

A Probiotic Amylase Blend Positively Impacts Gut Microbiota Modulation in a Randomized, Placebo-Controlled, Double-Blind Study

Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Design
2.2. Study Participants
2.3. Stool Kits
2.4. Microbiota Analyses
2.4.1. DNA Extraction and PCR Amplification
2.4.2. Library Preparation and Sequencing
2.5. Bioinformatics
2.5.1. Pre-Processing
2.5.2. Data Preparation
2.5.3. Data Cleaning and QC
2.5.4. Data Analysis
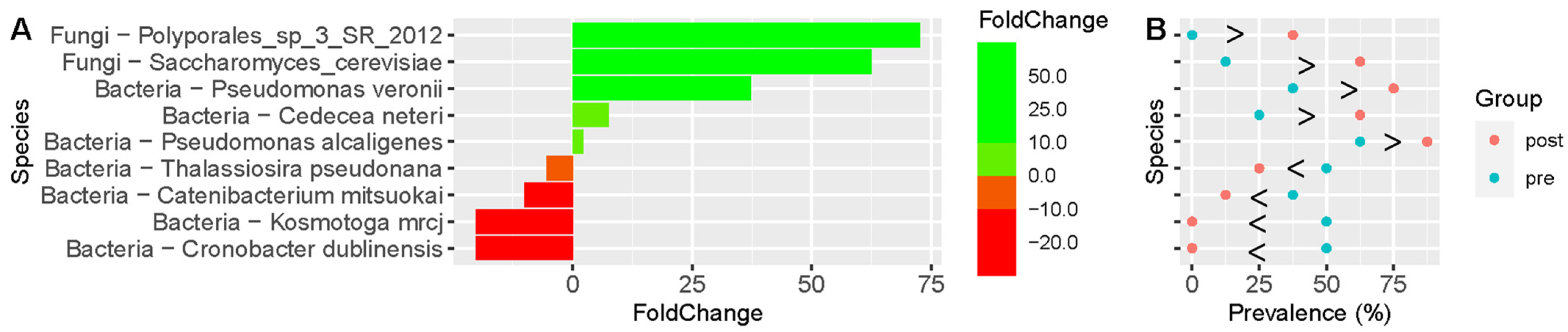
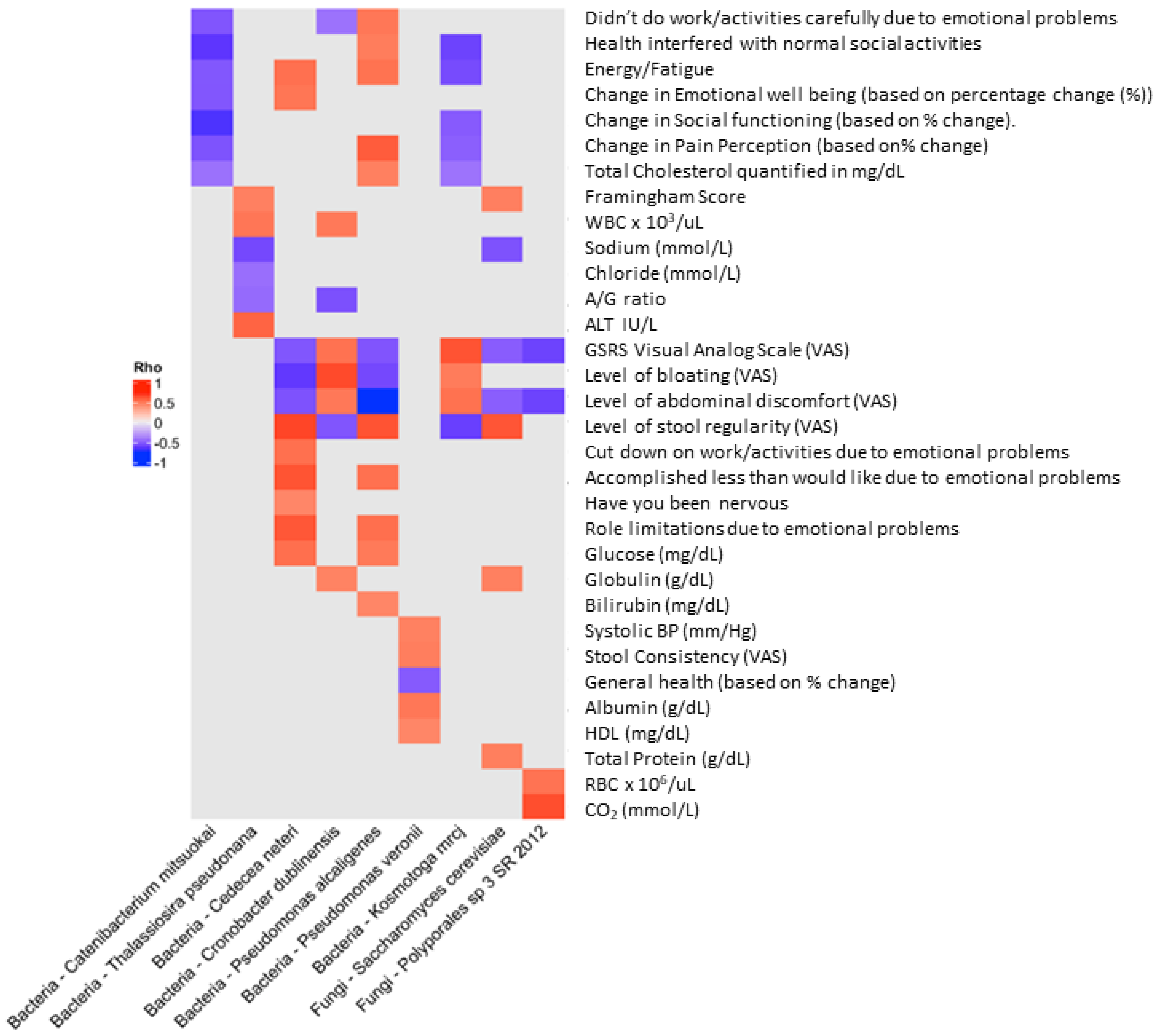
3. Results
Microbiota Analyses
4. Discussion
5. Limitations and Considerations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mukherjee, P.K.; Sendid, B.; Hoarau, G.; Colombel, J.-F.; Poulain, D.; Ghannoum, M.A. Mycobiota in gastrointestinal diseases. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S.; et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Zmora, N.; Soffer, E.; Elinav, E. Transforming medicine with the microbiome. Sci. Transl. Med. 2019, 11, eaaw1815. [Google Scholar] [CrossRef]
- Butel, M.J. Probiotics, gut microbiota and health. Med. Mal. Infect. 2014, 44, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wallace, C.J.K.; Milev, R. The effects of probiotics on depressive symptoms in humans: A systematic review. Ann. Gen. Psychiatry 2017, 16, 14. [Google Scholar] [CrossRef] [PubMed]
- Bubnov, R.V.; Babenko, L.P.; Lazarenko, L.M.; Mokrozub, V.V.; Spivak, M.Y. Specific properties of probiotic strains: Relevance and benefits for the host. EPMA J. 2018, 9, 205–223. [Google Scholar] [CrossRef] [PubMed]
- La Monica, M.B.; Raub, B.; Lopez, H.L.; Ziegenfuss, T.N. A probiotic amylase blend reduces gastrointestinal symptoms in a randomised clinical study. Benef. Microbes 2023, 14, 459–476. [Google Scholar] [CrossRef] [PubMed]
- Hager, C.L.; Isham, N.; Schrom, K.P.; Chandra, J.; McCormick, T.; Miyagi, M.; Ghannoum, M.A. Effects of a Novel Probiotic Combination on Pathogenic Bacterial-Fungal Polymicrobial Biofilms. mBio 2019, 10, e00338-19. [Google Scholar] [CrossRef] [PubMed]
- Argolo-Filho, R.C.; Loguercio, L.L. Bacillus thuringiensis Is an Environmental Pathogen and Host-Specificity Has Developed as an Adaptation to Human-Generated Ecological Niches. Insects 2013, 5, 62–91. [Google Scholar] [CrossRef]
- Zhang, Y.; Min, S.; Sun, Y.; Ye, J.; Zhou, Z.; Li, H. Characteristics of population structure, antimicrobial resistance, virulence factors, and morphology of methicillin-resistant Macrococcus caseolyticus in global clades. BMC Microbiol. 2022, 22, 266. [Google Scholar] [CrossRef]
