
Myeloid Cells in Myocardial Ischemic Injury: The Role of the Macrophage Migration Inhibitory Factor

 , ,
, , 
Abstract
:1. Introduction
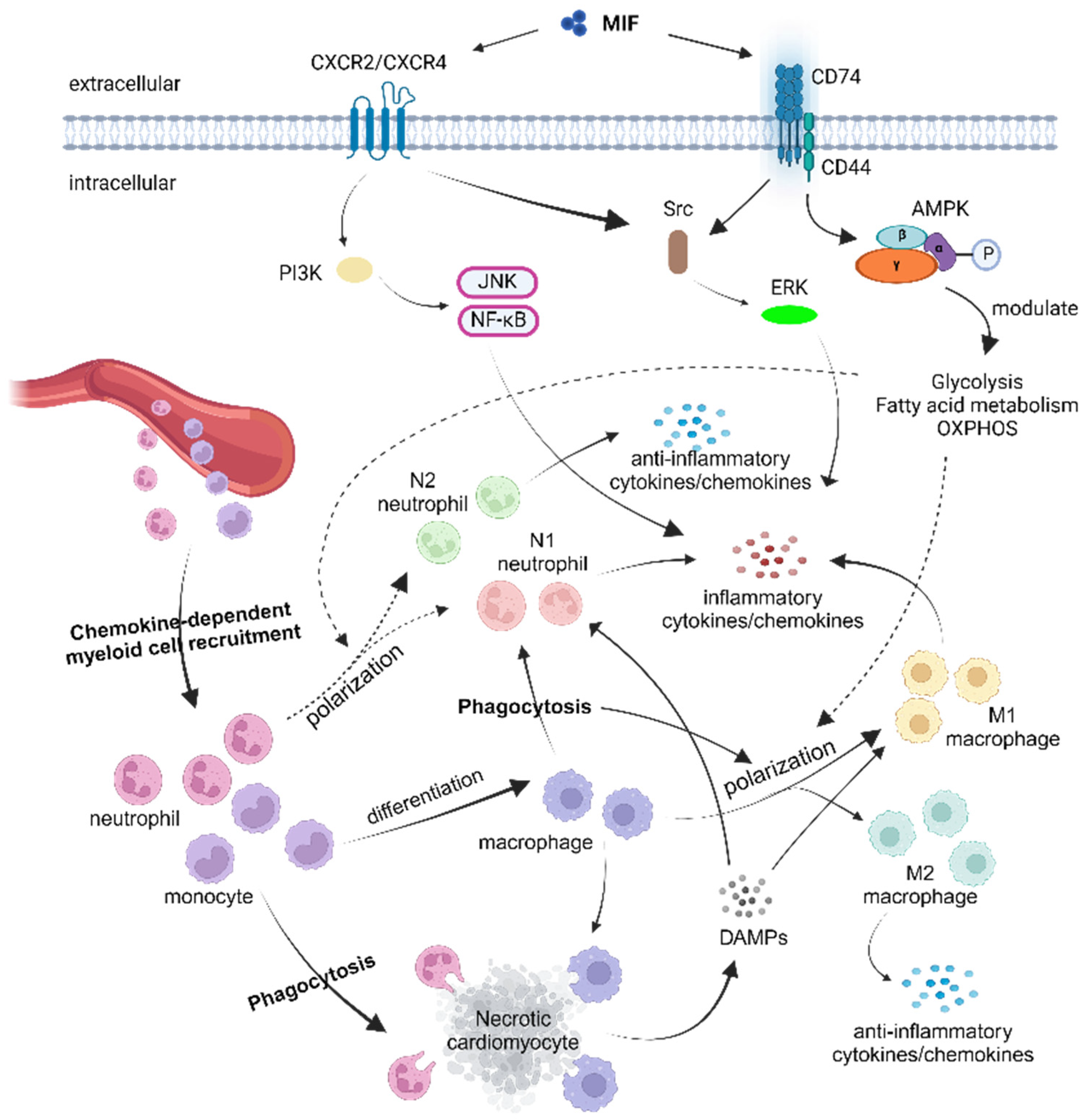
2. Myeloid Cell Subsets in MI or I/R
2.1. Cardiac Resident Myeloid Dendritic Cells (DCs)
2.2. Monocytes and Macrophages
2.3. Neutrophils

{kind=link}
| (A) | ||||
|---|---|---|---|---|
| Myeloid Cells | Key Processes | Mechanisms/Key Molecules | Functional Outcome | References |
| Macrophages | Inflammation, Polarization, Phagocytosis | PDK1,4, Hexokinase, 6-PFK, Succinate, OXPHOS, NAD+, TNF-α, IL-1β, IL-6 | Healing, Regulation of Inflammation | [42,52,66] |
| Neutrophils | Oxidative burst, Phagocytosis, NET Formation | Ly6G+SiglecFHIGH, MPO, ROS, HOCl, superoxide, CXCR2, CXCR4, CXCR7, SiglecF, AnxA1, MerTK | Regulation of Inflammation | [40,42,50,62,64,67,68] |
| Modulation of Macrophage Activity | Serine Proteases, MMPs, Lipocalin | Tissue Repair | ||
| Monocytes | Chemotaxis, Infiltration, Differentiation | MCP-1, CCR2, CCR2, CXCR2, JNK Pathway, HIF-1α, PGC-1β, GLUT1, Ly-6Ghigh | Modulation of Inflammation, Tissue Remodeling | [25,26,68,69,70] |
| Dendritic Cells | Antigen Presentation, T Cell Activation | Exosomes, HMGB1/TLR4 Signaling Pathway | Activation of CD4+ T Cells, Induction of Inflammation | [18] |
| (B) | ||||
| Processes and Factors | Key Processes | Mechanisms/Key Molecules | Functional Outcome | References |
| MIF Involvement | Inflammation, Cell Survival | MIF-CD74 Signaling Axis Pathway, CD74, Src Kinase, PI3K, AKT, ERK, p38 MAPK |
| [2,68,71,72,73,74] |
| Metabolic Processes | Energy Production, Cellular Energetics | Glycolytic Enzymes AMPK, PGC-1α, Succinate, OXPHOS | Modulation of ATP Generation via Multiple Metabolic Pathways, Regain Metabolic Homeostasis | [11,22,23,63,69,70,75,76,77,78,79] |
| Regulatory Factors | Gene Expression, Immunometabolism, Cell Signaling | NF-κB, STAT3 | Cellular Adaptation, Tissue Homeostasis | [18,50,54,62,80,81] |
3. Metabolic Regulation of Myeloid Cells in MI or I/R
4. Role of the Macrophage Migration Inhibitory Factor (MIF) in MI or I/R
4.1. Cardioprotective Effects of the MIF
4.2. Pro-Inflammatory Effects of the MIF
4.3. Future Directions of MIF Study and the Potential Therapeutic Implications
5. Modulation of Myeloid Cell Responses by the MIF
5.1. The MIF and Dendritic Cells
5.2. The MIF and Macrophage Polarization
5.3. The MIF and Neutrophils
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated protein kinase |
| DAMPs | Damage-associated molecular pattern molecules |
| DCs | Dendritic cells |
| HOCl | Hypochlorous acid |
| I/R | Ischemia and reperfusion |
| MI | Myocardial infarction |
| MIF | Macrophage migration inhibitory factor |
| NETs | Neutrophil extracellular traps |
| OXPHOS | Oxidative phosphorylation |
References
- Neri, M.; Riezzo, I.; Pascale, N.; Pomara, C.; Turillazzi, E. Ischemia/Reperfusion Injury following Acute Myocardial Infarction: A Critical Issue for Clinicians and Forensic Pathologists. Mediat. Inflamm. 2017, 2017, 7018393. [Google Scholar] [CrossRef]
- Anzai, A.; Anzai, T.; Nagai, S.; Maekawa, Y.; Naito, K.; Kaneko, H.; Sugano, Y.; Takahashi, T.; Abe, H.; Mochizuki, S.; et al. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation 2012, 125, 1234–1245. [Google Scholar] [CrossRef]
