
Machine Learning Reveals Impacts of Smoking on Gene Profiles of Different Cell Types in Lung

Abstract
:1. Introduction
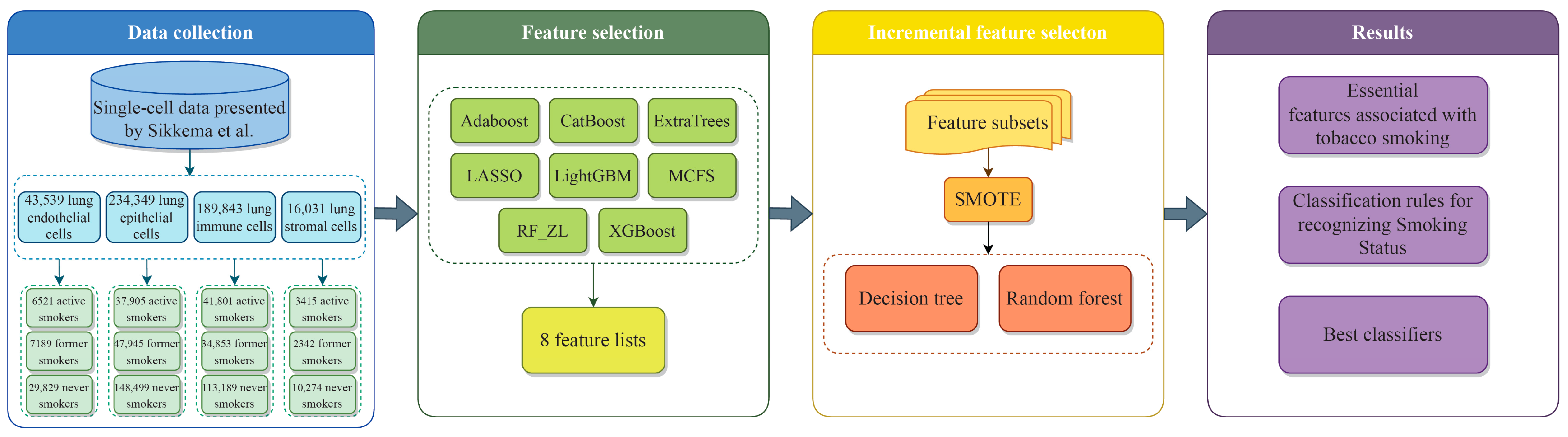
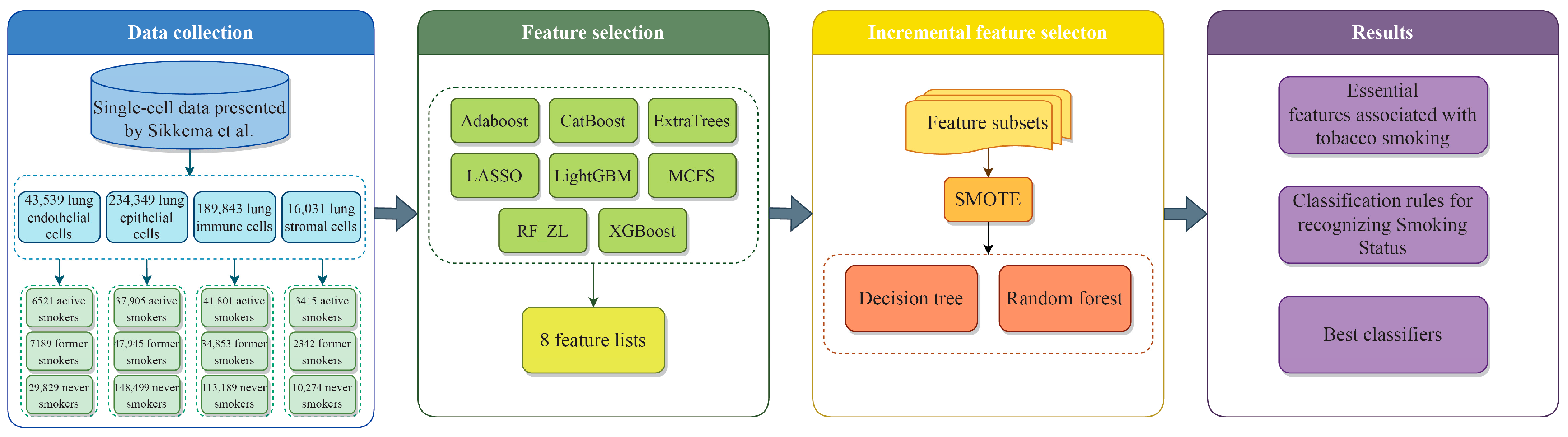
2. Materials and Methods
2.1. Gene Expression Data in Lung Cells Related to Smoking Status
2.2. Feature Ranking Methods for Prioritizing Features Based on Their Importance
2.2.1. Adaptive Boosting
2.2.2. Categorical Boosting
2.2.3. Extremely Randomized Trees
2.2.4. Least Absolute Shrinkage and Selection Operator
2.2.5. Light Gradient Boosting Machine
2.2.6. Monte Carlo Feature Selection
2.2.7. Random Forest
2.2.8. Extreme Gradient Boosting
2.3. Incremental Feature Selection
2.4. Synthetic Minority Oversampling Technique
2.5. Classification Algorithm for Establishing Classification Rules
2.5.1. Decision Tree
2.5.2. Random Forest
2.6. Performance Evaluation
2.7. Outline of the Analysis Procedure
3. Results
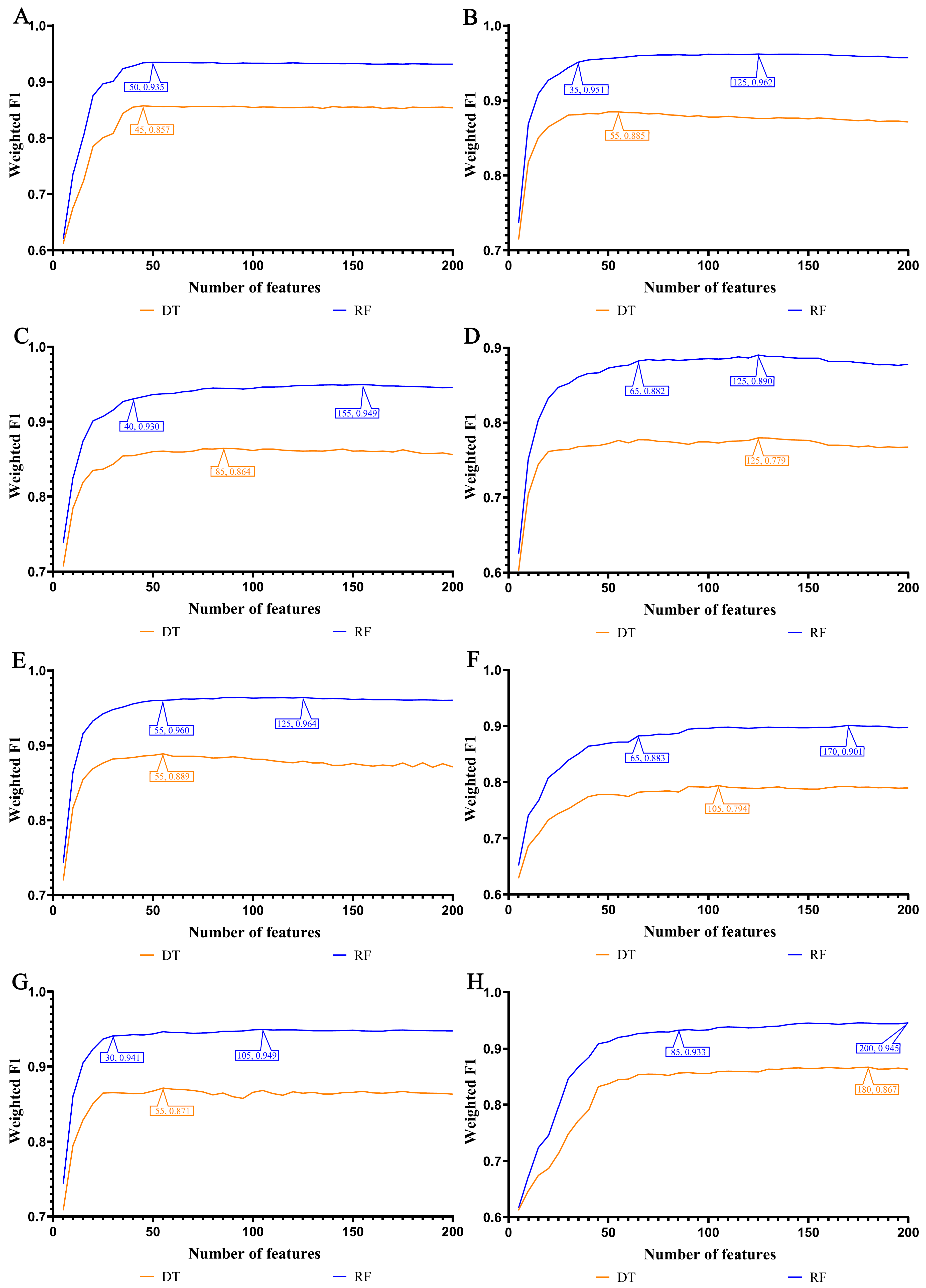
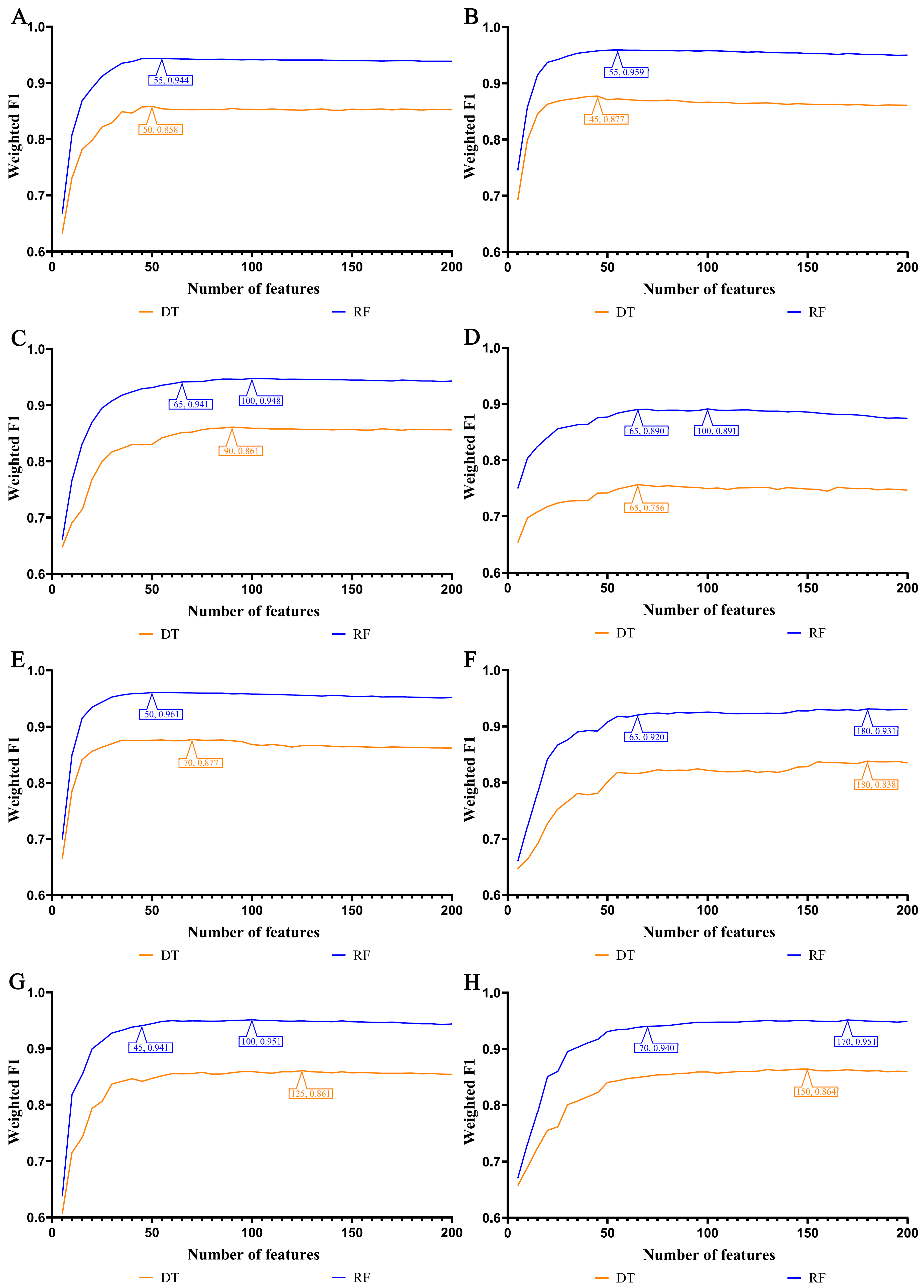
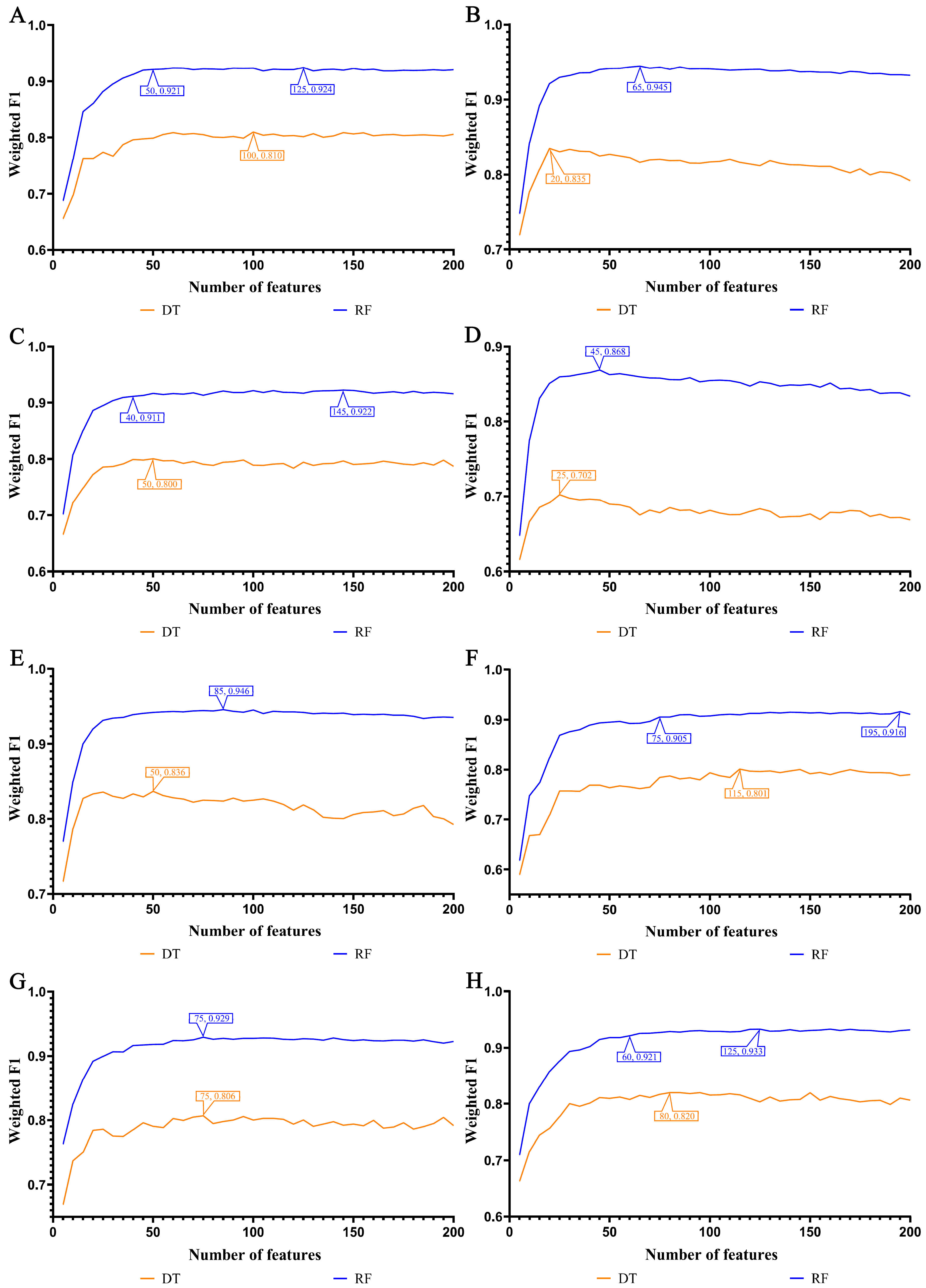
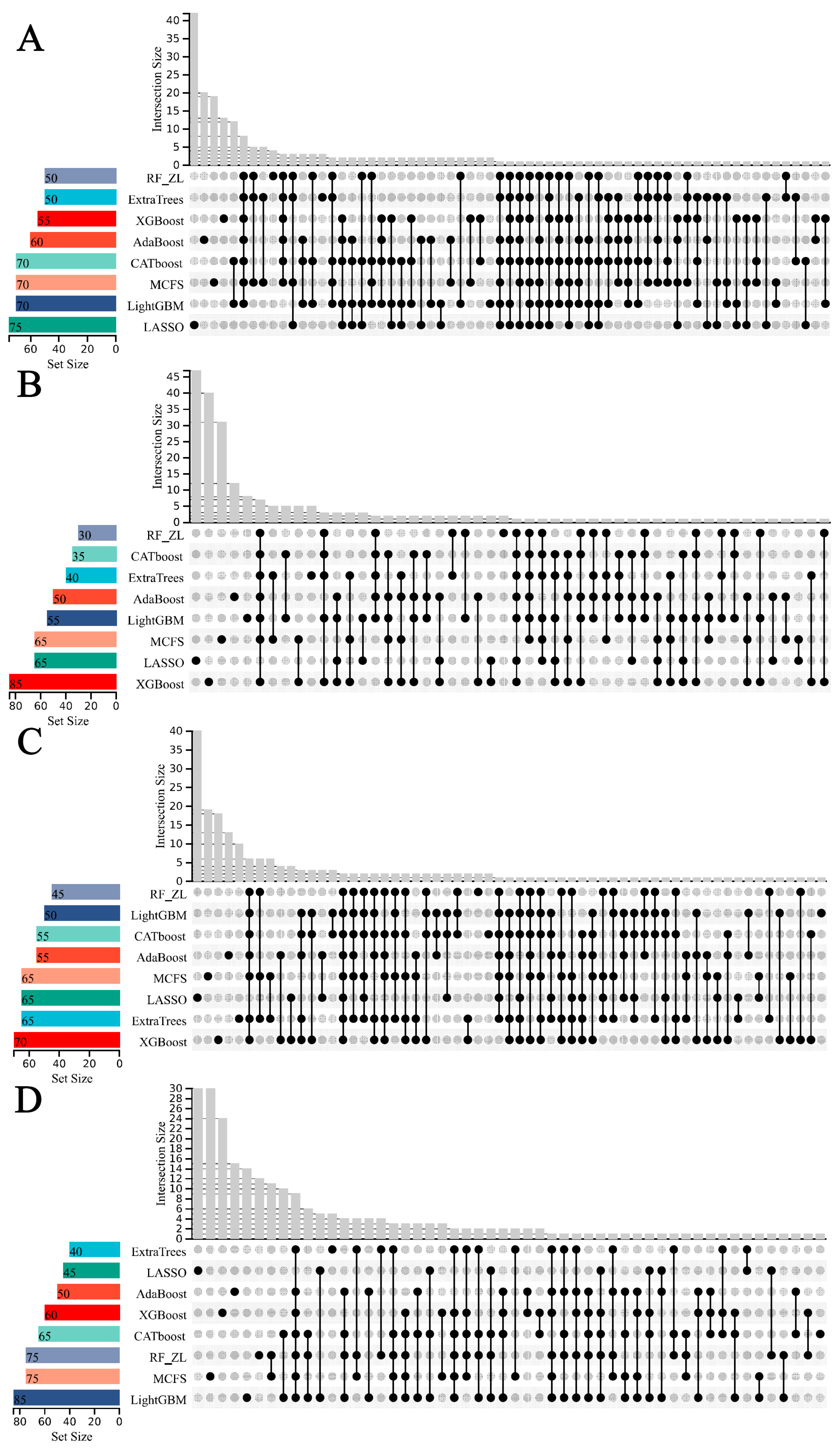
3.1. Feature Ranking Results of Features in Order of Importance
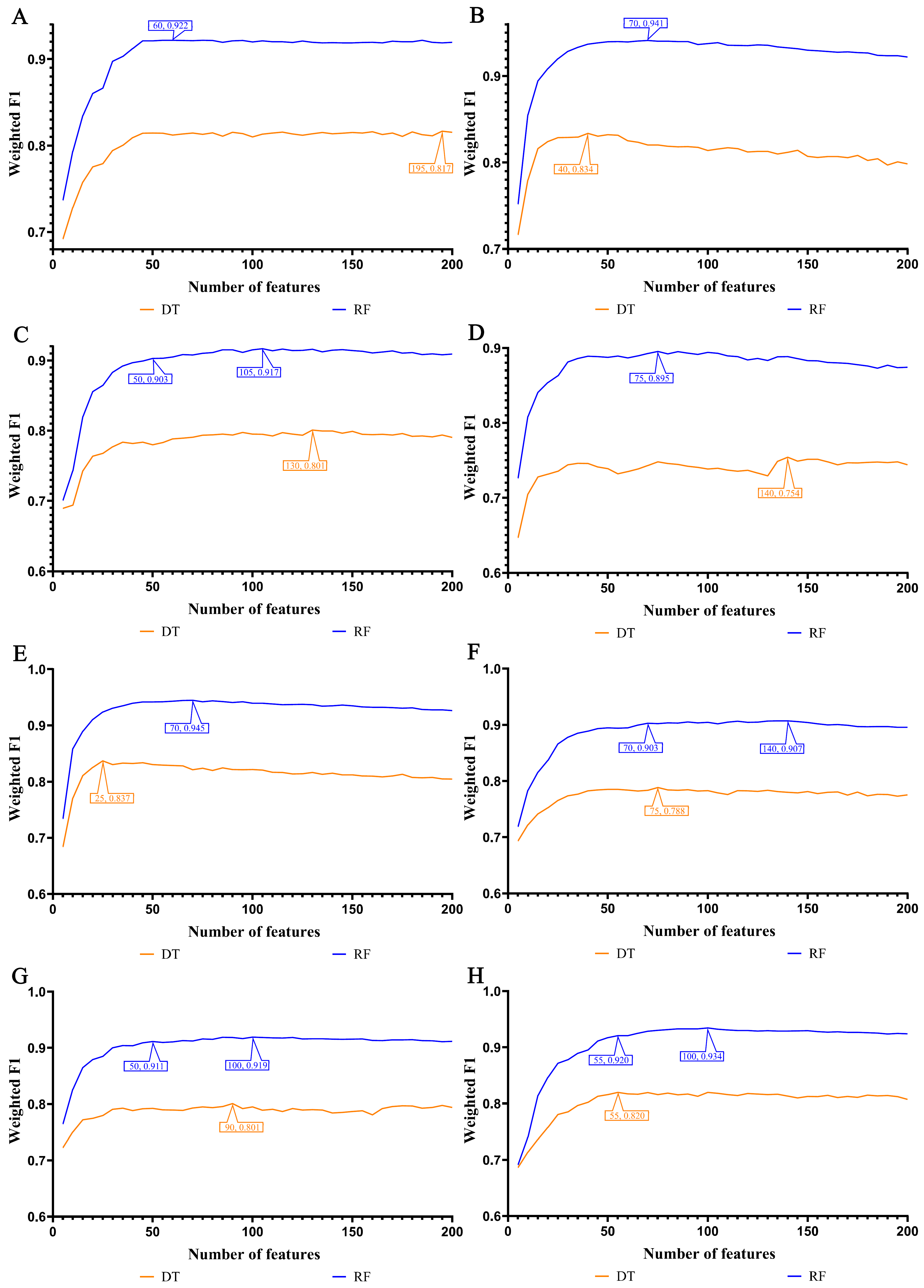
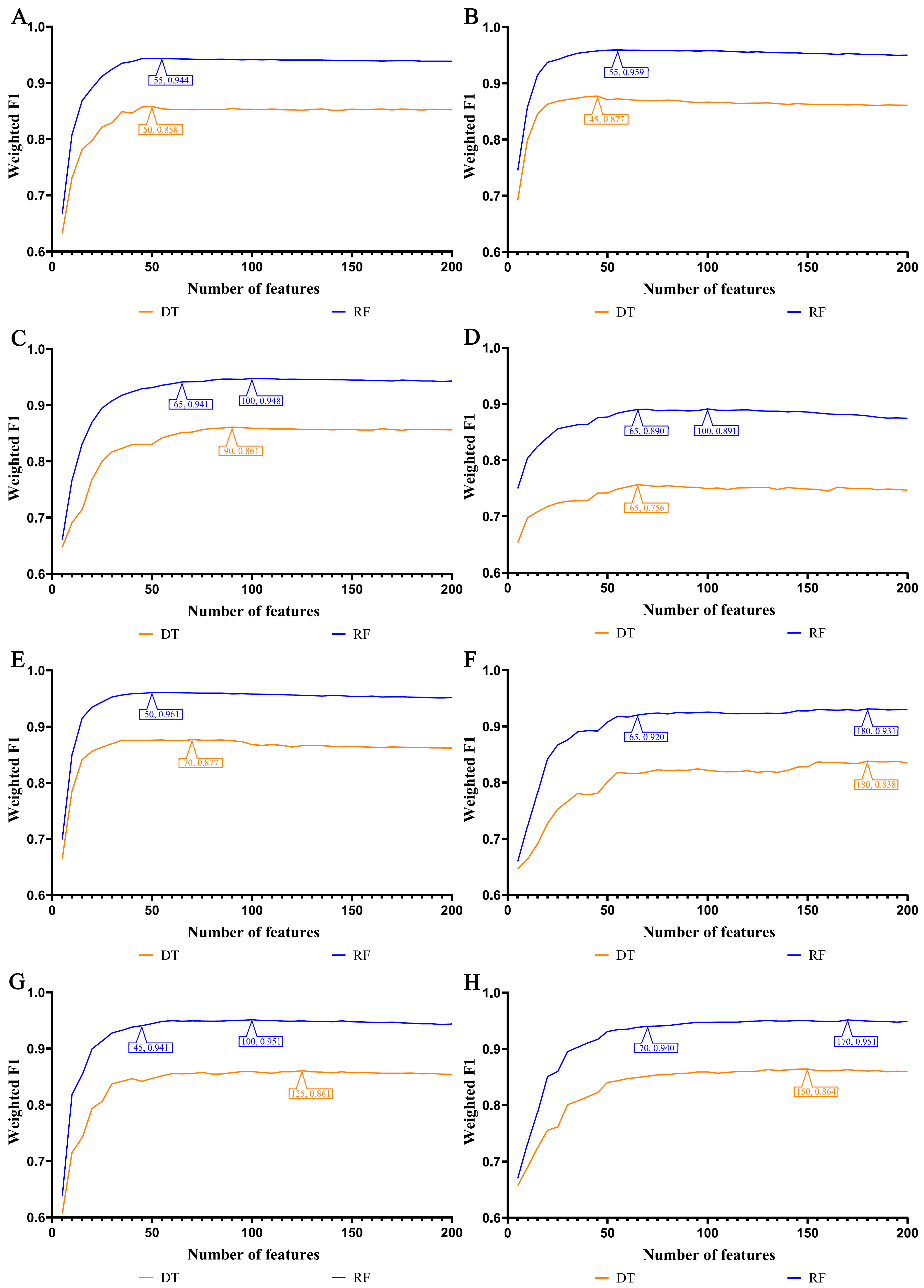
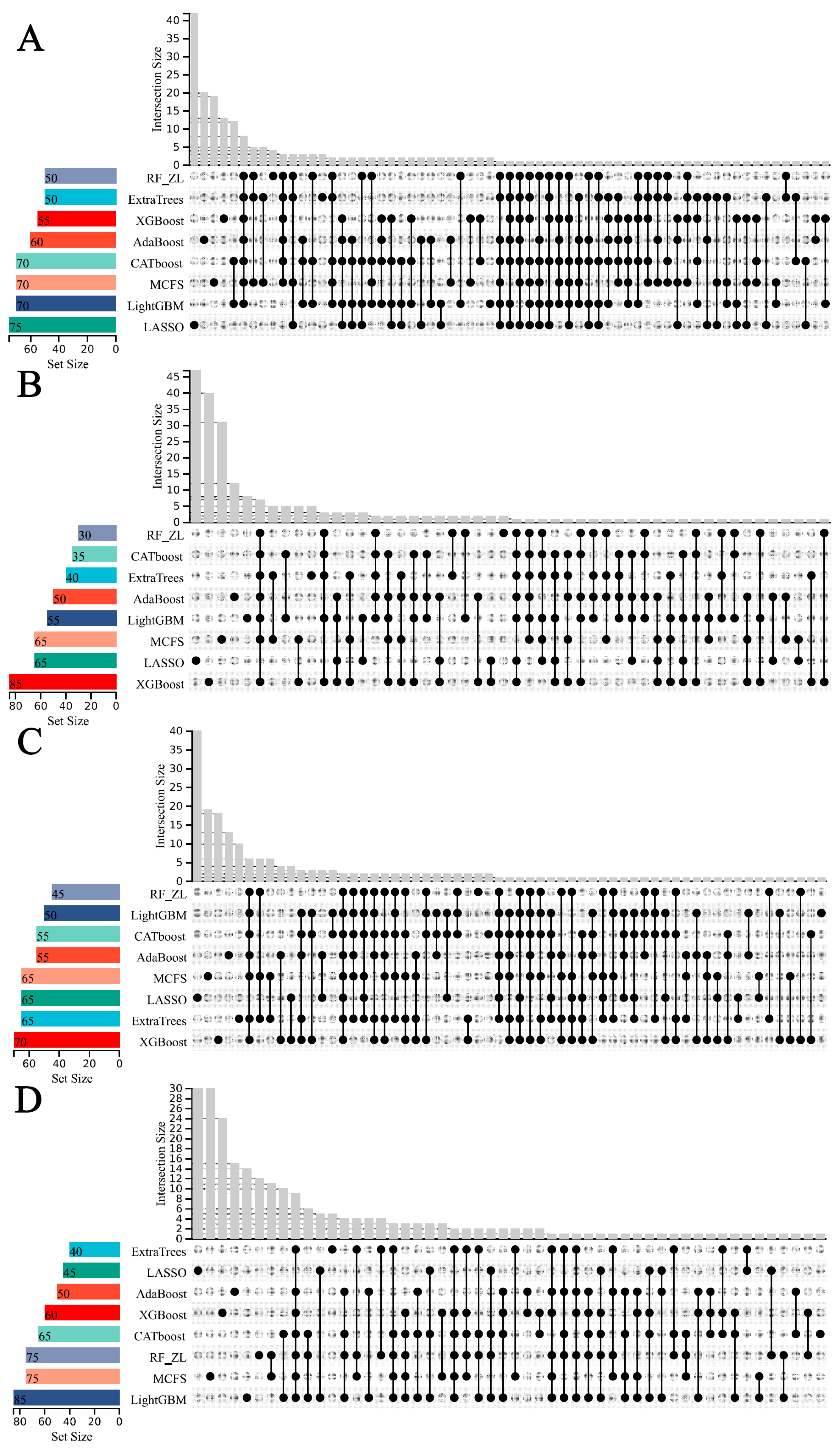
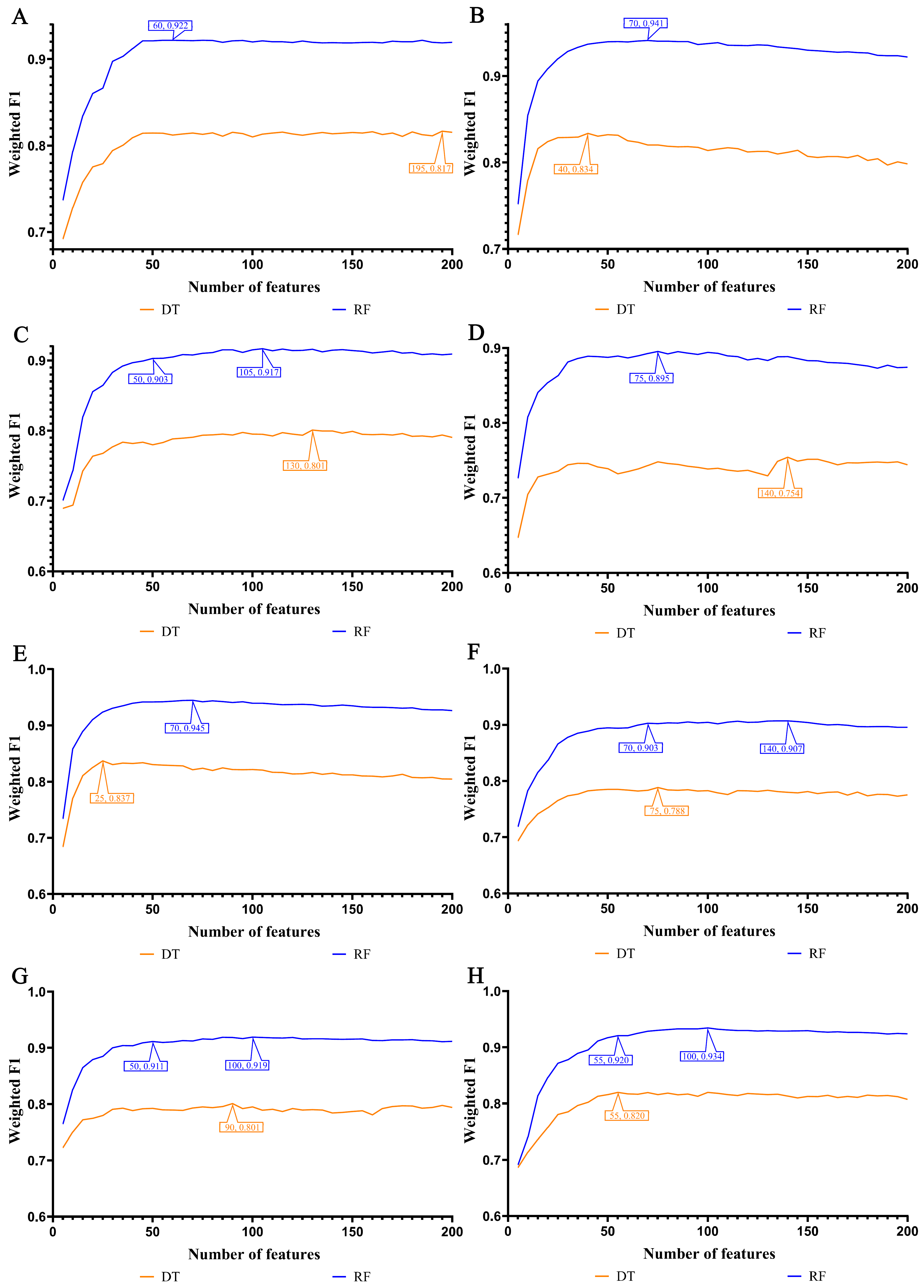
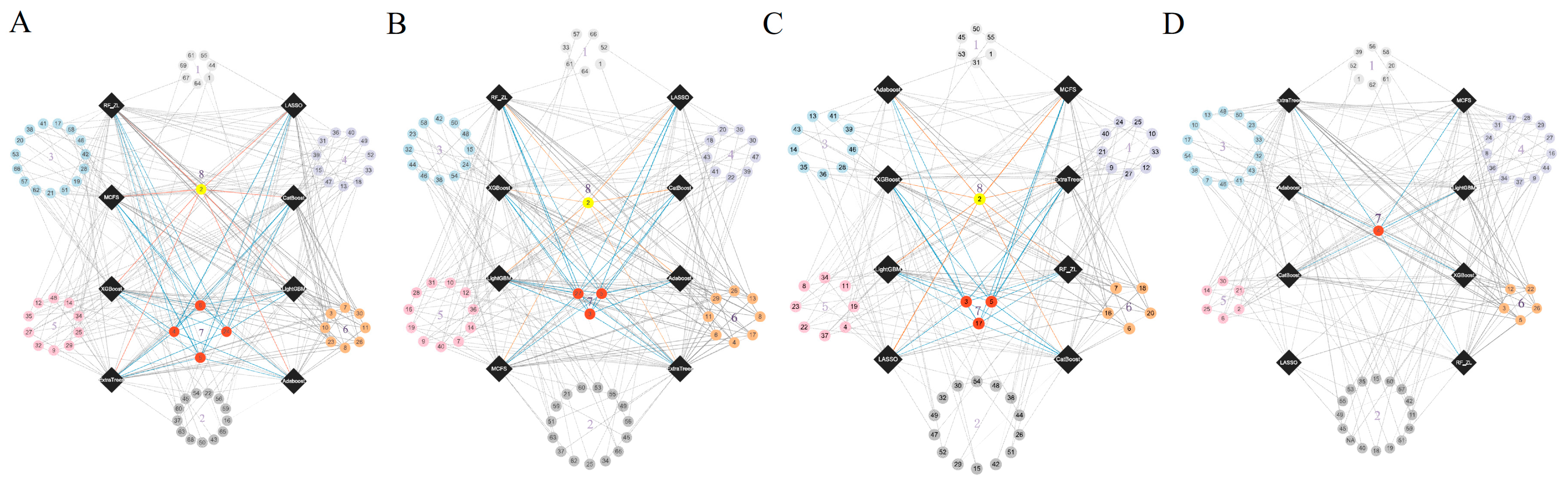
3.2. IFS Results and Feature Intersections for Finding Key Features Associated with Smoking Status
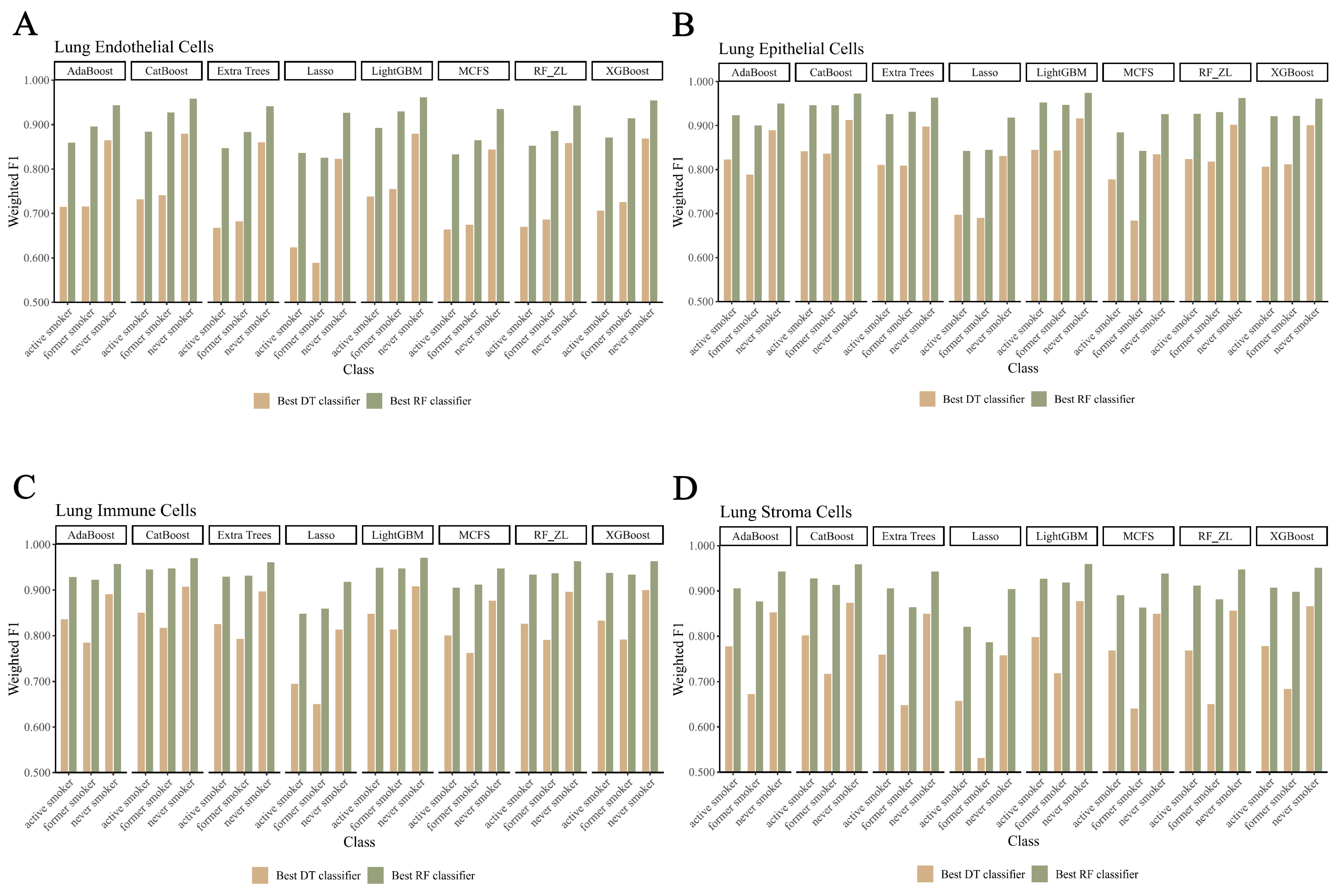
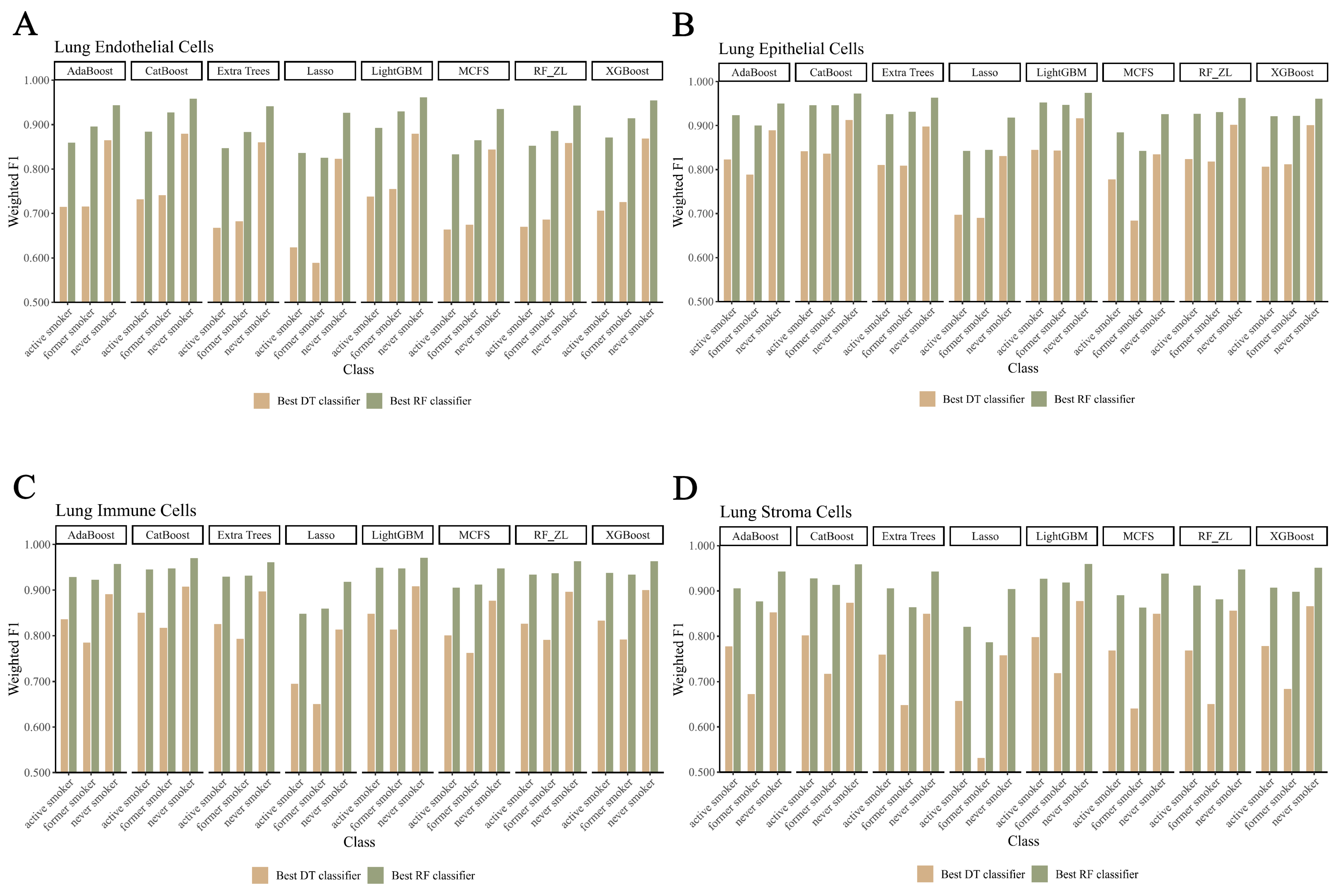
3.3. Establishing Classification Rules for Identifying Smoking Status
4. Discussion
4.1. Analysis of Key Features Associated with Smoking Status
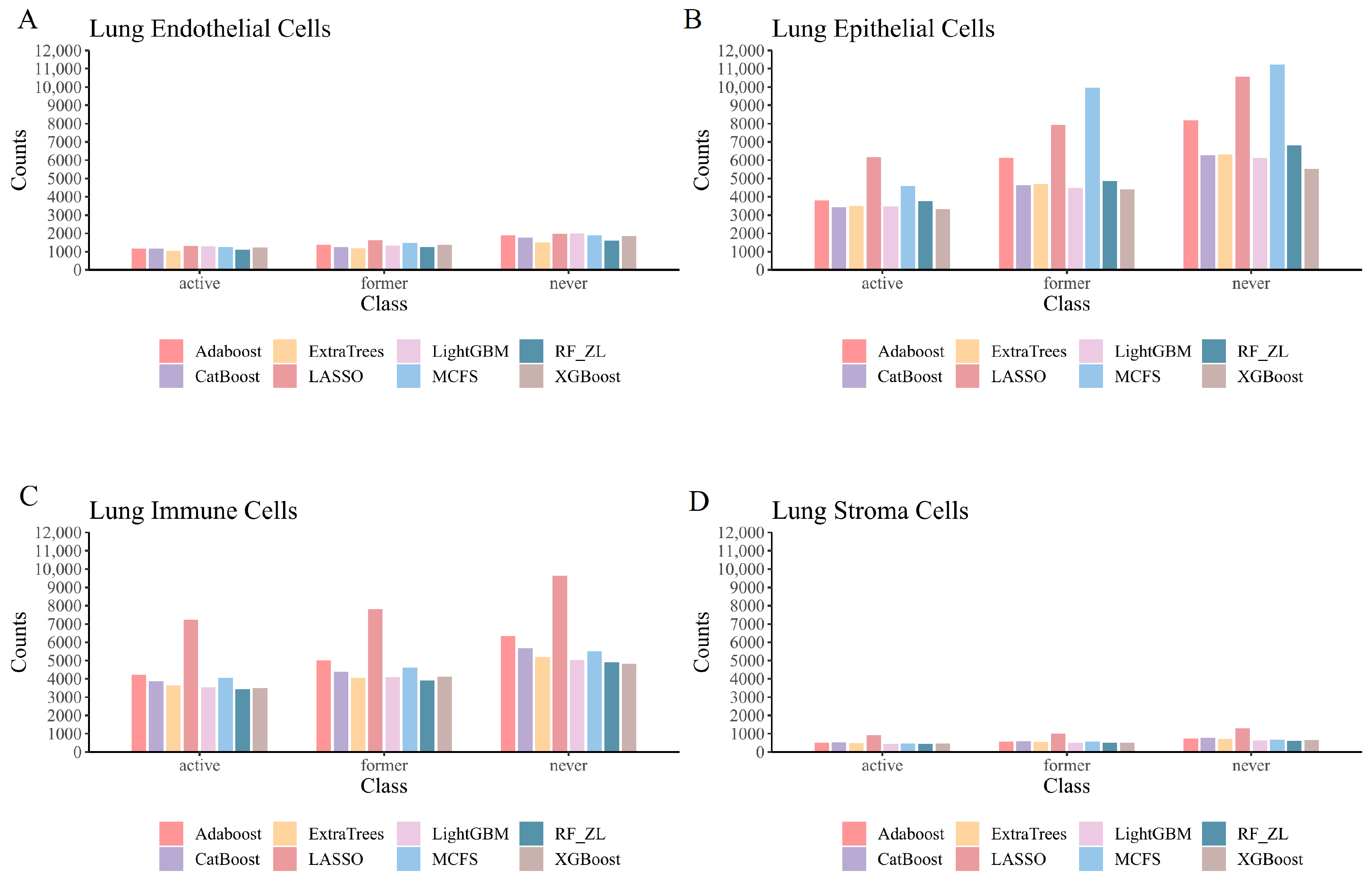
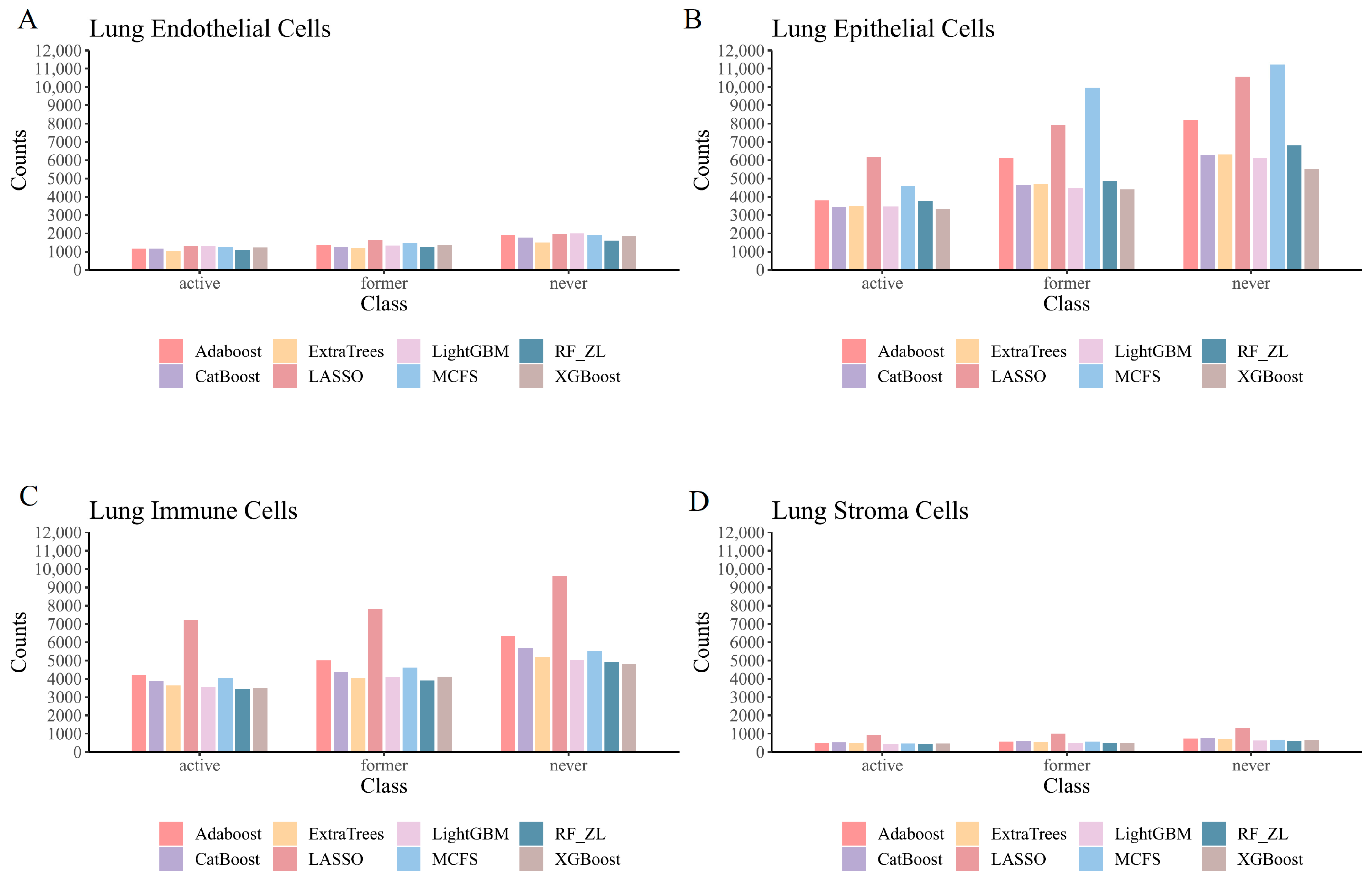
4.1.1. Qualitative Features in Lung Endothelial Cells
4.1.2. Qualitative Features in Lung Epithelial Cells
4.1.3. Qualitative Features in Lung Immune Cells
4.1.4. Qualitative Features in Lung Stroma Cells
4.2. Analysis of Decision Rules for Indicating Smoking Status in Different Lung Cell Types
4.2.1. Qualitative Rule Parameters in Lung Endothelial Cells
4.2.2. Qualitative Rule Parameters in Lung Epithelial Cells
4.2.3. Qualitative Rule Parameters in Lung Immune Cells
4.2.4. Qualitative Rule Parameters in Lung Stroma Cells
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Beck, G.J.; Doyle, C.A.; Schachter, E.N. Smoking and lung function. Am. Rev. Respir. Dis. 1981, 123, 149–155. [Google Scholar] [PubMed]
- Gold, D.R.; Wang, X.; Wypij, D.; Speizer, F.E.; Ware, J.H.; Dockery, D.W. Effects of cigarette smoking on lung function in adolescent boys and girls. N. Engl. J. Med. 1996, 335, 931–937. [Google Scholar] [CrossRef]
- Doll, R.; Hill, A.B. Smoking and carcinoma of the lung. Br. Med. J. 1950, 2, 739. [Google Scholar] [CrossRef] [PubMed]
- Forey, B.A.; Thornton, A.J.; Lee, P.N. Systematic review with meta-analysis of the epidemiological evidence relating smoking to copd, chronic bronchitis and emphysema. BMC Pulm. Med. 2011, 11, 36. [Google Scholar] [CrossRef]
- Duijts, L.; Reiss, I.K.; Brusselle, G.; de Jongste, J.C. Early origins of chronic obstructive lung diseases across the life course. Eur. J. Epidemiol. 2014, 29, 871–885. [Google Scholar] [CrossRef]
- Martinez-Pitre, P.J.; Sabbula, B.R.; Cascella, M. Restrictive lung disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Nia, P.S.; Weyler, J.; Colpaert, C.; Vermeulen, P.; Van Marck, E.; Van Schil, P. Prognostic value of smoking status in operated non-small cell lung cancer. Lung Cancer 2005, 47, 351–359. [Google Scholar]