- Ghannoum, M.A.; McCormick, T.S.; Retuerto, M.; Bebek, G.; Cousineau, S.; Hartman, L.; Barth, C.; Schrom, K. Evaluation of Microbiome Alterations Following Consumption of BIOHM, a Novel Probiotic. Curr. Issues Mol. Biol. 2021, 43, 2135–2146. [Google Scholar] [CrossRef] [PubMed]
- Palm, K.M.; Abrams, M.K.; Sears, S.B.; Wherley, S.D.; Alfahmy, A.M.; Kamumbu, S.A.; Chakraborty, N.N.; Mahajan, S.T.; El-Nashar, S.A.; Henderson, J.W.; et al. The Response of the Urinary Microbiome to Botox. Int. Urogynecol. J. 2024, 35, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Ross, I. R: A Language for Data Analysis and Graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.A.; Hugenholtz, F.; Lahti, L.; Smidt, H.; de Vos, W.M. Intestinal microbiome landscaping: Insight in community assemblage and implications for microbial modulation strategies. FEMS Microbiol. Rev. 2017, 41, 182–199. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Kuhbacher, A.; Burger-Kentischer, A.; Rupp, S. Interaction of Candida Species with the Skin. Microorganisms 2017, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Abarenkov, K.; Kristiansson, E.; Ryberg, M.; Nogal-Prata, S.; Gomez-Martinez, D.; Stuer-Patowsky, K.; Jansson, T.; Polme, S.; Ghobad-Nejhad, M.; Corcoll, N.; et al. The curse of the uncultured fungus. MycoKeys 2022, 86, 177–194. [Google Scholar] [CrossRef]
- Hallen-Adams, H.E.; Suhr, M.J. Fungi in the healthy human gastrointestinal tract. Virulence 2017, 8, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, M.; Abawi, G.S.; Smart, C.D.; Hodge, K.T. Diversity and Aggressiveness of Rhizoctonia solani and Rhizoctonia-like Fungi on Vegetables in New York. Plant Dis. 2009, 93, 615–624. [Google Scholar] [CrossRef]
- Souza, P.M.; Werneck, G.; Aliakbarian, B.; Siqueira, F.; Ferreira Filho, E.X.; Perego, P.; Converti, A.; Magalhaes, P.O.; Junior, A.P. Production, purification and characterization of an aspartic protease from Aspergillus foetidus. Food Chem. Toxicol. 2017, 109, 1103–1110. [Google Scholar] [CrossRef]
- Zhang, W.; Zhuo, X.; Hu, L.; Zhang, X. Effects of Crude beta-Glucosidases from Issatchenkia terricola, Pichia kudriavzevii, Metschnikowia pulcherrima on the Flavor Complexity and Characteristics of Wines. Microorganisms 2020, 8, 953. [Google Scholar] [CrossRef]
- Abid, R.; Waseem, H.; Ali, J.; Ghazanfar, S.; Muhammad Ali, G.; Elasbali, A.M.; Alharethi, S.H. Probiotic Yeast Saccharomyces: Back to Nature to Improve Human Health. J. Fungi 2022, 8, 444. [Google Scholar] [CrossRef]
- Garcia, G.; Dogi, C.; de Moreno de LeBlanc, A.; Greco, C.; Cavaglieri, L. Gut-borne Saccharomyces cerevisiae, a promising candidate for the formulation of feed additives, modulates immune system and gut microbiota. Benef. Microbes 2016, 7, 659–668. [Google Scholar] [CrossRef]