- Bonaventura, A.; Montecucco, F.; Dallegri, F.; Carbone, F.; Lüscher, T.F.; Camici, G.G.; Liberale, L. Novel findings in neutrophil biology and their impact on cardiovascular disease. Cardiovasc. Res. 2019, 115, 1266–1285. [Google Scholar] [CrossRef]
- Epelman, S.; Liu, P.P.; Mann, D.L. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat. Rev. Immunol. 2015, 15, 117–129. [Google Scholar] [CrossRef]
- Feng, Q.; Li, Q.; Zhou, H.; Sun, L.; Lin, C.; Jin, Y.; Wang, D.; Guo, G. The role of major immune cells in myocardial infarction. Front. Immunol. 2022, 13, 1084460. [Google Scholar] [CrossRef]
- Liehn, E.A.; Kanzler, I.; Konschalla, S.; Kroh, A.; Simsekyilmaz, S.; Sonmez, T.T.; Bucala, R.; Bernhagen, J.; Weber, C. Compartmentalized protective and detrimental effects of endogenous macrophage migration-inhibitory factor mediated by CXCR2 in a mouse model of myocardial ischemia/reperfusion. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2180–2186. [Google Scholar] [CrossRef] [PubMed]
- Tilstam, P.V.; Qi, D.; Leng, L.; Young, L.; Bucala, R. MIF family cytokines in cardiovascular diseases and prospects for precision-based therapeutics. Expert Opin. Ther. Targets 2017, 21, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Voss, S.; Kruger, S.; Scherschel, K.; Warnke, S.; Schwarzl, M.; Schrage, B.; Girdauskas, E.; Meyer, C.; Blankenberg, S.; Westermann, D.; et al. Macrophage Migration Inhibitory Factor (MIF) Expression Increases during Myocardial Infarction and Supports Pro-Inflammatory Signaling in Cardiac Fibroblasts. Biomolecules 2019, 9, 38. [Google Scholar] [CrossRef]
- Zernecke, A.; Bernhagen, J.; Weber, C. Macrophage migration inhibitory factor in cardiovascular disease. Circulation 2008, 117, 1594–1602. [Google Scholar] [CrossRef]
- Ma, H.; Wang, J.; Thomas, D.P.; Tong, C.; Leng, L.; Wang, W.; Merk, M.; Zierow, S.; Bernhagen, J.; Ren, J.; et al. Impaired macrophage migration inhibitory factor-AMP-activated protein kinase activation and ischemic recovery in the senescent heart. Circulation 2010, 122, 282–292. [Google Scholar] [CrossRef]
- Wang, H.; Slotabec, L.; Didik, S.; Li, Z.; Leng, L.; Zhao, B.; Bucala, R.; Li, J. A small molecule macrophage migration inhibitory factor agonist ameliorates age-related myocardial intolerance to ischemia-reperfusion insults via metabolic regulation. Metabolism 2024, 153, 155792. [Google Scholar] [CrossRef]
- Wang, J.; Tong, C.; Yan, X.; Yeung, E.; Gandavadi, S.; Hare, A.A.; Du, X.; Chen, Y.; Xiong, H.; Ma, C.; et al. Limiting cardiac ischemic injury by pharmacological augmentation of macrophage migration inhibitory factor-AMP-activated protein kinase signal transduction. Circulation 2013, 128, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.; Zhang, A.; Yang, B.; Charles, E.J.; Kron, I.L.; Yang, Z. Plasmacytoid Dendritic Cells Mediate Myocardial Ischemia/Reperfusion Injury by Secreting Type I Interferons. J. Am. Heart Assoc. 2021, 10, e020754. [Google Scholar] [CrossRef]
- Pluijmert, N.J.; Atsma, D.E.; Quax, P.H.A. Post-ischemic Myocardial Inflammatory Response: A Complex and Dynamic Process Susceptible to Immunomodulatory Therapies. Front. Cardiovasc. Med. 2021, 8, 647785. [Google Scholar] [CrossRef]
