]. | |
| SCA27B | Severe cerebellar vermis atrophy on MRI and post-mortem tissue. Loss of Purkinje neurons on post-mortem tissue [29]. | ||
| Voltage-gated potassium channels | Kv1.1 | EA1 | No cerebellar pathology was reported. |
| Kv1.2 | Ataxia associated with epileptic encephalopathy | Cerebellar atrophy on MRI in a subset of patients [32]. | |
| Kv1.6 | SCA3 | See above. | |
| Kv3.3 | SCA13 | Cerebellar atrophy on MRI [33]. | |
| SCA3 | Mild cerebellar atrophy with enlarged 4th ventricle [4]; Degeneration of the cerebellar fastigial nucleus [3]. | ||
| SCA1,2 | See above. | ||
| Kv4.3 | SCA19, 22 | Severe Purkinje neuron degeneration in cerebellar autopsy. Mild cerebellar atrophy on MRI in some patients [34,35]. | |
| SCA1 | See above. | ||
| Calcium-activated potassium channels | BK | Liang-Wang syndrome | Cerebral atrophy involving the vermis and hemisphere on MRI [36,37]. |
| SCA1 | Global cerebellar volume loss on MRI [4]. Cerebellar atrophy on biopsy and degeneration of cerebellar Purkinje neurons, and the cerebellar fastigial nucleus [3]. | ||
| SCA2 | Cerebellar atrophy on MRI [38]; Global cerebellar volume loss involving both the vermis and cerebellar hemispheres on MRI [4]; Degeneration of cerebellar Purkinje neurons and the cerebellar fastigial nucleus [3]. | ||
| SCA7 | Cerebellar atrophy mainly involves the superior part of the vermis on MRI [4]; Degeneration of cerebellar Purkinje neurons and the cerebellar fastigial nucleus [3]. | ||
| SK2 | Dominant neurodevelopmental movement disorders; autosomal-dominant tremulous myoclonus-dystonia | Cerebellar atrophy on MRI in one case [39] (Mochel, personal communication) | |
| Voltage-gated calcium channels | Cav2.1 | SCA6 | Cerebellar atrophy involving the vermis and the hemisphere [4]; Degeneration of cerebellar Purkinje neurons. |
| EA2 | Cerebellar vermis atrophy on MRI [40]. | ||
| Cav3.1 | SCA42 | Cerebellar atrophy on MRI [41,42]. | |
| SCA1,2,7 | See above. | ||
| Other calcium channels and calcium pumps | TRPC3 | SCA41 | Mild cerebellar vermis atrophy on MRI [43]. |
| IP3R1 | SCA15 | Cerebellar atrophy on MRI and CT [44]. | |
| SCA29 | Cerebellar atrophy on MRI [45]. | ||
| SCA2, 3 | See above. | ||
| PMCA2 | Congenital cerebellar ataxia | Cerebellar atrophy on MRI [46]. | |
| PMCA3 | X-linked congenital cerebellar ataxia | Volume loss of cerebellar hemisphere and vermis on MRI [47]. |
2.2. Voltage-Gated Potassium Channels
2.2.1. Kv1 Channel Family
Kv1.1
Kv1.2
Kv1.6
2.2.2. Kv3.3
2.2.3. Kv4.3
2.3. Calcium-Activated Potassium Channels
2.3.1. Large-Conductance Calcium-Activated Potassium (BK) Channels
2.3.2. Small-Conductance Calcium-Activated Potassium (SK2) Channels
2.4. Voltage-Gated Calcium Channels
2.4.1. Cav2.1
2.4.2. Cav3.1
2.5. Other Calcium Channels and Calcium Pumps
2.5.1. TRPC3
2.5.2. IP3R1
2.5.3. PMCAs
3. Emerging Therapies for Cerebellar Ataxia Impinge on Ion Channels
3.1. Non-Pharmacological Approaches in Cerebellar Ataxia
3.1.1. Rehabilitation
3.1.2. Gene Suppression Strategies
3.2. Pharmacological Approaches in Cerebellar Ataxia
3.2.1. Omaveloxolone
3.2.2. 4-Aminopyridine
3.2.3. Chlorzoxazone and Baclofen
3.3. Other Approaches
Cerebellar Stimulation: Deep Brain Stimulation and Non-Invasive Cerebellar Stimulation
4. Discussion: Is There a “Levodopa” for Cerebellar Ataxia?
4.1. Voltage-Gated Sodium Channels
4.2. Voltage-Gated Potassium Channels
4.3. Calcium-Activated Potassium Channels
4.4. Calcium Channels and Calcium Pumps
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Durr, A. Autosomal dominant cerebellar ataxias: Polyglutamine expansions and beyond. Lancet Neurol. 2010, 9, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T. Purkinje Neurons: Development, Morphology, and Function. Cerebellum 2018, 17, 699–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidel, K.; Siswanto, S.; Brunt, E.R.; den Dunnen, W.; Korf, H.W.; Rub, U. Brain pathology of spinocerebellar ataxias. Acta Neuropathol. 2012, 124, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Cocozza, S.; Pontillo, G.; De Michele, G.; Di Stasi, M.; Guerriero, E.; Perillo, T.; Pane, C.; De Rosa, A.; Ugga, L.; Brunetti, A. Conventional MRI findings in hereditary degenerative ataxias: A pictorial review. Neuroradiology 2021, 63, 983–999. [Google Scholar] [CrossRef]
- Hourez, R.; Servais, L.; Orduz, D.; Gall, D.; Millard, I.; de Kerchove d’Exaerde, A.; Cheron, G.; Orr, H.T.; Pandolfo, M.; Schiffmann, S.N. Aminopyridines correct early dysfunction and delay neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 11795–11807. [Google Scholar] [CrossRef] [Green Version]
- Dell’Orco, J.M.; Wasserman, A.H.; Chopra, R.; Ingram, M.A.; Hu, Y.S.; Singh, V.; Wulff, H.; Opal, P.; Orr, H.T.; Shakkottai, V.G. Neuronal Atrophy Early in Degenerative Ataxia Is a Compensatory Mechanism to Regulate Membrane Excitability. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 11292–11307. [Google Scholar] [CrossRef] [Green Version]
- Stoyas, C.A.; Bushart, D.D.; Switonski, P.M.; Ward, J.M.; Alaghatta, A.; Tang, M.B.; Niu, C.; Wadhwa, M.; Huang, H.; Savchenko, A.; et al. Nicotinamide Pathway-Dependent Sirt1 Activation Restores Calcium Homeostasis to Achieve Neuroprotection in Spinocerebellar Ataxia Type 7. Neuron 2020, 105, 630–644.e9. [Google Scholar] [CrossRef]
- Bushart, D.D.; Chopra, R.; Singh, V.; Murphy, G.G.; Wulff, H.; Shakkottai, V.G. Targeting potassium channels to treat cerebellar ataxia. Ann. Clin. Transl. Neurol. 2018, 5, 297–314. [Google Scholar] [CrossRef]
- Bushart, D.D.; Huang, H.; Man, L.J.; Morrison, L.M.; Shakkottai, V.G. A Chlorzoxazone-Baclofen Combination Improves Cerebellar Impairment in Spinocerebellar Ataxia Type 1. Mov. Disord. Off. J. Mov. Disord. Soc. 2021, 36, 622–631. [Google Scholar] [CrossRef]
- Jayabal, S.; Chang, H.H.; Cullen, K.E.; Watt, A.J. 4-aminopyridine reverses ataxia and cerebellar firing deficiency in a mouse model of spinocerebellar ataxia type 6. Sci. Rep. 2016, 6, 29489. [Google Scholar] [CrossRef] [Green Version]
- Kasumu, A.W.; Hougaard, C.; Rode, F.; Jacobsen, T.A.; Sabatier, J.M.; Eriksen, B.L.; Strobaek, D.; Liang, X.; Egorova, P.; Vorontsova, D.; et al. Selective positive modulator of calcium-activated potassium channels exerts beneficial effects in a mouse model of spinocerebellar ataxia type 2. Chem. Biol. 2012, 19, 1340–1353. [Google Scholar] [CrossRef] [Green Version]