- Sun, S.; Xu, X.; Liang, L.; Wang, X.; Bai, X.; Zhu, L.; He, Q.; Liang, H.; Xin, X.; Wang, L.; et al. Lactic Acid-Producing Probiotic Saccharomyces cerevisiae Attenuates Ulcerative Colitis via Suppressing Macrophage Pyroptosis and Modulating Gut Microbiota. Front. Immunol. 2021, 12, 777665. [Google Scholar] [CrossRef]
- Govrins, M.; Lass-Florl, C. Candida parapsilosis complex in the clinical setting. Nat. Rev. Microbiol. 2024, 22, 46–59. [Google Scholar] [CrossRef]
- Duysburgh, C.; Miclotte, L.; Green, J.B.; Watts, K.T.; Sardi, M.I.; Chakrabarti, A.; Khafipour, E.; Marzorati, M. Saccharomyces cerevisiae derived postbiotic alters gut microbiome metabolism in the human distal colon resulting in immunomodulatory potential in vitro. Front. Microbiol. 2024, 15, 1358456. [Google Scholar] [CrossRef]
- Zoumpourtikoudi, V.; Pyrgelis, N.; Chatzigrigoriou, M.; Tasakis, R.N.; Touraki, M. Interactions among yeast and probiotic bacteria enhance probiotic properties and metabolism offering augmented protection to Artemia franciscana against Vibrio anguillarum. Microb. Pathog. 2018, 125, 497–506. [Google Scholar] [CrossRef]
- Pais, P.; Oliveira, J.; Almeida, V.; Yilmaz, M.; Monteiro, P.T.; Teixeira, M.C. Transcriptome-wide differences between Saccharomyces cerevisiae and Saccharomyces cerevisiae var. boulardii: Clues on host survival and probiotic activity based on promoter sequence variability. Genomics 2021, 113, 530–539. [Google Scholar] [CrossRef]
- Ansari, F.; Alian Samakkhah, S.; Bahadori, A.; Jafari, S.M.; Ziaee, M.; Khodayari, M.T.; Pourjafar, H. Health-promoting properties of Saccharomyces cerevisiae var. boulardii as a probiotic; characteristics, isolation, and applications in dairy products. Crit. Rev. Food Sci. Nutr. 2023, 63, 457–485. [Google Scholar] [CrossRef]
- Badia, R.; Brufau, M.T.; Guerrero-Zamora, A.M.; Lizardo, R.; Dobrescu, I.; Martin-Venegas, R.; Ferrer, R.; Salmon, H.; Martinez, P.; Brufau, J. beta-Galactomannan and Saccharomyces cerevisiae var. boulardii modulate the immune response against Salmonella enterica serovar Typhimurium in porcine intestinal epithelial and dendritic cells. Clin. Vaccine Immunol. 2012, 19, 368–376. [Google Scholar] [CrossRef]
- Jawhara, S.; Habib, K.; Maggiotto, F.; Pignede, G.; Vandekerckove, P.; Maes, E.; Dubuquoy, L.; Fontaine, T.; Guerardel, Y.; Poulain, D. Modulation of intestinal inflammation by yeasts and cell wall extracts: Strain dependence and unexpected anti-inflammatory role of glucan fractions. PLoS ONE 2012, 7, e40648. [Google Scholar] [CrossRef]