- Lv, H.; Lipes, M.A. Role of impaired central tolerance to alpha-myosin in inflammatory heart disease. Trends Cardiovasc. Med. 2012, 22, 113–117. [Google Scholar] [CrossRef]
- Van der Borght, K.; Scott, C.L.; Nindl, V.; Bouche, A.; Martens, L.; Sichien, D.; Van Moorleghem, J.; Vanheerswynghels, M.; De Prijck, S.; Saeys, Y.; et al. Myocardial Infarction Primes Autoreactive T Cells through Activation of Dendritic Cells. Cell Rep. 2017, 18, 3005–3017. [Google Scholar] [CrossRef]
- Lee, J.S.; Jeong, S.J.; Kim, S.; Chalifour, L.; Yun, T.J.; Miah, M.A.; Li, B.; Majdoubi, A.; Sabourin, A.; Keler, T.; et al. Conventional Dendritic Cells Impair Recovery after Myocardial Infarction. J. Immunol. 2018, 201, 1784–1798. [Google Scholar] [CrossRef]
- Xue, J.; Ge, H.; Lin, Z.; Wang, H.; Lin, W.; Liu, Y.; Wu, G.; Xia, J.; Zhao, Q. The role of dendritic cells regulated by HMGB1/TLR4 signalling pathway in myocardial ischaemia reperfusion injury. J. Cell. Mol. Med. 2019, 23, 2849–2862. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Gao, W.; Yuan, J.; Wu, C.; Yao, K.; Zhang, L.; Ma, L.; Zhu, J.; Zou, Y.; Ge, J. Exosomes derived from dendritic cells improve cardiac function via activation of CD4(+) T lymphocytes after myocardial infarction. J. Mol. Cell. Cardiol. 2016, 91, 123–133. [Google Scholar] [CrossRef]
- Basit, F.; Mathan, T.; Sancho, D.; de Vries, I.J.M. Human Dendritic Cell Subsets Undergo Distinct Metabolic Reprogramming for Immune Response. Front. Immunol. 2018, 9, 2489. [Google Scholar] [CrossRef]
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; van der Laan, A.M.; et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 2014, 115, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, G.; Bredemeyer, A.; Li, W.; Zaitsev, K.; Koenig, A.L.; Lokshina, I.; Mohan, J.; Ivey, B.; Hsiao, H.M.; Weinheimer, C.; et al. Tissue Resident CCR2− and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ. Res. 2019, 124, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Mouton, A.J.; DeLeon-Pennell, K.Y.; Rivera Gonzalez, O.J.; Flynn, E.R.; Freeman, T.C.; Saucerman, J.J.; Garrett, M.R.; Ma, Y.; Harmancey, R.; Lindsey, M.L. Mapping macrophage polarization over the myocardial infarction time continuum. Basic. Res. Cardiol. 2018, 113, 26. [Google Scholar] [CrossRef] [PubMed]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; Althagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2019, 20, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Leuschner, F.; Rauch, P.J.; Ueno, T.; Gorbatov, R.; Marinelli, B.; Lee, W.W.; Dutta, P.; Wei, Y.; Robbins, C.; Iwamoto, Y.; et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J. Exp. Med. 2012, 209, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Nahrendorf, M.; Swirski, F.K. Monocyte and macrophage heterogeneity in the heart. Circ. Res. 2013, 112, 1624–1633. [Google Scholar] [CrossRef] [PubMed]
- Hilgendorf, I.; Gerhardt, L.M.; Tan, T.C.; Winter, C.; Holderried, T.A.; Chousterman, B.G.; Iwamoto, Y.; Liao, R.; Zirlik, A.; Scherer-Crosbie, M.; et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ. Res. 2014, 114, 1611–1622. [Google Scholar] [CrossRef] [PubMed]