- Toh, C.-K.; Wong, E.-H.; Lim, W.-T.; Leong, S.-S.; Fong, K.-W.; Wee, J.; Tan, E.-H. The impact of smoking status on the behavior and survival outcome of patients with advanced non-small cell lung cancer: A retrospective analysis. Chest 2004, 126, 1750–1756. [Google Scholar] [CrossRef]
- Klebe, S.; Leigh, J.; Henderson, D.W.; Nurminen, M. Asbestos, smoking and lung cancer: An update. Int. J. Environ. Res. Public Health 2020, 17, 258. [Google Scholar] [CrossRef]
- Huang, F.; Ma, Q.; Ren, J.; Li, J.; Wang, F.; Huang, T.; Cai, Y.D. Identification of smoking-associated transcriptome aberration in blood with machine learning methods. BioMed Res. Int. 2023, 2023, 5333361. [Google Scholar] [CrossRef]
- Shu, Y.; Zhu, L.; Yuan, F.; Kong, X.; Huang, T.; Cai, Y.-D. Analysis of the relationship between PM2.5 and lung cancer based on protein-protein interactions. Comb. Chem. High Throughput Screen. 2016, 19, 100–108. [Google Scholar] [CrossRef]
- Pfeifer, G.P. Smoke Signals in the DNA of Normal Lung Cells; Nature Publishing Group: London, UK, 2020. [Google Scholar]
- Sarker, A.H.; Trego, K.S.; Zhang, W.; Jacob, P., III; Snijders, A.M.; Mao, J.H.; Schick, S.F.; Cooper, P.K.; Hang, B. Thirdhand smoke exposure causes replication stress and impaired transcription in human lung cells. Environ. Mol. Mutagen. 2020, 61, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.A.; Paszkiewicz, G.M.; Hutson, A.D.; Pauly, J.L. Inflammatory response of lung macrophages and epithelial cells to tobacco smoke: A literature review of ex vivo investigations. Immunol. Res. 2010, 46, 94–126. [Google Scholar] [CrossRef]
- Ramage, L.; Jones, A.C.; Whelan, C.J. Induction of apoptosis with tobacco smoke and related products in a549 lung epithelial cells in vitro. J. Inflamm. 2006, 3, 3. [Google Scholar] [CrossRef]
- Liu, Y.; Di, Y.P. Effects of second hand smoke on airway secretion and mucociliary clearance. Front. Physiol. 2012, 3, 342. [Google Scholar] [CrossRef]
- Sharma, J.; Young, D.M.; Marentette, J.O.; Rastogi, P.; Turk, J.; McHowat, J. Lung endothelial cell platelet-activating factor production and inflammatory cell adherence are increased in response to cigarette smoke component exposure. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2012, 302, L47–L55. [Google Scholar] [CrossRef]
- Schweitzer, K.S.; Hatoum, H.; Brown, M.B.; Gupta, M.; Justice, M.J.; Beteck, B.; Van Demark, M.; Gu, Y.; Presson, R.G., Jr.; Hubbard, W.C. Mechanisms of lung endothelial barrier disruption induced by cigarette smoke: Role of oxidative stress and ceramides. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2011, 301, L836–L846. [Google Scholar] [CrossRef] [PubMed]
- Gaurav, R.; Mikuls, T.R.; Thiele, G.M.; Nelson, A.J.; Niu, M.; Guda, C.; Eudy, J.D.; Barry, A.E.; Wyatt, T.A.; Romberger, D.J. High-throughput analysis of lung immune cells in a combined murine model of agriculture dust-triggered airway inflammation with rheumatoid arthritis. PLoS ONE 2021, 16, e0240707. [Google Scholar] [CrossRef] [PubMed]
- Pontis, F.; Roz, L.; Mensah, M.; Segale, M.; Moro, M.; Bertolini, G.; Petraroia, I.; Centonze, G.; Ferretti, A.M.; Suatoni, P. Circulating extracellular vesicles from individuals at high-risk of lung cancer induce pro-tumorigenic conversion of stromal cells through transfer of mir-126 and mir-320. J. Exp. Clin. Cancer Res. 2021, 40, 237. [Google Scholar] [CrossRef] [PubMed]
- Kreus, M.; Lehtonen, S.; Hinttala, R.; Salonen, J.; Porvari, K.; Kaarteenaho, R. Nhlrc2 expression is increased in idiopathic pulmonary fibrosis. Respir. Res. 2022, 23, 206. [Google Scholar] [CrossRef]
- Huang, G.-H.; Zhang, Y.-H.; Chen, L.; Li, Y.; Huang, T.; Cai, Y.-D. Identifying lung cancer cell markers with machine learning methods and single-cell rna-seq data. Life 2021, 11, 940. [Google Scholar] [CrossRef]