- Moslehi-Jenabian, S.; Pedersen, L.L.; Jespersen, L. Beneficial effects of probiotic and food borne yeasts on human health. Nutrients 2010, 2, 449–473. [Google Scholar] [CrossRef]
- Duina, A.A.; Miller, M.E.; Keeney, J.B. Budding yeast for budding geneticists: A primer on the Saccharomyces cerevisiae model system. Genetics 2014, 197, 33–48. [Google Scholar] [CrossRef]
- Edwards-Ingram, L.; Gitsham, P.; Burton, N.; Warhurst, G.; Clarke, I.; Hoyle, D.; Oliver, S.G.; Stateva, L. Genotypic and physiological characterization of Saccharomyces boulardii, the probiotic strain of Saccharomyces cerevisiae. Appl. Environ. Microbiol. 2007, 73, 2458–2467. [Google Scholar] [CrossRef]
- Fernandez-Pacheco, P.; Pintado, C.; Briones Perez, A.; Arevalo-Villena, M. Potential Probiotic Strains of Saccharomyces and Non-Saccharomyces: Functional and Biotechnological Characteristics. J. Fungi 2021, 7, 177. [Google Scholar] [CrossRef]
- Garcia-Mazcorro, J.F.; Rodriguez-Herrera, M.V.; Marroquin-Cardona, A.G.; Kawas, J.R. The health enhancer yeast Saccharomyces cerevisiae in two types of commercial products for animal nutrition. Lett. Appl. Microbiol. 2019, 68, 472–478. [Google Scholar] [CrossRef]
- Justo, A.; Miettinen, O.; Floudas, D.; Ortiz-Santana, B.; Sjökvist, E.; Lindner, D.; Nakasone, K.; Niemelä, T.; Larsson, K.-H.; Ryvarden, L.; et al. A revised family-level classification of the Polyporales (Basidiomycota). Fungal Biol. 2017, 121, 798–824. [Google Scholar] [CrossRef]
- Suzuki, M.; Suzuki, S.; Matsui, M.; Hiraki, Y.; Kawano, F.; Shibayama, K. Genome Sequence of a Strain of the Human Pathogenic Bacterium Pseudomonas alcaligenes That Caused Bloodstream Infection. Genome Announc. 2013, 1, e00919-13. [Google Scholar] [CrossRef]
- Thompson, D.K.; Sharkady, S.M. Expanding spectrum of opportunistic Cedecea infections: Current clinical status and multidrug resistance. Int. J. Infect. Dis. 2020, 100, 461–469. [Google Scholar] [CrossRef]
- Kageyama, A.; Benno, Y. Catenibacterium mitsuokai gen. nov., sp. nov., a gram-positive anaerobic bacterium isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2000, 50 Pt 4, 1595–1599. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef]
- Han, S.; Van Treuren, W.; Fischer, C.R.; Merrill, B.D.; DeFelice, B.C.; Sanchez, J.M.; Higginbottom, S.K.; Guthrie, L.; Fall, L.A.; Dodd, D.; et al. A metabolomics pipeline for the mechanistic interrogation of the gut microbiome. Nature 2021, 595, 415–420. [Google Scholar] [CrossRef]