- Tsujioka, H.; Imanishi, T.; Ikejima, H.; Kuroi, A.; Takarada, S.; Tanimoto, T.; Kitabata, H.; Okochi, K.; Arita, Y.; Ishibashi, K.; et al. Impact of heterogeneity of human peripheral blood monocyte subsets on myocardial salvage in patients with primary acute myocardial infarction. J. Am. Coll. Cardiol. 2009, 54, 130–138. [Google Scholar] [CrossRef] [PubMed]
- van der Laan, A.M.; Hirsch, A.; Robbers, L.F.; Nijveldt, R.; Lommerse, I.; Delewi, R.; van der Vleuten, P.A.; Biemond, B.J.; Zwaginga, J.J.; van der Giessen, W.J.; et al. A proinflammatory monocyte response is associated with myocardial injury and impaired functional outcome in patients with ST-segment elevation myocardial infarction: Monocytes and myocardial infarction. Am. Heart J. 2012, 163, 57–65.e2. [Google Scholar] [CrossRef]
- de Jong, R.C.M.; Pluijmert, N.J.; de Vries, M.R.; Pettersson, K.; Atsma, D.E.; Jukema, J.W.; Quax, P.H.A. Annexin A5 reduces infarct size and improves cardiac function after myocardial ischemia-reperfusion injury by suppression of the cardiac inflammatory response. Sci. Rep. 2018, 8, 6753. [Google Scholar] [CrossRef]
- Nakano, Y.; Matoba, T.; Tokutome, M.; Funamoto, D.; Katsuki, S.; Ikeda, G.; Nagaoka, K.; Ishikita, A.; Nakano, K.; Koga, J.; et al. Nanoparticle-Mediated Delivery of Irbesartan Induces Cardioprotection from Myocardial Ischemia-Reperfusion Injury by Antagonizing Monocyte-Mediated Inflammation. Sci. Rep. 2016, 6, 29601. [Google Scholar] [CrossRef]
- Pluijmert, N.J.; de Jong, R.C.M.; de Vries, M.R.; Pettersson, K.; Atsma, D.E.; Jukema, J.W.; Quax, P.H.A. Phosphorylcholine Antibodies Preserve Cardiac Function and Reduce Infarct Size by Attenuating the Post-Ischemic Inflammatory Response. JACC Basic Transl. Sci. 2020, 5, 1228–1239. [Google Scholar] [CrossRef]
- Shiraishi, M.; Shintani, Y.; Shintani, Y.; Ishida, H.; Saba, R.; Yamaguchi, A.; Adachi, H.; Yashiro, K.; Suzuki, K. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J. Clin. Investig. 2016, 126, 2151–2166. [Google Scholar] [CrossRef]
- Tokutome, M.; Matoba, T.; Nakano, Y.; Okahara, A.; Fujiwara, M.; Koga, J.I.; Nakano, K.; Tsutsui, H.; Egashira, K. Peroxisome proliferator-activated receptor-gamma targeting nanomedicine promotes cardiac healing after acute myocardial infarction by skewing monocyte/macrophage polarization in preclinical animal models. Cardiovasc. Res. 2019, 115, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Huang, S.; Sheng, Y.; Peng, X.; Liu, H.; Jin, N.; Cai, J.; Shu, Y.; Li, T.; Li, P.; et al. Topiramate modulates post-infarction inflammation primarily by targeting monocytes or macrophages. Cardiovasc. Res. 2017, 113, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.S.; Zhao, G.L.; Liu, Q.; Jiang, S.C.; Wang, Y.; Zhang, D.M. Silencing collapsin response mediator protein-2 reprograms macrophage phenotype and improves infarct healing in experimental myocardial infarction model. J. Inflamm. 2015, 12, 11. [Google Scholar] [CrossRef]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef]
- Puhl, S.L.; Steffens, S. Neutrophils in Post-myocardial Infarction Inflammation: Damage vs. Resolution? Front. Cardiovasc. Med. 2019, 6, 25. [Google Scholar] [CrossRef]
- Daseke, M.J., 2nd; Chalise, U.; Becirovic-Agic, M.; Salomon, J.D.; Cook, L.M.; Case, A.J.; Lindsey, M.L. Neutrophil signaling during myocardial infarction wound repair. Cell. Signal. 2021, 77, 109816. [Google Scholar] [CrossRef]