- Dohmen, J.; Baranovskii, A.; Ronen, J.; Uyar, B.; Franke, V.; Akalin, A. Identifying tumor cells at the single-cell level using machine learning. Genome Biol. 2022, 23, 123. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Cao, Y.; Han, X.; Ma, X.; Li, R.; Wang, R.; Xiao, L.; Xie, L. Revealing exph5 as a potential diagnostic gene biomarker of the late stage of copd based on machine learning analysis. Comput. Biol. Med. 2023, 154, 106621. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, D.; Liao, H.; Zhang, S.; Guo, W.; Chen, L.; Lu, L.; Huang, T.; Cai, Y.-D. Exploring the genomic patterns in human and mouse cerebellums via single-cell sequencing and machine learning method. Front. Genet. 2022, 13, 857851. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.-K.; Park, S.; Lee, K.-H.; Jeong, D.; Woo, J.; Park, J.; Yi, S.-M.; Han, D.; Yoo, C.-G.; Kim, S. Machine learning-based proteomics reveals ferroptosis in copd patient-derived airway epithelial cells upon smoking exposure. J. Korean Med. Sci. 2023, 38, e220. [Google Scholar] [CrossRef]
- Sikkema, L.; Ramírez-Suástegui, C.; Strobl, D.C.; Gillett, T.E.; Zappia, L.; Madissoon, E.; Markov, N.S.; Zaragosi, L.E.; Ji, Y.; Ansari, M.; et al. An integrated cell atlas of the lung in health and disease. Nat. Med. 2023, 29, 1563–1577. [Google Scholar] [CrossRef] [PubMed]
- Freund, Y.; Schapire, R.E. A decision-theoretic generalization of on-line learning and an application to boosting. J. Comput. Syst. Sci. 1997, 55, 119–139. [Google Scholar] [CrossRef]
- Dorogush, A.V.; Ershov, V.; Gulin, A. Catboost: Gradient boosting with categorical features support. arXiv 2018, arXiv:1810.11363. [Google Scholar]
- Geurts, P.; Ernst, D.; Wehenkel, L. Extremely randomized trees. Mach. Learn. 2006, 63, 3–42. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B (Methodol.) 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.-Y. Lightgbm: A highly efficient gradient boosting decision tree. Adv. Neural Inf. Process. Syst. 2017, 30, 3146–3154. [Google Scholar]
- Draminski, M.; Rada-Iglesias, A.; Enroth, S.; Wadelius, C.; Koronacki, J.; Komorowski, J. Monte carlo feature selection for supervised classification. Bioinformatics 2008, 24, 110–117. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; Association for Computing Machinery: New York, NY, USA, 2016; pp. 785–794. [Google Scholar]
- Liu, H.A.; Setiono, R. Incremental feature selection. Appl. Intell. 1998, 9, 217–230. [Google Scholar] [CrossRef]
- Safavian, S.R.; Landgrebe, D. A survey of decision tree classifier methodology. IEEE Trans. Syst. Man Cybern. 1991, 21, 660–674. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Kohavi, R. A study of cross-validation and bootstrap for accuracy estimation and model selection. In Proceedings of the International Joint Conference on Artificial Intelligence, Montreal, QC, Canada, 20–25 August 1995; Lawrence Erlbaum Associates Ltd: Mahwah, NJ, USA; pp. 1137–1145. [Google Scholar]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. Smote: Synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- ElKarami, B.; Deebajah, M.; Polk, S.; Peabody, J.; Shahrrava, B.; Menon, M.; Alkhateeb, A.; Alanee, S. Machine learning-based prediction of upgrading on magnetic resonance imaging targeted biopsy in patients eligible for active surveillance. Urol. Oncol. 2022, 40, 191.e115–191.e120. [Google Scholar] [CrossRef] [PubMed]
- Yagin, F.H.; Alkhateeb, A.; Raza, A.; Samee, N.A.; Mahmoud, N.F.; Colak, C.; Yagin, B. An explainable artificial intelligence model proposed for the prediction of myalgic encephalomyelitis/chronic fatigue syndrome and the identification of distinctive metabolites. Diagnostics 2023, 13, 3495. [Google Scholar] [CrossRef]