- Cook, A.A.; Jayabal, S.; Sheng, J.; Fields, E.; Leung, T.C.S.; Quilez, S.; McNicholas, E.; Lau, L.; Huang, S.; Watt, A.J. Activation of TrkB-Akt signaling rescues deficits in a mouse model of SCA6. Sci. Adv. 2022, 8, eabh3260. [Google Scholar] [CrossRef]
- Cook, A.A.; Fields, E.; Watt, A.J. Losing the Beat: Contribution of Purkinje Cell Firing Dysfunction to Disease, and Its Reversal. Neuroscience 2021, 462, 247–261. [Google Scholar] [CrossRef]
- Raman, I.M.; Bean, B.P. Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J. Neurosci. Off. J. Soc. Neurosci. 1999, 19, 1663–1674. [Google Scholar] [CrossRef] [Green Version]
- Bushart, D.D.; Shakkottai, V.G. Ion channel dysfunction in cerebellar ataxia. Neurosci. Lett. 2019, 688, 41–48. [Google Scholar] [CrossRef]
- Kalume, F.; Yu, F.H.; Westenbroek, R.E.; Scheuer, T.; Catterall, W.A. Reduced sodium current in Purkinje neurons from Nav1.1 mutant mice: Implications for ataxia in severe myoclonic epilepsy in infancy. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 11065–11074. [Google Scholar] [CrossRef] [Green Version]
- Khaliq, Z.M.; Gouwens, N.W.; Raman, I.M. The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: An experimental and modeling study. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 4899–4912. [Google Scholar] [CrossRef] [Green Version]
- Veeramah, K.R.; O’Brien, J.E.; Meisler, M.H.; Cheng, X.; Dib-Hajj, S.D.; Waxman, S.G.; Talwar, D.; Girirajan, S.; Eichler, E.E.; Restifo, L.L.; et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am. J. Hum. Genet. 2012, 90, 502–510. [Google Scholar] [CrossRef] [Green Version]
- Kohrman, D.C.; Harris, J.B.; Meisler, M.H. Mutation detection in the med and medJ alleles of the sodium channel Scn8a. Unusual splicing due to a minor class AT-AC intron. J. Biol. Chem. 1996, 271, 17576–17581. [Google Scholar] [CrossRef] [Green Version]
- Raman, I.M.; Sprunger, L.K.; Meisler, M.H.; Bean, B.P. Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron 1997, 19, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Dravet, C.; Bureau, M.; Oguni, H.; Fukuyama, Y.; Cokar, O. Severe myoclonic epilepsy in infancy: Dravet syndrome. Adv. Neurol. 2005, 95, 71–102. [Google Scholar] [PubMed]
- Selmer, K.K.; Eriksson, A.S.; Brandal, K.; Egeland, T.; Tallaksen, C.; Undlien, D.E. Parental SCN1A mutation mosaicism in familial Dravet syndrome. Clin. Genet. 2009, 76, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Weuring, W.J.; Singh, S.; Volkers, L.; Rook, M.B.; van ‘t Slot, R.H.; Bosma, M.; Inserra, M.; Vetter, I.; Verhoeven-Duif, N.M.; Braun, K.P.J.; et al. NaV1.1 and NaV1.6 selective compounds reduce the behavior phenotype and epileptiform activity in a novel zebrafish model for Dravet Syndrome. PLoS ONE 2020, 15, e0219106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brusse, E.; de Koning, I.; Maat-Kievit, A.; Oostra, B.A.; Heutink, P.; van Swieten, J.C. Spinocerebellar ataxia associated with a mutation in the fibroblast growth factor 14 gene (SCA27): A new phenotype. Mov. Disord. Off. J. Mov. Disord. Soc. 2006, 21, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Bardgett, M.E.; Wong, M.; Wozniak, D.F.; Lou, J.; McNeil, B.D.; Chen, C.; Nardi, A.; Reid, D.C.; Yamada, K.; et al. Ataxia and paroxysmal dyskinesia in mice lacking axonally transported FGF14. Neuron 2002, 35, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Shakkottai, V.G.; Xiao, M.; Xu, L.; Wong, M.; Nerbonne, J.M.; Ornitz, D.M.; Yamada, K.A. FGF14 regulates the intrinsic excitability of cerebellar Purkinje neurons. Neurobiol. Dis. 2009, 33, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Bosch, M.K.; Carrasquillo, Y.; Ransdell, J.L.; Kanakamedala, A.; Ornitz, D.M.; Nerbonne, J.M. Intracellular FGF14 (iFGF14) Is Required for Spontaneous and Evoked Firing in Cerebellar Purkinje Neurons and for Motor Coordination and Balance. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 6752–6769. [Google Scholar] [CrossRef] [Green Version]
- Laezza, F.; Gerber, B.R.; Lou, J.Y.; Kozel, M.A.; Hartman, H.; Craig, A.M.; Ornitz, D.M.; Nerbonne, J.M. The FGF14(F145S) mutation disrupts the interaction of FGF14 with voltage-gated Na+ channels and impairs neuronal excitability. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 12033–12044. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, D.; Danzi, M.C.; Wilke, C.; Renaud, M.; Fazal, S.; Dicaire, M.J.; Scriba, C.K.; Ashton, C.; Yanick, C.; Beijer, D.; et al. Deep Intronic FGF14 GAA Repeat Expansion in Late-Onset Cerebellar Ataxia. N. Engl. J. Med. 2023, 388, 128–141. [Google Scholar] [CrossRef]
- Catarino, C.B.; Liu, J.Y.; Liagkouras, I.; Gibbons, V.S.; Labrum, R.W.; Ellis, R.; Woodward, C.; Davis, M.B.; Smith, S.J.; Cross, J.H.; et al. Dravet syndrome as epileptic encephalopathy: Evidence from long-term course and neuropathology. Brain 2011, 134, 2982–3010. [Google Scholar] [CrossRef] [Green Version]
- Ohba, C.; Kato, M.; Takahashi, S.; Lerman-Sagie, T.; Lev, D.; Terashima, H.; Kubota, M.; Kawawaki, H.; Matsufuji, M.; Kojima, Y.; et al. Early onset epileptic encephalopathy caused by de novo SCN8A mutations. Epilepsia 2014, 55, 994–1000. [Google Scholar] [CrossRef]
- Masnada, S.; Hedrich, U.B.S.; Gardella, E.; Schubert, J.; Kaiwar, C.; Klee, E.W.; Lanpher, B.C.; Gavrilova, R.H.; Synofzik, M.; Bast, T.; et al. Clinical spectrum and genotype-phenotype associations of KCNA2-related encephalopathies. Brain 2017, 140, 2337–2354. [Google Scholar] [CrossRef] [Green Version]
- Figueroa, K.P.; Minassian, N.A.; Stevanin, G.; Waters, M.; Garibyan, V.; Forlani, S.; Strzelczyk, A.; Burk, K.; Brice, A.; Durr, A.; et al. KCNC3: Phenotype, mutations, channel biophysics-a study of 260 familial ataxia patients. Hum. Mutat. 2010, 31, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.C.; Durr, A.; Majczenko, K.; Huang, Y.H.; Liu, Y.C.; Lien, C.C.; Tsai, P.C.; Ichikawa, Y.; Goto, J.; Monin, M.L.; et al. Mutations in KCND3 cause spinocerebellar ataxia type 22. Ann. Neurol. 2012, 72, 859–869. [Google Scholar] [CrossRef] [Green Version]
- Duarri, A.; Jezierska, J.; Fokkens, M.; Meijer, M.; Schelhaas, H.J.; den Dunnen, W.F.; van Dijk, F.; Verschuuren-Bemelmans, C.; Hageman, G.; van de Vlies, P.; et al. Mutations in potassium channel kcnd3 cause spinocerebellar ataxia type 19. Ann. Neurol. 2012, 72, 870–880. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Li, X.; Moutton, S.; Schrier Vergano, S.A.; Cogne, B.; de Saint-Martin, A.; Hurst, A.C.E.; Hu, Y.; Bodamer, O.; Thevenon, J.; et al. De novo loss-of-function KCNMA1 variants are associated with a new multiple malformation syndrome and a broad spectrum of developmental and neurological phenotypes. Hum. Mol. Genet. 2019, 28, 2937–2951. [Google Scholar] [CrossRef]