- Ma, Y. Role of Neutrophils in Cardiac Injury and Repair Following Myocardial Infarction. Cells 2021, 10, 1676. [Google Scholar] [CrossRef]
- Vinten-Johansen, J. Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc. Res. 2004, 61, 481–497. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.D. Cytokine-induced modulation of cardiac function. Circ. Res. 2004, 95, 1140–1153. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Zhou, X.; Ji, W.J.; Lu, R.Y.; Zhang, Y.; Zhang, Y.D.; Ma, Y.Q.; Zhao, J.H.; Li, Y.M. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: Therapeutic potential of DNase-based reperfusion strategy. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H500–H509. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Shi, H.; Zeng, T.; Liu, H.; Su, Y.; Cheng, X.; Ye, J.; Yin, Y.; Liu, M.; Zheng, H.; et al. Increased neutrophil extracellular traps activate NLRP3 and inflammatory macrophages in adult-onset Still’s disease. Arthritis Res. Ther. 2019, 21, 9. [Google Scholar] [CrossRef] [PubMed]
- Kahlenberg, J.M.; Carmona-Rivera, C.; Smith, C.K.; Kaplan, M.J. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J. Immunol. 2013, 190, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Helseth, R.; Shetelig, C.; Andersen, G.O.; Langseth, M.S.; Limalanathan, S.; Opstad, T.B.; Arnesen, H.; Hoffmann, P.; Eritsland, J.; Seljeflot, I. Neutrophil Extracellular Trap Components Associate with Infarct Size, Ventricular Function, and Clinical Outcome in STEMI. Mediat. Inflamm. 2019, 2019, 7816491. [Google Scholar] [CrossRef] [PubMed]
- Hofbauer, T.M.; Mangold, A.; Scherz, T.; Seidl, V.; Panzenböck, A.; Ondracek, A.S.; Müller, J.; Schneider, M.; Binder, T.; Hell, L.; et al. Neutrophil extracellular traps and fibrocytes in ST-segment elevation myocardial infarction. Basic Res. Cardiol. 2019, 114, 33. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, D.; Wang, X.; Zhu, Z.; Wang, T.; Ma, A.; Liu, P. Neutrophil extracellular traps and dsDNA predict outcomes among patients with ST-elevation myocardial infarction. Sci. Rep. 2019, 9, 11599. [Google Scholar] [CrossRef] [PubMed]
- Mangold, A.; Alias, S.; Scherz, T.; Hofbauer, M.; Jakowitsch, J.; Panzenbock, A.; Simon, D.; Laimer, D.; Bangert, C.; Kammerlander, A.; et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ. Res. 2015, 116, 1182–1192. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, M.; Xu, Y.; Yan, S.; Jin, E.; Li, X. Neutrophil extracellular trap burden correlates with the stenosis of coronary atherosclerosis. PeerJ 2023, 11, e15471. [Google Scholar] [CrossRef]
- Loh, W.; Vermeren, S. Anti-Inflammatory Neutrophil Functions in the Resolution of Inflammation and Tissue Repair. Cells 2022, 11, 4076. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yabluchanskiy, A.; Iyer, R.P.; Cannon, P.L.; Flynn, E.R.; Jung, M.; Henry, J.; Cates, C.A.; Deleon-Pennell, K.Y.; Lindsey, M.L. Temporal neutrophil polarization following myocardial infarction. Cardiovasc. Res. 2016, 110, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Peiseler, M.; Kubes, P. More friend than foe: The emerging role of neutrophils in tissue repair. J. Clin. Investig. 