- Powers, D. Evaluation: From precision, recall and f-measure to roc., informedness, markedness & correlation. J. Mach. Learn. Technol. 2011, 2, 37–63. [Google Scholar]
- Chen, L.; Chen, Y. Rmtlysptm: Recognizing multiple types of lysine ptm sites by deep analysis on sequences. Brief. Bioinform. 2024, 25, bbad450. [Google Scholar] [CrossRef]
- Ren, J.; Gao, Q.; Zhou, X.; Chen, L.; Guo, W.; Feng, K.; Huang, T.; Cai, Y.-D. Identification of key gene expression associated with quality of life after recovery from covid-19. Med. Biol. Eng. Comput. 2024, 62, 1031–1048. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Chen, L.; Guo, W.; Feng, K.; Huang, T.; Cai, Y.-D. Patterns of gene expression profiles associated with colorectal cancer in colorectal mucosa by using machine learning methods. Comb. Chem. High Throughput Screen. 2024. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhou, X.; Huang, K.; Chen, L.; Guo, W.; Feng, K.; Huang, T.; Cai, Y.-D. Identification of key genes associated with persistent immune changes and secondary immune activation responses induced by influenza vaccination after COVID-19 recovery by machine learning methods. Comput. Biol. Med. 2024, 169, 107883. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, L. Prediction of drug pathway-based disease classes using multiple properties of drugs. Curr. Bioinform. 2024, in press. [CrossRef]
- Chen, L.; Chen, K.; Zhou, B. Inferring drug-disease associations by a deep analysis on drug and disease networks. Math. Biosci. Eng. 2023, 20, 14136–14157. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhao, X. Pcda-hnmp: Predicting circrna-disease association using heterogeneous network and meta-path. Math. Biosci. Eng. 2023, 20, 20553–20575. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, C.; Xu, J. Predictefc: A fast and efficient multi-label classifier for predicting enzyme family classes. BMC Bioinform. 2024, 25, 50. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Ran, B.; Chen, L. A graphsage-based model with fingerprints only to predict drug-drug interactions. Math. Biosci. Eng. 2024, 21, 2922–2942. [Google Scholar] [CrossRef] [PubMed]
- Matthews, B. Comparison of the predicted and observed secondary structure of t4 phage lysozyme. Biochim. Biophys. Acta (BBA)-Protein Struct. 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Gorodkin, J. Comparing two k-category assignments by a k-category correlation coefficient. Comput. Biol. Chem. 2004, 28, 367–374. [Google Scholar] [CrossRef]
- Amighi, J.; Hoke, M.; Mlekusch, W.; Schlager, O.; Exner, M.; Haumer, M.; Pernicka, E.; Koppensteiner, R.; Minar, E.; Rumpold, H.; et al. Beta 2 microglobulin and the risk for cardiovascular events in patients with asymptomatic carotid atherosclerosis. Stroke 2011, 42, 1826–1833. [Google Scholar] [CrossRef] [PubMed]
- Stanga, Z.; Nock, S.; Medina-Escobar, P.; Nydegger, U.E.; Risch, M.; Risch, L. Factors other than the glomerular filtration rate that determine the serum beta-2-microglobulin level. PLoS ONE 2013, 8, e72073. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Yun, K.J.; Chun, H.J.; Jang, E.H.; Han, K.D.; Park, Y.M.; Baek, K.H.; Song, K.H.; Cha, B.Y.; Park, C.S.; et al. Clinical utility of serum beta-2-microglobulin as a predictor of diabetic complications in patients with type 2 diabetes without renal impairment. Diabetes Metab. 2014, 40, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Lin, Y.; Zhai, X.; Cheng, L. Association of beta-2-microglobulin with coronary heart disease and all-cause mortality in the united states general population. Front. Cardiovasc. Med. 2022, 9, 834150. [Google Scholar] [CrossRef] [PubMed]