- Liang, L.; Liu, H.; Bartholdi, D.; van Haeringen, A.; Fernandez-Jaen, A.; Peeters, E.E.A.; Xiong, H.; Bai, X.; Xu, C.; Ke, T.; et al. Identification and functional analysis of two new de novo KCNMA1 variants associated with Liang-Wang syndrome. Acta Physiol. 2022, 235, e13800. [Google Scholar] [CrossRef]
- Nigri, A.; Sarro, L.; Mongelli, A.; Pinardi, C.; Porcu, L.; Castaldo, A.; Ferraro, S.; Grisoli, M.; Bruzzone, M.G.; Gellera, C.; et al. Progression of Cerebellar Atrophy in Spinocerebellar Ataxia Type 2 Gene Carriers: A Longitudinal MRI Study in Preclinical and Early Disease Stages. Front. Neurol. 2020, 11, 616419. [Google Scholar] [CrossRef]
- Mochel, F.; Rastetter, A.; Ceulemans, B.; Platzer, K.; Yang, S.; Shinde, D.N.; Helbig, K.L.; Lopergolo, D.; Mari, F.; Renieri, A.; et al. Variants in the SK2 channel gene (KCNN2) lead to dominant neurodevelopmental movement disorders. Brain 2020, 143, 3564–3573. [Google Scholar] [CrossRef]
- Yuan, X.; Zheng, Y.; Gao, F.; Sun, W.; Wang, Z.; Zhao, G. Case Report: A Novel CACNA1A Mutation Caused Flunarizine-Responsive Type 2 Episodic Ataxia and Hemiplegic Migraine with Abnormal MRI of Cerebral White Matter. Front. Neurol. 2022, 13, 899813. [Google Scholar] [CrossRef]
- Li, X.; Zhou, C.; Cui, L.; Zhu, L.; Du, H.; Liu, J.; Wang, C.; Fang, S. A case of a novel CACNA1G mutation from a Chinese family with SCA42: A case report and literature review. Medicine 2018, 97, e12148. [Google Scholar] [CrossRef] [PubMed]
- Son, H.; Yoon, J.G.; Kim, M.J.; Moon, J.; Kim, H.J. First Cases of Spinocerebellar Ataxia 42 in Two Korean Families. J. Mov. Disord. 2023, 16, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Fogel, B.L.; Hanson, S.M.; Becker, E.B. Do mutations in the murine ataxia gene TRPC3 cause cerebellar ataxia in humans? Mov. Disord. Off. J. Mov. Disord. Soc. 2015, 30, 284–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marelli, C.; van de Leemput, J.; Johnson, J.O.; Tison, F.; Thauvin-Robinet, C.; Picard, F.; Tranchant, C.; Hernandez, D.G.; Huttin, B.; Boulliat, J.; et al. SCA15 due to large ITPR1 deletions in a cohort of 333 white families with dominant ataxia. Arch. Neurol. 2011, 68, 637–643. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, M.; Ohba, C.; Iai, M.; Hirabayashi, S.; Osaka, H.; Hiraide, T.; Saitsu, H.; Matsumoto, N. Sporadic infantile-onset spinocerebellar ataxia caused by missense mutations of the inositol 1,4,5-triphosphate receptor type 1 gene. J. Neurol. 2015, 262, 1278–1284. [Google Scholar] [CrossRef]
- Vicario, M.; Zanni, G.; Vallese, F.; Santorelli, F.; Grinzato, A.; Cieri, D.; Berto, P.; Frizzarin, M.; Lopreiato, R.; Zonta, F.; et al. A V1143F mutation in the neuronal-enriched isoform 2 of the PMCA pump is linked with ataxia. Neurobiol. Dis. 2018, 115, 157–166. [Google Scholar] [CrossRef]
- Feyma, T.; Ramsey, K.; Group, C.R.R.; Huentelman, M.J.; Craig, D.W.; Padilla-Lopez, S.; Narayanan, V.; Kruer, M.C. Dystonia in ATP2B3-associated X-linked spinocerebellar ataxia. Mov. Disord. Off. J. Mov. Disord. Soc. 2016, 31, 1752–1753. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.H.; Shin, C.M.; Kim, M.J.; Cha, C.I. Immunohistochemical study on the distribution of six members of the Kv1 channel subunits in the rat basal ganglia. Brain Res. 2000, 875, 164–170. [Google Scholar] [CrossRef]
- Bushart, D.D.; Zalon, A.J.; Zhang, H.; Morrison, L.M.; Guan, Y.; Paulson, H.L.; Shakkottai, V.G.; McLoughlin, H.S. Antisense Oligonucleotide Therapy Targeted Against ATXN3 Improves Potassium Channel-Mediated Purkinje Neuron Dysfunction in Spinocerebellar Ataxia Type 3. Cerebellum 2021, 20, 41–53. [Google Scholar] [CrossRef]
- Graves, T.D.; Cha, Y.H.; Hahn, A.F.; Barohn, R.; Salajegheh, M.K.; Griggs, R.C.; Bundy, B.N.; Jen, J.C.; Baloh, R.W.; Hanna, M.G.; et al. Episodic ataxia type 1: Clinical characterization, quality of life and genotype-phenotype correlation. Brain 2014, 137, 1009–1018. [Google Scholar] [CrossRef] [Green Version]
- D’Adamo, M.C.; Hasan, S.; Guglielmi, L.; Servettini, I.; Cenciarini, M.; Catacuzzeno, L.; Franciolini, F. New insights into the pathogenesis and therapeutics of episodic ataxia type 1. Front. Cell. Neurosci. 2015, 9, 317. [Google Scholar] [CrossRef] [Green Version]
- Miceli, F.; Guerrini, R.; Nappi, M.; Soldovieri, M.V.; Cellini, E.; Gurnett, C.A.; Parmeggiani, L.; Mei, D.; Taglialatela, M. Distinct epilepsy phenotypes and response to drugs in KCNA1 gain- and loss-of function variants. Epilepsia 2022, 63, e7–e14. [Google Scholar] [CrossRef]
- Muller, P.; Takacs, D.S.; Hedrich, U.B.S.; Coorg, R.; Masters, L.; Glinton, K.E.; Dai, H.; Cokley, J.A.; Riviello, J.J.; Lerche, H.; et al. KCNA1 gain-of-function epileptic encephalopathy treated with 4-aminopyridine. Ann. Clin. Transl. Neurol. 2023, 10, 656–663. [Google Scholar] [CrossRef]
- Syrbe, S.; Hedrich, U.B.S.; Riesch, E.; Djemie, T.; Muller, S.; Moller, R.S.; Maher, B.; Hernandez-Hernandez, L.; Synofzik, M.; Caglayan, H.S.; et al. De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat. Genet. 2015, 47, 393–399. [Google Scholar] [CrossRef]
- Hedrich, U.B.S.; Lauxmann, S.; Wolff, M.; Synofzik, M.; Bast, T.; Binelli, A.; Serratosa, J.M.; Martinez-Ulloa, P.; Allen, N.M.; King, M.D.; et al. 4-Aminopyridine is a promising treatment option for patients with gain-of-function KCNA2-encephalopathy. Sci. Transl. Med. 2021, 13, eaaz4957. [Google Scholar] [CrossRef]
- Xie, G.; Harrison, J.; Clapcote, S.J.; Huang, Y.; Zhang, J.Y.; Wang, L.Y.; Roder, J.C. A new Kv1.2 channelopathy underlying cerebellar ataxia. J. Biol. Chem. 2010, 285, 32160–32173. [Google Scholar] [CrossRef] [Green Version]
- Salpietro, V.; Galassi Deforie, V.; Efthymiou, S.; O’Connor, E.; Marce-Grau, A.; Maroofian, R.; Striano, P.; Zara, F.; Morrow, M.M.; Group, S.S.; et al. De novo KCNA6 variants with attenuated K(V) 1.6 channel deactivation in patients with epilepsy. Epilepsia 2023, 64, 443–455. [Google Scholar] [CrossRef]
- Waters, M.F.; Minassian, N.A.; Stevanin, G.; Figueroa, K.P.; Bannister, J.P.; Nolte, D.; Mock, A.F.; Evidente, V.G.; Fee, D.B.; Muller, U.; et al. Mutations in voltage-gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat. Genet. 2006, 38, 447–451. [Google Scholar] [CrossRef]