2019, 129, 2629–2639. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, B.; Leoni, G.; Hinkel, R.; Ormanns, S.; Paulin, N.; Ortega-Gomez, A.; Viola, J.R.; de Jong, R.; Bongiovanni, D.; Bozoglu, T.; et al. Pro-Angiogenic Macrophage Phenotype to Promote Myocardial Repair. J. Am. Coll. Cardiol. 2019, 73, 2990–3002. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.; Schauer, C.; Czegley, C.; Kling, L.; Petru, L.; Schmid, B.; Weidner, D.; Reinwald, C.; Biermann, M.H.C.; Blunder, S.; et al. Aggregated neutrophil extracellular traps resolve inflammation by proteolysis of cytokines and chemokines and protection from antiproteases. FASEB J. 2019, 33, 1401–1414. [Google Scholar] [CrossRef]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhofer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Eghbalzadeh, K.; Georgi, L.; Louis, T.; Zhao, H.; Keser, U.; Weber, C.; Mollenhauer, M.; Conforti, A.; Wahlers, T.; Paunel-Gorgulu, A. Compromised Anti-inflammatory Action of Neutrophil Extracellular Traps in PAD4-Deficient Mice Contributes to Aggravated Acute Inflammation After Myocardial Infarction. Front. Immunol. 2019, 10, 2313. [Google Scholar] [CrossRef] [PubMed]
- Mangold, A.; Hofbauer, T.M.; Ondracek, A.S.; Artner, T.; Scherz, T.; Speidl, W.S.; Krychtiuk, K.A.; Sadushi-Kolici, R.; Jakowitsch, J.; Lang, I.M. Neutrophil extracellular traps and monocyte subsets at the culprit lesion site of myocardial infarction patients. Sci. Rep. 2019, 9, 16304. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.L., III; Kuethe, J.W.; Caldwell, C.C. Neutrophil derived microvesicles: Emerging role of a key mediator to the immune response. Endocr. Metab. Immune Disord. Drug Targets 2014, 14, 210–217. [Google Scholar] [CrossRef]
- Calcagno, D.M.; Zhang, C.; Toomu, A.; Huang, K.; Ninh, V.K.; Miyamoto, S.; Aguirre, A.D.; Fu, Z.; Heller Brown, J.; King, K.R. SiglecF(HI) Marks Late-Stage Neutrophils of the Infarcted Heart: A Single-Cell Transcriptomic Analysis of Neutrophil Diversification. J. Am. Heart Assoc. 2021, 10, e019019. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Regulation of the inflammatory response in cardiac repair. Circ. Res. 2012, 110, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef]
- Zaremba, J.; Skrobanski, P.; Losy, J. The level of chemokine CXCL5 in the cerebrospinal fluid is increased during the first 24 hours of ischaemic stroke and correlates with the size of early brain damage. Folia Morphol. 2006, 65, 1–5. [Google Scholar]
- Cai, W.; Liu, S.; Hu, M.; Huang, F.; Zhu, Q.; Qiu, W.; Hu, X.; Colello, J.; Zheng, S.G.; Lu, Z. Functional Dynamics of Neutrophils After Ischemic Stroke. Transl. Stroke Res. 2020, 11, 108–121. [Google Scholar] [CrossRef]
- Hou, Y.; Yang, D.; Zhang, Q.; Wang, X.; Yang, J.; Wu, C. Pseudoginsenoside-F11 ameliorates ischemic neuron injury by regulating the polarization of neutrophils and macrophages in vitro. Int. Immunopharmacol. 2020, 85, 106564. [Google Scholar] [CrossRef]
- Ohms, M.; Moller, S.; Laskay, T. An Attempt to Polarize Human Neutrophils Toward N1 and N2 Phenotypes in vitro. Front. Immunol. 2020, 11, 532. [Google Scholar] [CrossRef]
- Mesri, M.; Altieri, D.C. Endothelial cell activation by leukocyte microparticles. J. Immunol. 1998, 161, 4382–4387. [Google Scholar] [CrossRef]