- Sultana, A.; Alam, M.S.; Liu, X.; Sharma, R.; Singla, R.K.; Gundamaraju, R.; Shen, B. Single-cell rna-seq analysis to identify potential biomarkers for diagnosis, and prognosis of non-small cell lung cancer by using comprehensive bioinformatics approaches. Transl. Oncol. 2023, 27, 101571. [Google Scholar] [CrossRef] [PubMed]
- Warren, G.W.; Cummings, K.M. Tobacco and lung cancer: Risks, trends, and outcomes in patients with cancer. In American Society of Clinical Oncology Educational Book; American Society of Clinical Oncology: Alexandria, VA, USA, 2013; pp. 359–364. [Google Scholar]
- Xu, B.; Liu, L.; Song, G. Functions and regulation of translation elongation factors. Front. Mol. Biosci. 2022, 8, 816398. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhuan, B.; Yan, Y.; Jiang, S.; Wang, T. Identification of gene markers in the development of smoking-induced lung cancer. Gene 2016, 576, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Hangai, S.; Kawamura, T.; Kimura, Y.; Chang, C.Y.; Hibino, S.; Yamamoto, D.; Nakai, Y.; Tateishi, R.; Oshima, M.; Oshima, H.; et al. Orchestration of myeloid-derived suppressor cells in the tumor microenvironment by ubiquitous cellular protein tctp released by tumor cells. Nat. Immunol. 2021, 22, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.E.; Cober, N.D.; Dai, Z.; Stewart, D.J.; Zhao, Y.Y. Endothelial cells in the pathogenesis of pulmonary arterial hypertension. Eur. Respir. J. 2021, 58, 2003957. [Google Scholar] [CrossRef] [PubMed]
- Mostafaei, S.; Kazemnejad, A.; Azimzadeh Jamalkandi, S.; Amirhashchi, S.; Donnelly, S.C.; Armstrong, M.E.; Doroudian, M. Identification of novel genes in human airway epithelial cells associated with chronic obstructive pulmonary disease (copd) using machine-based learning algorithms. Sci. Rep. 2018, 8, 15775. [Google Scholar] [CrossRef]
- Li, L.; Wang, X. Ferroptosis-associated cholesterol metabolism regulated by p85α in human bronchial epithelial cells with smoking. Clin. Transl. Discov. 2022, 2, e30. [Google Scholar] [CrossRef]
- Rathnayake, S.N.H.; Ditz, B.; Willemse, B.W.M.; Timens, W.; Kooistra, W.; Heijink, I.H.; Oliver, B.G.G.; van den Berge, M.; Faiz, A. Longitudinal effects of 1-year smoking cessation on human bronchial epithelial transcriptome. Chest 2023, 164, 85–89. [Google Scholar] [CrossRef]
- Jonckheere, A.I.; Smeitink, J.A.; Rodenburg, R.J. Mitochondrial atp synthase: Architecture, function and pathology. J. Inherit. Metab. Dis. 2012, 35, 211–225. [Google Scholar] [CrossRef]
- Rehbini, O. Mitochondrial epigenetic roles in cigarette smoke and copd. EC Pulmonol. Respir. Med. 2020, 9, 120–145. [Google Scholar]
- Seow, W.J.; Hu, W.; Dai, Y.; Vermeulen, R.; Byun, H.M.; Wong, J.Y.Y.; Bassig, B.A.; Blechter, B.; Duan, H.; Niu, Y.; et al. Association between diesel exhaust exposure and mitochondrial DNA methylation. Carcinogenesis 2022, 43, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Shankarkumar, U. The human leukocyte antigen (hla) system. Int. J. Hum. Genet. 2004, 4, 91–103. [Google Scholar] [CrossRef]
- Karlson, E.W.; Chang, S.-C.; Cui, J.; Chibnik, L.B.; Fraser, P.A.; De Vivo, I.; Costenbader, K.H. Gene–environment interaction between hla-drb1 shared epitope and heavy cigarette smoking in predicting incident rheumatoid arthritis. Ann. Rheum. Dis. 2010, 69, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Vassallo, R.; Luckey, D.; Behrens, M.; Madden, B.; Luthra, H.; David, C.; Taneja, V. Cellular and humoral immunity in arthritis are profoundly influenced by the interaction between cigarette smoke effects and host hla-dr and dq genes. Clin. Immunol. 2014, 152, 25–35. [Google Scholar] [CrossRef]