- Zagha, E.; Manita, S.; Ross, W.N.; Rudy, B. Dendritic Kv3.3 potassium channels in cerebellar purkinje cells regulate generation and spatial dynamics of dendritic Ca2+ spikes. J. Neurophysiol. 2010, 103, 3516–3525. [Google Scholar] [CrossRef] [Green Version]
- Hurlock, E.C.; McMahon, A.; Joho, R.H. Purkinje-cell-restricted restoration of Kv3.3 function restores complex spikes and rescues motor coordination in Kcnc3 mutants. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 4640–4648. [Google Scholar] [CrossRef] [Green Version]
- Hurlock, E.C.; Bose, M.; Pierce, G.; Joho, R.H. Rescue of motor coordination by Purkinje cell-targeted restoration of Kv3.3 channels in Kcnc3-null mice requires Kcnc1. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 15735–15744. [Google Scholar] [CrossRef] [Green Version]
- Joho, R.H.; Street, C.; Matsushita, S.; Knopfel, T. Behavioral motor dysfunction in Kv3-type potassium channel-deficient mice. Genes Brain Behav. 2006, 5, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Quraishi, I.H.; McClure, H.; Williams, L.A.; Cheng, Y.; Kale, S.; Dempsey, G.T.; Agrawal, S.; Gerber, D.J.; McManus, O.B.; et al. Suppression of Kv3.3 channels by antisense oligonucleotides reverses biochemical effects and motor impairment in spinocerebellar ataxia type 13 mice. FASEB J. 2021, 35, e22053. [Google Scholar] [CrossRef]
- Zhang, Y.; Kaczmarek, L.K. Kv3.3 potassium channels and spinocerebellar ataxia. J. Physiol. 2016, 594, 4677–4684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chopra, R.; Bushart, D.D.; Cooper, J.P.; Yellajoshyula, D.; Morrison, L.M.; Huang, H.; Handler, H.P.; Man, L.J.; Dansithong, W.; Scoles, D.R.; et al. Altered Capicua expression drives regional Purkinje neuron vulnerability through ion channel gene dysregulation in spinocerebellar ataxia type 1. Hum. Mol. Genet. 2020, 29, 3249–3265. [Google Scholar] [CrossRef] [PubMed]
- Shakkottai, V.G.; do Carmo Costa, M.; Dell’Orco, J.M.; Sankaranarayanan, A.; Wulff, H.; Paulson, H.L. Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 13002–13014. [Google Scholar] [CrossRef]
- Dell’Orco, J.M.; Pulst, S.M.; Shakkottai, V.G. Potassium channel dysfunction underlies Purkinje neuron spiking abnormalities in spinocerebellar ataxia type 2. Hum. Mol. Genet. 2017, 26, 3935–3945. [Google Scholar] [CrossRef] [Green Version]
- Alfaro-Ruiz, R.; Aguado, C.; Martin-Belmonte, A.; Moreno-Martinez, A.E.; Lujan, R. Cellular and Subcellular Localisation of Kv4-Associated KChIP Proteins in the Rat Cerebellum. Int. J. Mol. Sci. 2020, 21, 6403. [Google Scholar] [CrossRef]
- Hsiao, C.T.; Tropea, T.F.; Fu, S.J.; Bardakjian, T.M.; Gonzalez-Alegre, P.; Soong, B.W.; Tang, C.Y.; Jeng, C.J. Rare Gain-of-Function KCND3 Variant Associated with Cerebellar Ataxia, Parkinsonism, Cognitive Dysfunction, and Brain Iron Accumulation. Int. J. Mol. Sci. 2021, 22, 8247. [Google Scholar] [CrossRef]
- Du, X.; Carvalho-de-Souza, J.L.; Wei, C.; Carrasquel-Ursulaez, W.; Lorenzo, Y.; Gonzalez, N.; Kubota, T.; Staisch, J.; Hain, T.; Petrossian, N.; et al. Loss-of-function BK channel mutation causes impaired mitochondria and progressive cerebellar ataxia. Proc. Natl. Acad. Sci. USA 2020, 117, 6023–6034. [Google Scholar] [CrossRef]
- Srinivasan, S.R.; Huang, H.; Chang, W.C.; Nasburg, J.A.; Nguyen, H.M.; Strassmaier, T.; Wulff, H.; Shakkottai, V.G. Discovery of Novel Activators of Large-Conductance Calcium-Activated Potassium Channels for the Treatment of Cerebellar Ataxia. Mol. Pharm. 2022, 102, 438–449. [Google Scholar] [CrossRef]
- Chen, X.; Kovalchuk, Y.; Adelsberger, H.; Henning, H.A.; Sausbier, M.; Wietzorrek, G.; Ruth, P.; Yarom, Y.; Konnerth, A. Disruption of the olivo-cerebellar circuit by Purkinje neuron-specific ablation of BK channels. Proc. Natl. Acad. Sci. USA 2010, 107, 12323–12328. [Google Scholar] [CrossRef] [Green Version]
- Cheron, G.; Marquez-Ruiz, J.; Cheron, J.; Prigogine, C.; Ammann, C.; Lukowski, R.; Ruth, P.; Dan, B. Purkinje cell BKchannel ablation induces abnormal rhythm in deep cerebellar nuclei and prevents LTD. Sci. Rep. 2018, 8, 4220. [Google Scholar] [CrossRef] [Green Version]
- Du, W.; Bautista, J.F.; Yang, H.; Diez-Sampedro, A.; You, S.A.; Wang, L.; Kotagal, P.; Luders, H.O.; Shi, J.; Cui, J.; et al. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat. Genet. 2005, 37, 733–738. [Google Scholar] [CrossRef]
- Zhang, G.; Gibson, R.A.; McDonald, M.; Liang, P.; Kang, P.W.; Shi, J.; Yang, H.; Cui, J.; Mikati, M.A. A Gain-of-Function Mutation in KCNMA1 Causes Dystonia Spells Controlled with Stimulant Therapy. Mov. Disord. Off. J. Mov. Disord. Soc. 2020, 35, 1868–1873. [Google Scholar] [CrossRef]
- Li, X.; Poschmann, S.; Chen, Q.; Fazeli, W.; Oundjian, N.J.; Snoeijen-Schouwenaars, F.M.; Fricke, O.; Kamsteeg, E.J.; Willemsen, M.; Wang, Q.K. De novo BK channel variant causes epilepsy by affecting voltage gating but not Ca(2+) sensitivity. Eur. J. Hum. Genet. 2018, 26, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Moldenhauer, H.J.; Matychak, K.K.; Meredith, A.L. Comparative gain-of-function effects of the KCNMA1-N999S mutation on human BK channel properties. J. Neurophysiol. 2020, 123, 560–570. [Google Scholar] [CrossRef] [Green Version]
- Brenner, R.; Chen, Q.H.; Vilaythong, A.; Toney, G.M.; Noebels, J.L.; Aldrich, R.W. BK channel beta4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nat. Neurosci. 2005, 8, 1752–1759. [Google Scholar] [CrossRef]
- Balint, B.; Guerreiro, R.; Carmona, S.; Dehghani, N.; Latorre, A.; Cordivari, C.; Bhatia, K.P.; Bras, J. KCNN2 mutation in autosomal-dominant tremulous myoclonus-dystonia. Eur. J. Neurol. 2020, 27, 1471–1477. [Google Scholar] [CrossRef] [Green Version]
- Szatanik, M.; Vibert, N.; Vassias, I.; Guenet, J.L.; Eugene, D.; de Waele, C.; Jaubert, J. Behavioral effects of a deletion in Kcnn2, the gene encoding the SK2 subunit of small-conductance Ca2+-activated K+ channels. Neurogenetics 2008, 9, 237–248. [Google Scholar] [CrossRef]
- Shakkottai, V.G.; Chou, C.H.; Oddo, S.; Sailer, C.A.; Knaus, H.G.; Gutman, G.A.; Barish, M.E.; LaFerla, F.M.; Chandy, K.G. Enhanced neuronal excitability in the absence of neurodegeneration induces cerebellar ataxia. J. Clin. Investig. 2004, 113, 582–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Tang, T.S.; Tu, H.; Nelson, O.; Herndon, E.; Huynh, D.P.; Pulst, S.M.; Bezprozvanny, I. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 9148–9162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuramoto, T.; Yokoe, M.; Kunisawa, N.; Ohashi, K.; Miyake, T.; Higuchi, Y.; Yoshimi, K.; Mashimo, T.; Tanaka, M.; Kuwamura, M.; et al. Tremor dominant Kyoto (Trdk) rats carry a missense mutation in the gene encoding the SK2 subunit of small-conductance Ca(2+)-activated K(+) channel. Brain Res. 2017, 1676, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Jun, K.; Piedras-Renteria, E.S.; Smith, S.M.; Wheeler, D.B.; Lee, S.B.; Lee, T.G.; Chin, H.; Adams, M.E.; Scheller, R.H.; Tsien, R.W.; et al. Ablation of P/Q-type Ca(2+) channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the alpha(1A)-subunit. Proc. Natl. Acad. Sci. USA 1999, 96, 15245–15250. [Google Scholar] [CrossRef] [Green Version]