- Forsythe, S.J.; Dickins, B.; Jolley, K.A. Cronobacter, the emergent bacterial pathogen Enterobacter sakazakii comes of age; MLST and whole genome sequence analysis. BMC Genom. 2014, 15, 1121. [Google Scholar] [CrossRef]
- Yan, Q.; Fanning, S. Strategies for the identification and tracking of cronobacter species: An opportunistic pathogen of concern to neonatal health. Front. Pediatr. 2015, 3, 38. [Google Scholar] [CrossRef]
- Etchebehere, C.; Castello, E.; Wenzel, J.; del Pilar Anzola-Rojas, M.; Borzacconi, L.; Buitron, G.; Cabrol, L.; Carminato, V.M.; Carrillo-Reyes, J.; Cisneros-Perez, C.; et al. Microbial communities from 20 different hydrogen-producing reactors studied by 454 pyrosequencing. Appl. Microbiol. Biotechnol. 2016, 100, 3371–3384. [Google Scholar] [CrossRef]
- Fuka, M.M.; Wallisch, S.; Engel, M.; Welzl, G.; Havranek, J.; Schloter, M. Dynamics of bacterial communities during the ripening process of different Croatian cheese types derived from raw ewe’s milk cheeses. PLoS ONE 2013, 8, e80734. [Google Scholar] [CrossRef]
- Martino, P.; Micozzi, A.; Venditti, M.; Gentile, G.; Girmenia, C.; Raccah, R.; Santilli, S.; Alessandri, N.; Mandelli, F. Catheter-related right-sided endocarditis in bone marrow transplant recipients. Rev. Infect. Dis. 1990, 12, 250–257. [Google Scholar] [CrossRef]
- Ono, E.; Tohya, M.; Tada, T.; Hishinuma, T.; Watanabe, S.; Kuwahara-Arai, K.; Kirikae, T. Emergence of carbapenem-resistant Pseudomonas alcaligenes and Pseudomonas paralcaligenes clinical isolates with plasmids harbouring bla (IMP-1) in Japan. J. Med. Microbiol. 2023, 72, 001684. [Google Scholar] [CrossRef]
- Pintado, A.; Perez-Martinez, I.; Aragon, I.M.; Gutierrez-Barranquero, J.A.; de Vicente, A.; Cazorla, F.M.; Ramos, C. The Rhizobacterium Pseudomonas alcaligenes AVO110 Induces the Expression of Biofilm-Related Genes in Response to Rosellinia necatrix Exudates. Microorganisms 2021, 9, 1388. [Google Scholar] [CrossRef]
- Lugli, G.A.; Milani, C.; Mancabelli, L.; Turroni, F.; Ferrario, C.; Duranti, S.; van Sinderen, D.; Ventura, M. Ancient bacteria of the Otzi’s microbiome: A genomic tale from the Copper Age. Microbiome 2017, 5, 5. [Google Scholar] [CrossRef]
- Kharkova, A.; Perchikov, R.; Kurbanalieva, S.; Osina, K.; Popova, N.; Machulin, A.; Kamanina, O.; Saverina, E.; Saltanov, I.; Melenkov, S.; et al. Targeted Formation of Biofilms on the Surface of Graphite Electrodes as an Effective Approach to the Development of Biosensors for Early Warning Systems. Biosensors 2024, 14, 239. [Google Scholar] [CrossRef] [PubMed]