- Mkorombindo, T.; Tran-Nguyen, T.K.; Yuan, K.; Zhang, Y.; Xue, J.; Criner, G.J.; Kim, Y.I.; Pilewski, J.M.; Gaggar, A.; Cho, M.H.; et al. Hla-c and kir permutations influence chronic obstructive pulmonary disease risk. JCI Insight 2021, 6, e150187. [Google Scholar] [CrossRef]
- Liu, L.; Song, X.; Li, X.; Xue, L.; Ding, S.; Niu, L.; Xie, L.; Song, X. A three-platelet mrna set: Max, mturn and hla-b as biomarker for lung cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 2713–2723. [Google Scholar] [CrossRef]
- Luo, W.; Zeng, Z.; Jin, Y.; Yang, L.; Fan, T.; Wang, Z.; Pan, Y.; Yang, Y.; Yao, M.; Li, Y.; et al. Distinct immune microenvironment of lung adenocarcinoma in never-smokers from smokers. Cell Rep. Med. 2023, 4, 101078. [Google Scholar] [CrossRef]
- Chen, Y.; Feng, Y.; Chen, S. Hsp90b1 overexpression is associated with poor prognosis in tongue squamous cell carcinoma. J. Stomatol. Oral Maxillofac. Surg. 2022, 123, e833–e838. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Hopke, P.K. Concentration of reactive oxygen species (ros) in mainstream and sidestream cigarette smoke. Aerosol Sci. Technol. 2012, 46, 191–197. [Google Scholar] [CrossRef]
- Liu, K.; Kang, M.; Li, J.; Qin, W.; Wang, R. Prognostic value of the mrna expression of members of the hsp90 family in non-small cell lung cancer. Exp. Ther. Med. 2019, 17, 2657–2665. [Google Scholar] [CrossRef]
- Xiao, X.; Yeoh, B.S.; Vijay-Kumar, M. Lipocalin 2: An emerging player in iron homeostasis and inflammation. Annu. Rev. Nutr. 2017, 37, 103–130. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Asai, K.; Gi, M.; Kojima, K.; Kakehashi, A.; Oishi, Y.; Matsue, T.; Yukimatsu, N.; Hirata, K.; Kawaguchi, T.; et al. Response biomarkers of inhalation exposure to cigarette smoke in the mouse lung. J. Toxicol. Pathol. 2022, 35, 247–254. [Google Scholar] [CrossRef]
- Gordon, W.; Galitovskiy, V.; Edwards, R.; Andersen, B.; Grando, S.A. The tobacco carcinogen nitrosamine induces a differential gene expression response in tumour susceptible a/j and resistant c3h mouse lungs. Eur. J. Cancer 2013, 49, 725–733. [Google Scholar] [CrossRef]
- Gebel, S.; Gerstmayer, B.; Kuhl, P.; Borlak, J.; Meurrens, K.; Müller, T. The kinetics of transcriptomic changes induced by cigarette smoke in rat lungs reveals a specific program of defense, inflammation, and circadian clock gene expression. Toxicol. Sci. Off. J. Soc. Toxicol. 2006, 93, 422–431. [Google Scholar] [CrossRef]
- Tyagi, A.; Sharma, S.; Wu, K.; Wu, S.Y.; Xing, F.; Liu, Y.; Zhao, D.; Deshpande, R.P.; D’Agostino, R.B., Jr.; Watabe, K. Nicotine promotes breast cancer metastasis by stimulating n2 neutrophils and generating pre-metastatic niche in lung. Nat. Commun. 2021, 12, 474. [Google Scholar] [CrossRef]
- Fuior, E.V.; Gafencu, A.V. Apolipoprotein c1: Its pleiotropic effects in lipid metabolism and beyond. Int. J. Mol. Sci. 2019, 20, 5939. [Google Scholar] [CrossRef]
- Shi, X.; Wang, J.; Dai, S.; Qin, L.; Zhou, J.; Chen, Y. Apolipoprotein c1 (apoc1): A novel diagnostic and prognostic biomarker for cervical cancer. Onco Targets Ther. 2020, 13, 12881–12891. [Google Scholar] [CrossRef] [PubMed]
- Hussein Ali, E.; Hussein Abod Al-Khafaji, K.; Hassan Abood, A. Effect of smoking on low-density lipoproteins level in human. Arch. Razi Inst. 2022, 77, 1971–1974. [Google Scholar]
- Nakamura, M.; Yamamoto, Y.; Imaoka, W.; Kuroshima, T.; Toragai, R.; Ito, Y.; Kanda, E.; Schaefer, E.J.; Ai, M. Relationships between smoking status, cardiovascular risk factors, and lipoproteins in a large japanese population. J. Atheroscler. Thromb. 2021, 28, 942–953. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Liu, Q.; Li, M.; Yao, Y.; Qi, F.; Xu, Y.; Lu, S.; Yang, Z.; Guan, Y.; Li, M.D.; et al. Determination of genetic correlation between tobacco smoking and coronary artery disease. Front. Psychiatry 2023, 14, 1279962. [Google Scholar] [CrossRef]