- Zhuchenko, O.; Bailey, J.; Bonnen, P.; Ashizawa, T.; Stockton, D.W.; Amos, C.; Dobyns, W.B.; Subramony, S.H.; Zoghbi, H.Y.; Lee, C.C. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat. Genet. 1997, 15, 62–69. [Google Scholar] [CrossRef]
- Jen, J.C.; Baloh, R.W. Genetics of episodic ataxia. Adv. Neurol. 2002, 89, 459–461. [Google Scholar]
- Ashizawa, T.; Figueroa, K.P.; Perlman, S.L.; Gomez, C.M.; Wilmot, G.R.; Schmahmann, J.D.; Ying, S.H.; Zesiewicz, T.A.; Paulson, H.L.; Shakkottai, V.G.; et al. Clinical characteristics of patients with spinocerebellar ataxias 1, 2, 3 and 6 in the US; a prospective observational study. Orphanet J. Rare Dis. 2013, 8, 177. [Google Scholar] [CrossRef] [Green Version]
- Stevanin, G.; Durr, A.; David, G.; Didierjean, O.; Cancel, G.; Rivaud, S.; Tourbah, A.; Warter, J.M.; Agid, Y.; Brice, A. Clinical and molecular features of spinocerebellar ataxia type 6. Neurology 1997, 49, 1243–1246. [Google Scholar] [CrossRef]
- Watase, K.; Barrett, C.F.; Miyazaki, T.; Ishiguro, T.; Ishikawa, K.; Hu, Y.; Unno, T.; Sun, Y.; Kasai, S.; Watanabe, M.; et al. Spinocerebellar ataxia type 6 knockin mice develop a progressive neuronal dysfunction with age-dependent accumulation of mutant CaV2.1 channels. Proc. Natl. Acad. Sci. USA 2008, 105, 11987–11992. [Google Scholar] [CrossRef] [Green Version]
- Saegusa, H.; Wakamori, M.; Matsuda, Y.; Wang, J.; Mori, Y.; Zong, S.; Tanabe, T. Properties of human Cav2.1 channel with a spinocerebellar ataxia type 6 mutation expressed in Purkinje cells. Mol. Cell. Neurosci. 2007, 34, 261–270. [Google Scholar] [CrossRef]
- Jen, J.; Kim, G.W.; Baloh, R.W. Clinical spectrum of episodic ataxia type 2. Neurology 2004, 62, 17–22. [Google Scholar] [CrossRef]
- Jen, J.C.; Wan, J. Episodic ataxias. Handb. Clin. Neurol. 2018, 155, 205–215. [Google Scholar] [CrossRef]
- Fletcher, C.F.; Tottene, A.; Lennon, V.A.; Wilson, S.M.; Dubel, S.J.; Paylor, R.; Hosford, D.A.; Tessarollo, L.; McEnery, M.W.; Pietrobon, D.; et al. Dystonia and cerebellar atrophy in Cacna1a null mice lacking P/Q calcium channel activity. FASEB J. 2001, 15, 1288–1290. [Google Scholar] [CrossRef]
- Hoebeek, F.E.; Stahl, J.S.; van Alphen, A.M.; Schonewille, M.; Luo, C.; Rutteman, M.; van den Maagdenberg, A.M.; Molenaar, P.C.; Goossens, H.H.; Frens, M.A.; et al. Increased noise level of purkinje cell activities minimizes impact of their modulation during sensorimotor control. Neuron 2005, 45, 953–965. [Google Scholar] [CrossRef] [Green Version]
- Stahl, J.S.; James, R.A.; Oommen, B.S.; Hoebeek, F.E.; De Zeeuw, C.I. Eye movements of the murine P/Q calcium channel mutant tottering, and the impact of aging. J. Neurophysiol. 2006, 95, 1588–1607. [Google Scholar] [CrossRef] [Green Version]
- Walter, J.T.; Alvina, K.; Womack, M.D.; Chevez, C.; Khodakhah, K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat. Neurosci. 2006, 9, 389–397. [Google Scholar] [CrossRef]
- Spacey, S.D.; Hildebrand, M.E.; Materek, L.A.; Bird, T.D.; Snutch, T.P. Functional implications of a novel EA2 mutation in the P/Q-type calcium channel. Ann. Neurol. 2004, 56, 213–220. [Google Scholar] [CrossRef]
- Gao, Z.; Todorov, B.; Barrett, C.F.; van Dorp, S.; Ferrari, M.D.; van den Maagdenberg, A.M.; De Zeeuw, C.I.; Hoebeek, F.E. Cerebellar ataxia by enhanced Ca(V)2.1 currents is alleviated by Ca2+-dependent K+-channel activators in Cacna1a(S218L) mutant mice. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 15533–15546. [Google Scholar] [CrossRef] [Green Version]
- van den Maagdenberg, A.M.; Pizzorusso, T.; Kaja, S.; Terpolilli, N.; Shapovalova, M.; Hoebeek, F.E.; Barrett, C.F.; Gherardini, L.; van de Ven, R.C.; Todorov, B.; et al. High cortical spreading depression susceptibility and migraine-associated symptoms in Ca(v)2.1 S218L mice. Ann. Neurol. 2010, 67, 85–98. [Google Scholar] [CrossRef]
- Coutelier, M.; Blesneac, I.; Monteil, A.; Monin, M.L.; Ando, K.; Mundwiller, E.; Brusco, A.; Le Ber, I.; Anheim, M.; Castrioto, A.; et al. A Recurrent Mutation in CACNA1G Alters Cav3.1 T-Type Calcium-Channel Conduction and Causes Autosomal-Dominant Cerebellar Ataxia. Am. J. Hum. Genet. 2015, 97, 726–737. [Google Scholar] [CrossRef] [Green Version]
- Morino, H.; Matsuda, Y.; Muguruma, K.; Miyamoto, R.; Ohsawa, R.; Ohtake, T.; Otobe, R.; Watanabe, M.; Maruyama, H.; Hashimoto, K.; et al. A mutation in the low voltage-gated calcium channel CACNA1G alters the physiological properties of the channel, causing spinocerebellar ataxia. Mol. Brain 2015, 8, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, N.; Morino, H.; Matsuda, Y.; Satoh, K.; Hashimoto, K.; Maruyama, H.; Kawakami, H. Zonisamide can ameliorate the voltage-dependence alteration of the T-type calcium channel Ca(V)3.1 caused by a mutation responsible for spinocerebellar ataxia. Mol. Brain 2020, 13, 163. [Google Scholar] [CrossRef] [PubMed]
- Dansithong, W.; Paul, S.; Figueroa, K.P.; Rinehart, M.D.; Wiest, S.; Pflieger, L.T.; Scoles, D.R.; Pulst, S.M. Ataxin-2 regulates RGS8 translation in a new BAC-SCA2 transgenic mouse model. PLoS Genet. 2015, 11, e1005182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, K.Y.; Park, Y.G.; Park, H.Y.; Homanics, G.E.; Kim, J.; Kim, D. Lack of CaV3.1 channels causes severe motor coordination defects and an age-dependent cerebellar atrophy in a genetic model of essential tremor. Biochem. Biophys. Res. Commun. 2011, 410, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Chemin, J.; Siquier-Pernet, K.; Nicouleau, M.; Barcia, G.; Ahmad, A.; Medina-Cano, D.; Hanein, S.; Altin, N.; Hubert, L.; Bole-Feysot, C.; et al. De novo mutation screening in childhood-onset cerebellar atrophy identifies gain-of-function mutations in the CACNA1G calcium channel gene. Brain 2018, 141, 1998–2013. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.C.; Young, J.S.; Glitsch, M.D. Changes in TRPC channel expression during postnatal development of cerebellar neurons. Cell Calcium 2007, 42, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Becker, E.B.; Oliver, P.L.; Glitsch, M.D.; Banks, G.T.; Achilli, F.; Hardy, A.; Nolan, P.M.; Fisher, E.M.; Davies, K.E. A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice. Proc. Natl. Acad. Sci. USA 2009, 106, 6706–6711. [Google Scholar] [CrossRef] [Green Version]