- Ansaldo, E.; Belkaid, Y. How microbiota improve immunotherapy. Science 2021, 373, 966–967. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, L.; Osme, A.; Ghannoum, M.; Cominelli, F. A Novel Probiotic Combination Ameliorates Crohn’s Disease-Like Ileitis by Increasing Short-Chain Fatty Acid Production and Modulating Essential Adaptive Immune Pathways. Inflamm. Bowel Dis. 2023, 29, 1105–1117. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Organism | Associated Clinical Features and Concomitant Increase (Inc) or Decrease (Dec) in Significantly Different Taxa | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GSRS | Inc/Dec | Flatulence | Inc/Dec | Bloating | Inc/Dec | Stool Regularity | Inc/Dec | Constipation | Inc/Dec | Abdominal Discomfort | Inc/Dec | |
| Saccharomyces cerevisiae ^ | Y |  | Y | | Y | | Y | | Y | | Y | |
| Bifidiobacterium pseudolongum | Y |  | Y | | Y | | Y | | Y | | ||
| Pseudomonas alcaligenes ^ | Y | | Y | | Y | | Y | | ||||
| Clostridium aldenense ^ | Y | | Y | | Y | | Y | | ||||
| Pseudomonas veronii | Y | | Y | | Y | | ||||||
| Cedecea neteri ^ | Y | | Y | | Y | | ||||||
| Thalassiosira pseudonana | Y | | Y | | Y | | ||||||
| Paenibacillus lautus | Y | | Y | | Y | | ||||||
| Cryobacterium psychrophilium | Y | | Y | | Y | | ||||||
| Prevotella tannerae | Y | | Y | | Y | | ||||||
| Eubacterium biforme | Y | | Y | | Y | | ||||||
| Rhanella aquatic ^ | Y | | Y | | Y | | ||||||
| Bacteroides caccae ^ | Y | | Y | | Y | | ||||||
| Shigella sonnei | Y | | Y | | ||||||||
| Pseudoalternomonas piscicida ^ | Y | | Y | | ||||||||
| Serratia rubidaea | Y | | Y | | ||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghannoum, M.A.; Elshaer, M.; Al-Shakhshir, H.; Retuerto, M.; McCormick, T.S. A Probiotic Amylase Blend Positively Impacts Gut Microbiota Modulation in a Randomized, Placebo-Controlled, Double-Blind Study. Life 2024, 14, 824. https://doi.org/10.3390/life14070824
Ghannoum MA, Elshaer M, Al-Shakhshir H, Retuerto M, McCormick TS. A Probiotic Amylase Blend Positively Impacts Gut Microbiota Modulation in a Randomized, Placebo-Controlled, Double-Blind Study. Life. 2024; 14(7):824. https://doi.org/10.3390/life14070824
Chicago/Turabian StyleGhannoum, Mahmoud A., Mohammed Elshaer, Hilmi Al-Shakhshir, Mauricio Retuerto, and Thomas S. McCormick. 2024. "A Probiotic Amylase Blend Positively Impacts Gut Microbiota Modulation in a Randomized, Placebo-Controlled, Double-Blind Study" Life 14, no. 7: 824. https://doi.org/10.3390/life14070824
APA StyleGhannoum, M. A., Elshaer, M., Al-Shakhshir, H., Retuerto, M., & McCormick, T. S. (2024). A Probiotic Amylase Blend Positively Impacts Gut Microbiota Modulation in a Randomized, Placebo-Controlled, Double-Blind Study. Life, 14(7), 824. https://doi.org/10.3390/life14070824