- Li, Y.-Z.; Xie, J.; Wang, R.-Q.; Gao, X.-Q.; Liu, P.-J.; Liu, J. Klf2 is a clinical diagnostic and treatment biomarker of breast cancer. Front. Cell Dev. Biol. 2023, 11, 1182123. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.; Jain, M. Role of krüppel-like factors in endothelial cell function and shear stress–mediated vasoprotection. In Modern Mechanobiology; Taylor & Francis Group: Oxford, UK, 2021; pp. 23–56. [Google Scholar]
- El-Mahdy, M.A.; Abdelghany, T.M.; Hemann, C.; Ewees, M.G.; Mahgoup, E.M.; Eid, M.S.; Shalaan, M.T.; Alzarie, Y.A.; Zweier, J.L. Chronic cigarette smoke exposure triggers a vicious cycle of leukocyte and endothelial-mediated oxidant stress that results in vascular dysfunction. Am. J. Physiol.-Heart Circ. Physiol. 2020, 319, H51–H65. [Google Scholar] [CrossRef]
- Angel, P.; Karin, M. The role of jun, fos and the ap-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta (BBA)-Rev. Cancer 1991, 1072, 129–157. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, Y.; Besnard, V.; Ikegami, M.; Wert, S.E.; Heffner, C.; Murray, S.A.; Donahue, L.R.; Whitsett, J.A. Transcriptional programs controlling perinatal lung maturation. PLoS ONE 2012, 7, e37046. [Google Scholar]
- Bhalla, D.K.; Hirata, F.; Rishi, A.K.; Gairola, C.G. Cigarette smoke, inflammation, and lung injury: A mechanistic perspective. J. Toxicol. Environ. Health Part B 2009, 12, 45–64. [Google Scholar] [CrossRef]
- Zhou, L.; Xue, C.; Chen, Z.; Jiang, W.; He, S.; Zhang, X. C-fos is a mechanosensor that regulates inflammatory responses and lung barrier dysfunction during ventilator-induced acute lung injury. BMC Pulm. Med. 2022, 22, 9. [Google Scholar] [CrossRef]
- Liang, Z.; Zhang, Y.; Xu, Y.; Zhang, X.; Wang, Y. Hesperidin inhibits tobacco smoke-induced pulmonary cell proliferation and emt in mouse lung tissues via the p38 signaling pathway. Oncol. Lett. 2023, 25, 30. [Google Scholar] [CrossRef] [PubMed]
- Shieh, T.M.; Liu, C.J.; Hsia, S.M.; Ningrum, V.; Liao, C.C.; Lan, W.C.; Shih, Y.H. Lack of salivary long non-coding rna xist expression is associated with increased risk of oral squamous cell carcinoma: A cross-sectional study. J. Clin. Med. 2021, 10, 4622. [Google Scholar] [CrossRef] [PubMed]
- Soldin, O.P.; Makambi, K.H.; Soldin, S.J.; O’Mara, D.M. Steroid hormone levels associated with passive and active smoking. Steroids 2011, 76, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Grimm, S.L.; Dong, X.; Zhang, Y.; Carisey, A.F.; Arnold, A.P.; Moorthy, B.; Coarfa, C.; Lingappan, K. Effect of sex chromosomes versus hormones in neonatal lung injury. JCI Insight 2021, 6, e146863. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Jiang, P.; Chen, J.; Yang, Y.; Guo, X. Xist promotes apoptosis and the inflammatory response in cse-stimulated cells via the mir-200c-3p/egr3 axis. BMC Pulm. Med. 2021, 21, 215. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ma, X.; An, Y.; Sun, Y.; Dou, W.; Li, M.; Bao, H.; Zhang, C. Green tea polyphenols alleviate hydrogen peroxide-induced oxidative stress, inflammation, and apoptosis in bovine mammary epithelial cells by activating erk1/2–nfe2l2–hmox1 pathways. Front. Vet. Sci. 2022, 8, 804241. [Google Scholar] [CrossRef]
- Inesta-Vaquera, F.; Navasumrit, P.; Henderson, C.J.; Frangova, T.G.; Honda, T.; Dinkova-Kostova, A.T.; Ruchirawat, M.; Wolf, C.R. Application of the in vivo oxidative stress reporter hmox1 as mechanistic biomarker of arsenic toxicity. Environ. Pollut. 2021, 270, 116053. [Google Scholar] [CrossRef]