- Sekerkova, G.; Kim, J.A.; Nigro, M.J.; Becker, E.B.; Hartmann, J.; Birnbaumer, L.; Mugnaini, E.; Martina, M. Early onset of ataxia in moonwalker mice is accompanied by complete ablation of type II unipolar brush cells and Purkinje cell dysfunction. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 19689–19694. [Google Scholar] [CrossRef] [Green Version]
- Hara, K.; Shiga, A.; Nozaki, H.; Mitsui, J.; Takahashi, Y.; Ishiguro, H.; Yomono, H.; Kurisaki, H.; Goto, J.; Ikeuchi, T.; et al. Total deletion and a missense mutation of ITPR1 in Japanese SCA15 families. Neurology 2008, 71, 547–551. [Google Scholar] [CrossRef]
- Huang, L.; Chardon, J.W.; Carter, M.T.; Friend, K.L.; Dudding, T.E.; Schwartzentruber, J.; Zou, R.; Schofield, P.W.; Douglas, S.; Bulman, D.E.; et al. Missense mutations in ITPR1 cause autosomal dominant congenital nonprogressive spinocerebellar ataxia. Orphanet J. Rare Dis. 2012, 7, 67. [Google Scholar] [CrossRef] [Green Version]
- Jarius, S.; Scharf, M.; Begemann, N.; Stocker, W.; Probst, C.; Serysheva, I.I.; Nagel, S.; Graus, F.; Psimaras, D.; Wildemann, B.; et al. Antibodies to the inositol 1,4,5-trisphosphate receptor type 1 (ITPR1) in cerebellar ataxia. J. Neuroinflamm. 2014, 11, 206. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Tang, T.S.; Tu, H.; Nelson, O.; Pook, M.; Hammer, R.; Nukina, N.; Bezprozvanny, I. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 3. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 12713–12724. [Google Scholar] [CrossRef] [Green Version]
- Di Leva, F.; Domi, T.; Fedrizzi, L.; Lim, D.; Carafoli, E. The plasma membrane Ca2+ ATPase of animal cells: Structure, function and regulation. Arch. Biochem. Biophys. 2008, 476, 65–74. [Google Scholar] [CrossRef]
- Empson, R.M.; Huang, H.; Nagaraja, R.Y.; Roome, C.J.; Knopfel, T. Enhanced synaptic inhibition in the cerebellar cortex of the ataxic PMCA2(−/−) knockout mouse. Cerebellum 2013, 12, 667–675. [Google Scholar] [CrossRef]
- Empson, R.M.; Akemann, W.; Knopfel, T. The role of the calcium transporter protein plasma membrane calcium ATPase PMCA2 in cerebellar Purkinje neuron function. Funct. Neurol. 2010, 25, 153–158. [Google Scholar]
- Zanni, G.; Cali, T.; Kalscheuer, V.M.; Ottolini, D.; Barresi, S.; Lebrun, N.; Montecchi-Palazzi, L.; Hu, H.; Chelly, J.; Bertini, E.; et al. Mutation of plasma membrane Ca2+ ATPase isoform 3 in a family with X-linked congenital cerebellar ataxia impairs Ca2+ homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 14514–14519. [Google Scholar] [CrossRef] [Green Version]
- Marquer, A.; Barbieri, G.; Perennou, D. The assessment and treatment of postural disorders in cerebellar ataxia: A systematic review. Ann. Phys. Rehabil. Med. 2014, 57, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Ilg, W.; Synofzik, M.; Brotz, D.; Burkard, S.; Giese, M.A.; Schols, L. Intensive coordinative training improves motor performance in degenerative cerebellar disease. Neurology 2009, 73, 1823–1830. [Google Scholar] [CrossRef]
- Ilg, W.; Brotz, D.; Burkard, S.; Giese, M.A.; Schols, L.; Synofzik, M. Long-term effects of coordinative training in degenerative cerebellar disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2010, 25, 2239–2246. [Google Scholar] [CrossRef]
- Miyai, I.; Ito, M.; Hattori, N.; Mihara, M.; Hatakenaka, M.; Yagura, H.; Sobue, G.; Nishizawa, M.; Cerebellar Ataxia Rehabilitation Trialists, C. Cerebellar ataxia rehabilitation trial in degenerative cerebellar diseases. Neurorehabil. Neural Repair 2012, 26, 515–522. [Google Scholar] [CrossRef]
- Sullivan, R.; Yau, W.Y.; O’Connor, E.; Houlden, H. Spinocerebellar ataxia: An update. J. Neurol. 2019, 266, 533–544. [Google Scholar] [CrossRef] [Green Version]
- Evers, M.M.; Toonen, L.J.; van Roon-Mom, W.M. Antisense oligonucleotides in therapy for neurodegenerative disorders. Adv. Drug Deliv. Rev. 2015, 87, 90–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, J.; Kordasiewicz, H.B.; O’Callaghan, B.; Handler, H.P.; Wagener, C.; Duvick, L.; Swayze, E.E.; Rainwater, O.; Hofstra, B.; Benneyworth, M.; et al. Antisense oligonucleotide-mediated ataxin-1 reduction prolongs survival in SCA1 mice and reveals disease-associated transcriptome profiles. JCI Insight 2018, 3, e123193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scoles, D.R.; Meera, P.; Schneider, M.D.; Paul, S.; Dansithong, W.; Figueroa, K.P.; Hung, G.; Rigo, F.; Bennett, C.F.; Otis, T.S.; et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature 2017, 544, 362–366. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, H.S.; Moore, L.R.; Chopra, R.; Komlo, R.; McKenzie, M.; Blumenstein, K.G.; Zhao, H.; Kordasiewicz, H.B.; Shakkottai, V.G.; Paulson, H.L. Oligonucleotide therapy mitigates disease in spinocerebellar ataxia type 3 mice. Ann. Neurol. 2018, 84, 64–77. [Google Scholar] [CrossRef]
- Xia, H.; Mao, Q.; Eliason, S.L.; Harper, S.Q.; Martins, I.H.; Orr, H.T.; Paulson, H.L.; Yang, L.; Kotin, R.M.; Davidson, B.L. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat. Med. 2004, 10, 816–820. [Google Scholar] [CrossRef]
- Keiser, M.S.; Boudreau, R.L.; Davidson, B.L. Broad therapeutic benefit after RNAi expression vector delivery to deep cerebellar nuclei: Implications for spinocerebellar ataxia type 1 therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 588–595. [Google Scholar] [CrossRef] [Green Version]
- Johnson, W.G. Friedreich ataxia. Clin. Neurosci. 1995, 3, 33–38. [Google Scholar]
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal.l recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- Gonzalez-Cabo, P.; Vazquez-Manrique, R.P.; Garcia-Gimeno, M.A.; Sanz, P.; Palau, F. Frataxin interacts functionally with mitochondrial electron transport chain proteins. Hum. Mol. Genet. 2005, 14, 2091–2098. [Google Scholar] [CrossRef] [Green Version]
- Clark, E.; Johnson, J.; Dong, Y.N.; Mercado-Ayon, E.; Warren, N.; Zhai, M.; McMillan, E.; Salovin, A.; Lin, H.; Lynch, D.R. Role of frataxin protein deficiency and metabolic dysfunction in Friedreich ataxia, an autosomal recessive mitochondrial disease. Neuronal Signal. 2018, 2, NS20180060. [Google Scholar] [CrossRef] [Green Version]