- Terrazas, C.A.; Huitron, E.; Vazquez, A.; Juarez, I.; Camacho, G.M.; Calleja, E.A.; Rodriguez-Sosa, M. MIF synergizes with Trypanosoma cruzi antigens to promote efficient dendritic cell maturation and IL-12 production via p38 MAPK. Int. J. Biol. Sci. 2011, 7, 1298–1310. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Dabritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e13. [Google Scholar] [CrossRef]
- Figueiredo, C.R.; Azevedo, R.A.; Mousdell, S.; Resende-Lara, P.T.; Ireland, L.; Santos, A.; Girola, N.; Cunha, R.; Schmid, M.C.; Polonelli, L.; et al. Blockade of MIF-CD74 Signalling on Macrophages and Dendritic Cells Restores the Antitumour Immune Response Against Metastatic Melanoma. Front. Immunol. 2018, 9, 1132. [Google Scholar] [CrossRef]
- Nakaya, K.; Nabata, Y.; Ichiyanagi, T.; An, W.W. Stimulation of dendritic cell maturation and induction of apoptosis in leukemia cells by a heat-stable extract from azuki bean (Vigna angularis), a promising immunopotentiating food and dietary supplement for cancer prevention. Asian Pac. J. Cancer Prev. 2012, 13, 607–611. [Google Scholar] [CrossRef]
- Rodrigues, D.A.S.; Prestes, E.B.; Gama, A.M.S.; Silva, L.S.; Pinheiro, A.A.S.; Ribeiro, J.M.C.; Campos, R.M.P.; Pimentel-Coelho, P.M.; De Souza, H.S.; Dicko, A.; et al. CXCR4 and MIF are required for neutrophil extracellular trap release triggered by Plasmodium-infected erythrocytes. PLoS Pathog. 2020, 16, e1008230. [Google Scholar] [CrossRef]
- Wu, Z.; Luan, H.; Huang, J.; Liao, B.; Xiao, F. Migration inhibitory factor and cluster of differentiation 74-mediated dendritic cell apoptosis exacerbates acute acetaminophen-induced liver injury. Immun. Inflamm. Dis. 2023, 11, e840. [Google Scholar] [CrossRef]
- DeBerge, M.; Chaudhary, R.; Schroth, S.; Thorp, E.B. Immunometabolism at the Heart of Cardiovascular Disease. JACC Basic Transl. Sci. 2023, 8, 884–904. [Google Scholar] [CrossRef]
- Fatmi, M.K.; Ren, D.; Fedorova, J.; Zoungrana, L.I.; Wang, H.; Davitt, K.; Li, Z.; Iglesias, M.; Lesnefsky, E.J.; Krause-Hauch, M.; et al. Cardiomyocyte Pdk4 response is associated with metabolic maladaptation in aging. Aging Cell 2023, 22, e13800. [Google Scholar] [CrossRef]
- Lavine, K.J.; Pinto, A.R.; Epelman, S.; Kopecky, B.J.; Clemente-Casares, X.; Godwin, J.; Rosenthal, N.; Kovacic, J.C. The Macrophage in Cardiac Homeostasis and Disease: JACC Macrophage in CVD Series (Part 4). J. Am. Coll. Cardiol. 2018, 72, 2213–2230. [Google Scholar] [CrossRef]
- Ma, Y.; Mouton, A.J.; Lindsey, M.L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 2018, 191, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Bories, G.; Lantz, C.; Emmons, R.; Becker, A.; Liu, E.; Abecassis, M.M.; Yvan-Charvet, L.; Thorp, E.B. Immunometabolism of Phagocytes and Relationships to Cardiac Repair. Front. Cardiovasc. Med. 2019, 6, 42. [Google Scholar] [CrossRef]
- Ajikumar, A.; Long, M.B.; Heath, P.R.; Wharton, S.B.; Ince, P.G.; Ridger, V.C.; Simpson, J.E. Neutrophil-Derived Microvesicle Induced Dysfunction of Brain Microvascular Endothelial Cells In Vitro. Int. J. Mol. Sci. 2019, 20, 5227. [Google Scholar] [CrossRef]