- van der Vaart, H.; Postma, D.S.; Timens, W.; Ten Hacken, N.H. Acute effects of cigarette smoke on inflammation and oxidative stress: A review. Thorax 2004, 59, 713–721. [Google Scholar] [CrossRef]
- Lee, J.S.; Park, S.J.; Cho, Y.S.; Huh, J.W.; Oh, Y.-M.; Lee, S.-D. Role of amp-activated protein kinase (ampk) in smoking-induced lung inflammation and emphysema. Tuberc. Respir. Dis. 2015, 78, 8–17. [Google Scholar] [CrossRef]
- Ryter, S.W. Heme oxygenase-1: An anti-inflammatory effector in cardiovascular, lung, and related metabolic disorders. Antioxidants 2022, 11, 555. [Google Scholar] [CrossRef]
- Xu, M.; Yang, W.; Wang, X.; Nayak, D.K. Lung secretoglobin scgb1a1 influences alveolar macrophage-mediated inflammation and immunity. Front. Immunol. 2020, 11, 584310. [Google Scholar] [CrossRef] [PubMed]
- Schamberger, A.C.; Staab-Weijnitz, C.A.; Mise-Racek, N.; Eickelberg, O. Cigarette smoke alters primary human bronchial epithelial cell differentiation at the air-liquid interface. Sci. Rep. 2015, 5, 8163. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, S.; Ito, S. Repeated whole cigarette smoke exposure alters cell differentiation and augments secretion of inflammatory mediators in air-liquid interface three-dimensional co-culture model of human bronchial tissue. Toxicol. Vitr. 2017, 38, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Malvin, N.P.; Kern, J.T.; Liu, T.-C.; Brody, S.L.; Stappenbeck, T.S. Autophagy proteins are required for club cell structure and function in airways. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2019, 317, L259–L270. [Google Scholar] [CrossRef] [PubMed]
- Firoz, A.; Ravanan, P.; Saha, P.; Prashar, T.; Talwar, P. Genome-wide screening and identification of potential kinases involved in endoplasmic reticulum stress responses. Life Sci. 2023, 317, 121452. [Google Scholar] [CrossRef]
- Somborac-Bačura, A.; van der Toorn, M.; Franciosi, L.; Slebos, D.J.; Žanić-Grubišić, T.; Bischoff, R.; van Oosterhout, A.J. Cigarette smoke induces endoplasmic reticulum stress response and proteasomal dysfunction in human alveolar epithelial cells. Exp. Physiol. 2013, 98, 316–325. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | ENSEMBL ID | Gene Symbol | Description |
|---|---|---|---|
| lung endothelial cells | ENSG00000166710 | B2M | beta-2-microglobulin |
| ENSG00000156508 | EEF1A1 | eukaryotic translation elongation factor 1 alpha 1 | |
| ENSG00000133112 | TPT1 | tumor protein, translationally-controlled 1 | |
| lung epithelial cells | ENSG00000087086 | FTL | ferritin light chain |
| ENSG00000228253 | MT-ATP8 | mitochondrially encoded ATP synthase 8 | |
| lung immune cells | ENSG00000234745 | HLA-B | major histocompatibility complex, class I, B |
| ENSG00000204525 | HLA-C | major histocompatibility complex, class I, C | |
| lung stroma cells | ENSG00000166598 | HSP90B1 | heat shock protein 90 beta family member 1 |
| ENSG00000148346 | LCN2 | lipocalin 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Q.; Shen, Y.; Guo, W.; Feng, K.; Huang, T.; Cai, Y. Machine Learning Reveals Impacts of Smoking on Gene Profiles of Different Cell Types in Lung. Life 2024, 14, 502. https://doi.org/10.3390/life14040502
Ma Q, Shen Y, Guo W, Feng K, Huang T, Cai Y. Machine Learning Reveals Impacts of Smoking on Gene Profiles of Different Cell Types in Lung. Life. 2024; 14(4):502. https://doi.org/10.3390/life14040502
Chicago/Turabian StyleMa, Qinglan, Yulong Shen, Wei Guo, Kaiyan Feng, Tao Huang, and Yudong Cai. 2024. "Machine Learning Reveals Impacts of Smoking on Gene Profiles of Different Cell Types in Lung" Life 14, no. 4: 502. https://doi.org/10.3390/life14040502