- Bolinches-Amoros, A.; Molla, B.; Pla-Martin, D.; Palau, F.; Gonzalez-Cabo, P. Mitochondrial dysfunction induced by frataxin deficiency is associated with cellular senescence and abnormal calcium metabolism. Front. Cell. Neurosci. 2014, 8, 124. [Google Scholar] [CrossRef] [Green Version]
- Pandolfo, M. Frataxin deficiency and mitochondrial dysfunction. Mitochondrion 2002, 2, 87–93. [Google Scholar] [CrossRef]
- Abeti, R.; Baccaro, A.; Esteras, N.; Giunti, P. Novel Nrf2-Inducer Prevents Mitochondrial Defects and Oxidative Stress in Friedreich’s Ataxia Models. Front. Cell. Neurosci. 2018, 12, 188. [Google Scholar] [CrossRef]
- Lynch, D.R.; Farmer, J.; Hauser, L.; Blair, I.A.; Wang, Q.Q.; Mesaros, C.; Snyder, N.; Boesch, S.; Chin, M.; Delatycki, M.B.; et al. Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Ann. Clin. Transl. Neurol. 2019, 6, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, K.A.; Sandiford, S.D.; Skerjanc, I.S.; Li, S.S. Reactive oxygen species and the neuronal fate. Cell. Mol. Life Sci. CMLS 2012, 69, 215–221. [Google Scholar] [CrossRef]
- Lynch, D.R.; Chin, M.P.; Delatycki, M.B.; Subramony, S.H.; Corti, M.; Hoyle, J.C.; Boesch, S.; Nachbauer, W.; Mariotti, C.; Mathews, K.D.; et al. Safety and Efficacy of Omaveloxolone in Friedreich Ataxia (MOXIe Study). Ann. Neurol. 2021, 89, 212–225. [Google Scholar] [CrossRef]
- Maltecca, F.; Magnoni, R.; Cerri, F.; Cox, G.A.; Quattrini, A.; Casari, G. Haploinsufficiency of AFG3L2, the gene responsible for spinocerebellar ataxia type 28, causes mitochondria-mediated Purkinje cell dark degeneration. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 9244–9254. [Google Scholar] [CrossRef] [Green Version]
- Pierson, T.M.; Adams, D.; Bonn, F.; Martinelli, P.; Cherukuri, P.F.; Teer, J.K.; Hansen, N.F.; Cruz, P.; James C. Mullikin For The Nisc Comparative Sequencing Program; Blakesley, R.W.; et al. Whole-exome sequencing identifies homozygous AFG3L2 mutations in a spastic ataxia-neuropathy syndrome linked to mitochondrial m-AAA proteases. PLoS Genet. 2011, 7, e1002325. [Google Scholar] [CrossRef] [Green Version]
- Nolden, M.; Ehses, S.; Koppen, M.; Bernacchia, A.; Rugarli, E.I.; Langer, T. The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell 2005, 123, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Steglich, G.; Neupert, W.; Langer, T. Prohibitins regulate membrane protein degradation by the m-AAA protease in mitochondria. Mol. Cell. Biol. 1999, 19, 3435–3442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maltecca, F.; Baseggio, E.; Consolato, F.; Mazza, D.; Podini, P.; Young, S.M., Jr.; Drago, I.; Bahr, B.A.; Puliti, A.; Codazzi, F.; et al. Purkinje neuron Ca2+ influx reduction rescues ataxia in SCA28 model. J. Clin. Investig. 2015, 125, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Mancini, C.; Hoxha, E.; Iommarini, L.; Brussino, A.; Richter, U.; Montarolo, F.; Cagnoli, C.; Parolisi, R.; Gondor Morosini, D.I.; Nicolo, V.; et al. Mice harbouring a SCA28 patient mutation in AFG3L2 develop late-onset ataxia associated with enhanced mitochondrial proteotoxicity. Neurobiol. Dis. 2019, 124, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Girard, M.; Lariviere, R.; Parfitt, D.A.; Deane, E.C.; Gaudet, R.; Nossova, N.; Blondeau, F.; Prenosil, G.; Vermeulen, E.G.; Duchen, M.R.; et al. Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Proc. Natl. Acad. Sci. USA 2012, 109, 1661–1666. [Google Scholar] [CrossRef] [Green Version]
- Ady, V.; Toscano-Marquez, B.; Nath, M.; Chang, P.K.; Hui, J.; Cook, A.; Charron, F.; Lariviere, R.; Brais, B.; McKinney, R.A.; et al. Altered synaptic and firing properties of cerebellar Purkinje cells in a mouse model of ARSACS. J. Physiol. 2018, 596, 4253–4267. [Google Scholar] [CrossRef]
- Strupp, M.; Kalla, R.; Claassen, J.; Adrion, C.; Mansmann, U.; Klopstock, T.; Freilinger, T.; Neugebauer, H.; Spiegel, R.; Dichgans, M.; et al. A randomized trial of 4-aminopyridine in EA2 and related familial episodic ataxias. Neurology 2011, 77, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Feil, K.; Bremova, T.; Muth, C.; Schniepp, R.; Teufel, J.; Strupp, M. Update on the Pharmacotherapy of Cerebellar Ataxia and Nystagmus. Cerebellum 2016, 15, 38–42. [Google Scholar] [CrossRef]
- Alvina, K.; Khodakhah, K. The therapeutic mode of action of 4-aminopyridine in cerebellar ataxia. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 7258–7268. [Google Scholar] [CrossRef] [Green Version]
- Davis, F.A.; Stefoski, D.; Rush, J. Orally administered 4-aminopyridine improves clinical signs in multiple sclerosis. Ann. Neurol. 1990, 27, 186–192. [Google Scholar] [CrossRef]
- Stefoski, D.; Davis, F.A.; Fitzsimmons, W.E.; Luskin, S.S.; Rush, J.; Parkhurst, G.W. 4-Aminopyridine in multiple sclerosis: Prolonged administration. Neurology 1991, 41, 1344–1348. [Google Scholar] [CrossRef]
- Dietrich, M.; Hartung, H.P.; Albrecht, P. Neuroprotective Properties of 4-Aminopyridine. Neurol.-Neuroimmunol. Neuroinflamm. 2021, 8, e976. [Google Scholar] [CrossRef]
- Trimmer, J.S.; Rhodes, K.J. Localization of voltage-gated ion channels in mammalian brain. Annu. Rev. Physiol. 2004, 66, 477–519. [Google Scholar] [CrossRef]
- Bostock, H.; Sears, T.A.; Sherratt, R.M. The effects of 4-aminopyridine and tetraethylammonium ions on normal and demyelinated mammalian nerve fibres. J. Physiol. 1981, 313, 301–315. [Google Scholar] [CrossRef]
- Cao, Y.; Dreixler, J.C.; Roizen, J.D.; Roberts, M.T.; Houamed, K.M. Modulation of recombinant small-conductance Ca(2+)-activated K(+) channels by the muscle relaxant chlorzoxazone and structurally related compounds. J. Pharmacol. Exp. Ther. 2001, 296, 683–689. [Google Scholar]
- Benton, M.D.; Lewis, A.H.; Bant, J.S.; Raman, I.M. Iberiotoxin-sensitive and -insensitive BK currents in Purkinje neuron somata. J. Neurophysiol. 2013, 109, 2528–2541. [Google Scholar] [CrossRef] [Green Version]
- Feil, K.; Claassen, J.; Bardins, S.; Teufel, J.; Krafczyk, S.; Schneider, E.; Schniepp, R.; Jahn, K.; Kalla, R.; Strupp, M. Effect of chlorzoxazone in patients with downbeat nystagmus: A pilot trial. Neurology 2013, 81, 1152–1158. [Google Scholar] [CrossRef] [Green Version]
- Egorova, P.A.; Zakharova, O.A.; Vlasova, O.L.; Bezprozvanny, I.B. In vivo analysis of cerebellar Purkinje cell activity in SCA2 transgenic mouse model. J. Neurophysiol. 2016, 115, 2840–2851. [Google Scholar] [CrossRef] [Green Version]
- Lake, W.; Shah, H. Intrathecal Baclofen Infusion for the Treatment of Movement Disorders. Neurosurg. Clin. N. Am. 2019, 30, 203–209. [Google Scholar] [CrossRef]
- Berntsson, S.G.; Gauffin, H.; Melberg, A.; Holtz, A.; Landtblom, A.M. Inherited Ataxia and Intrathecal Baclofen for the Treatment of Spasticity and Painful Spasms. Stereotact. Funct. Neurosurg. 2019, 97, 18–23. [Google Scholar] [CrossRef]
- Misgeld, U.; Bijak, M.; Jarolimek, W. A physiological role for GABAB receptors and the effects of baclofen in the mammalian central nervous system. Prog. Neurobiol. 1995, 46, 423–462. [Google Scholar] [CrossRef]
- Lujan, R.; Shigemoto, R. Localization of metabotropic GABA receptor subunits GABAB1 and GABAB2 relative to synaptic sites in the rat developing cerebellum. Eur. J. Neurosci. 2006, 23, 1479–1490. [Google Scholar] [CrossRef] [PubMed]
- Kulik, A.; Vida, I.; Lujan, R.; Haas, C.A.; Lopez-Bendito, G.; Shigemoto, R.; Frotscher, M. Subcellular localization of metabotropic GABA(B) receptor subunits GABA(B1a/b) and GABA(B2) in the rat hippocampus. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 11026–11035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Alacid, L.; Aguado, C.; Ciruela, F.; Martin, R.; Colon, J.; Cabanero, M.J.; Gassmann, M.; Watanabe, M.; Shigemoto, R.; Wickman, K.; et al. Subcellular compartment-specific molecular diversity of pre- and post-synaptic GABA-activated GIRK channels in Purkinje cells. J. Neurochem. 2009, 110, 1363–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hibino, H.; Inanobe, A.; Furutani, K.; Murakami, S.; Findlay, I.; Kurachi, Y. Inwardly rectifying potassium channels: Their structure, function, and physiological roles. Physiol. Rev. 2010, 90, 291–366. [Google Scholar] [CrossRef] [Green Version]
- Loeffler, M.A.; Synofzik, M.; Cebi, I.; Klocke, P.; Hormozi, M.; Gasser, T.; Gharabaghi, A.; Weiss, D. Case Report: Deep brain stimulation improves tremor in FGF-14 associated spinocerebellar ataxia. Front. Neurol. 2022, 13, 1048530. [Google Scholar] [CrossRef]
- Oyama, G.; Thompson, A.; Foote, K.D.; Limotai, N.; Abd-El-Barr, M.; Maling, N.; Malaty, I.A.; Rodriguez, R.L.; Subramony, S.H.; Ashizawa, T.; et al. Deep brain stimulation for tremor associated with underlying ataxia syndromes: A case series and discussion of issues. Tremor Other Hyperkinetic Mov. 2014, 4, 228. [Google Scholar] [CrossRef]
- Weiss, D.; Mielke, C.; Wachter, T.; Bender, B.; Liscic, R.M.; Scholten, M.; Naros, G.; Plewnia, C.; Gharabaghi, A.; Kruger, R. Long-term outcome of deep brain stimulation in fragile X-associated tremor/ataxia syndrome. Park. Relat. Disord. 2015, 21, 310–313. [Google Scholar] [CrossRef]
- Cury, R.G.; Franca, C.; Duarte, K.P.; Paraguay, I.; Diniz, J.M.; Cunha, P.; Galhardoni, R.; Silva, V.; Iglesio, R.; Bissoli, A.B.; et al. Safety and Outcomes of Dentate Nucleus Deep Brain Stimulation for Cerebellar Ataxia. Cerebellum 2022, 21, 861–865. [Google Scholar] [CrossRef]
- Kumar, G.; Asthana, P.; Yung, W.H.; Kwan, K.M.; Tin, C.; Ma, C.H.E. Deep Brain Stimulation of the Interposed Nucleus Reverses Motor Deficits and Stimulates Production of Anti-inflammatory Cytokines in Ataxia Mice. Mol. Neurobiol. 2022, 59, 4578–4592. [Google Scholar] [CrossRef]
- Miterko, L.N.; Lin, T.; Zhou, J.; van der Heijden, M.E.; Beckinghausen, J.; White, J.J.; Sillitoe, R.V. Neuromodulation of the cerebellum rescues movement in a mouse model of ataxia. Nat. Commun. 2021, 12, 1295. [Google Scholar] [CrossRef]
- Di Nuzzo, C.; Ruggiero, F.; Cortese, F.; Cova, I.; Priori, A.; Ferrucci, R. Non-invasive Cerebellar Stimulation in Cerebellar Disorders. CNS Neurol. Disord. Drug Targets 2018, 17, 193–198. [Google Scholar] [CrossRef]
- Kumar, G.; Ma, C.H.E. Toward a cerebello-thalamo-cortical computational model of spinocerebellar ataxia. Neural Netw. 2023, in press. [Google Scholar] [CrossRef]
- Voglis, G.; Tavernarakis, N. The role of synaptic ion channels in synaptic plasticity. EMBO Rep. 2006, 7, 1104–1110. [Google Scholar] [CrossRef] [Green Version]
- Breit, S.; Schulz, J.B.; Benabid, A.L. Deep brain stimulation. Cell Tissue Res. 2004, 318, 275–288. [Google Scholar] [CrossRef]
- Alrashdi, B.; Dawod, B.; Schampel, A.; Tacke, S.; Kuerten, S.; Marshall, J.S.; Cote, P.D. Nav1.6 promotes inflammation and neuronal degeneration in a mouse model of multiple sclerosis. J. Neuroinflamm. 2019, 16, 215. [Google Scholar] [CrossRef]
- Waxman, S.G. Mechanisms of disease: Sodium channels and neuroprotection in multiple sclerosis-current status. Nat. Clin. Pract. Neurol. 2008, 4, 159–169. [Google Scholar] [CrossRef]
- Guyton, M.K.; Wingrave, J.M.; Yallapragada, A.V.; Wilford, G.G.; Sribnick, E.A.; Matzelle, D.D.; Tyor, W.R.; Ray, S.K.; Banik, N.L. Upregulation of calpain correlates with increased neurodegeneration in acute experimental auto-immune encephalomyelitis. J. Neurosci. Res. 2005, 81, 53–61. [Google Scholar] [CrossRef]
- Grissmer, S.; Nguyen, A.N.; Aiyar, J.; Hanson, D.C.; Mather, R.J.; Gutman, G.A.; Karmilowicz, M.J.; Auperin, D.D.; Chandy, K.G. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol. Pharm. 1994, 45, 1227–1234. [Google Scholar]
- Giordano, I.; Bogdanow, M.; Jacobi, H.; Jahn, K.; Minnerop, M.; Schoels, L.; Synofzik, M.; Teufel, J.; Klockgether, T. Experience in a short-term trial with 4-aminopyridine in cerebellar ataxia. J. Neurol. 2013, 260, 2175–2176. [Google Scholar] [CrossRef]
- Marsden, C.D. Problems with long-term levodopa therapy for Parkinson’s disease. Clin. Neuropharmacol. 1994, 17 (Suppl. S2), S32–S44. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, H.; Shakkottai, V.G. Targeting Ion Channels and Purkinje Neuron Intrinsic Membrane Excitability as a Therapeutic Strategy for Cerebellar Ataxia. Life 2023, 13, 1350. https://doi.org/10.3390/life13061350
Huang H, Shakkottai VG. Targeting Ion Channels and Purkinje Neuron Intrinsic Membrane Excitability as a Therapeutic Strategy for Cerebellar Ataxia. Life. 2023; 13(6):1350. https://doi.org/10.3390/life13061350
Chicago/Turabian StyleHuang, Haoran, and Vikram G. Shakkottai. 2023. "Targeting Ion Channels and Purkinje Neuron Intrinsic Membrane Excitability as a Therapeutic Strategy for Cerebellar Ataxia" Life 13, no. 6: 1350. https://doi.org/10.3390/life13061350
APA StyleHuang, H., & Shakkottai, V. G. (2023). Targeting Ion Channels and Purkinje Neuron Intrinsic Membrane Excitability as a Therapeutic Strategy for Cerebellar Ataxia. Life, 13(6), 1350. https://doi.org/10.3390/life13061350