- Mesri, M.; Altieri, D.C. Leukocyte microparticles stimulate endothelial cell cytokine release and tissue factor induction in a JNK1 signaling pathway. J. Biol. Chem. 1999, 274, 23111–23118. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, S.; Vuckovic, I.; Jeon, R.; Lerman, A.; Folmes, C.D.; Dzeja, P.P.; Herrmann, J. Glycolytic Stimulation Is Not a Requirement for M2 Macrophage Differentiation. Cell Metab. 2018, 28, 463–475.e4. [Google Scholar] [CrossRef] [PubMed]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Wagner, R.A.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Zhao, M.; Zhou, B.; Yoshii, A.; Bugg, D.; Villet, O.; Sahu, A.; Olson, G.S.; Davis, J.; Tian, R. Mitochondrial dysfunction in macrophages promotes inflammation and suppresses repair after myocardial infarction. J. Clin. Investig. 2023, 133. [Google Scholar] [CrossRef] [PubMed]
- Nobre, C.C.; de Araujo, J.M.; Fernandes, T.A.; Cobucci, R.N.; Lanza, D.C.; Andrade, V.S.; Fernandes, J.V. Macrophage Migration Inhibitory Factor (MIF): Biological Activities and Relation with Cancer. Pathol. Oncol. Res. 2017, 23, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.M.; Liu, Y.; White, D.; Su, Y.; Drew, B.G.; Bruce, C.R.; Kiriazis, H.; Xu, Q.; Jennings, N.; Bobik, A.; et al. Deletion of macrophage migration inhibitory factor protects the heart from severe ischemia-reperfusion injury: A predominant role of anti-inflammation. J. Mol. Cell. Cardiol. 2011, 50, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.J.; Li, J.; Leng, L.; McDonald, C.; Atsumi, T.; Bucala, R.; Young, L.H. Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature 2008, 451, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Luedike, P.; Hendgen-Cotta, U.B.; Sobierajski, J.; Totzeck, M.; Reeh, M.; Dewor, M.; Lue, H.; Krisp, C.; Wolters, D.; Kelm, M.; et al. Cardioprotection through S-nitros(yl)ation of macrophage migration inhibitory factor. Circulation 2012, 125, 1880–1889. [Google Scholar] [CrossRef] [PubMed]
- Qi, D.; Hu, X.; Wu, X.; Merk, M.; Leng, L.; Bucala, R.; Young, L.H. Cardiac macrophage migration inhibitory factor inhibits JNK pathway activation and injury during ischemia/reperfusion. J. Clin. Investig. 2009, 119, 3807–3816. [Google Scholar] [CrossRef] [PubMed]
- Koga, K.; Kenessey, A.; Powell, S.R.; Sison, C.P.; Miller, E.J.; Ojamaa, K. Macrophage migration inhibitory factor provides cardioprotection during ischemia/reperfusion by reducing oxidative stress. Antioxid. Redox Signal 2011, 14, 1191–1202. [Google Scholar] [CrossRef]
- White, D.A.; Fang, L.; Chan, W.; Morand, E.F.; Kiriazis, H.; Duffy, S.J.; Taylor, A.J.; Dart, A.M.; Du, X.J.; Gao, X.M. Pro-inflammatory action of MIF in acute myocardial infarction via activation of peripheral blood mononuclear cells. PLoS ONE 2013, 8, e76206. [Google Scholar] [CrossRef]
- Zhang, R.Y.K.; Cochran, B.J.; Thomas, S.R.; Rye, K.A. Impact of Reperfusion on Temporal Immune Cell Dynamics After Myocardial Infarction. J. Am. Heart Assoc. 2023, 12, e027600. [Google Scholar] [CrossRef]
- Zuidema, M.Y.; Zhang, C. Ischemia/reperfusion injury: The role of immune cells. World J. Cardiol. 2010, 2, 325–332. [Google Scholar] [CrossRef]
- Ives, A.; Le Roy, D.; Theroude, C.; Bernhagen, J.; Roger, T.; Calandra, T. Macrophage migration inhibitory factor promotes the migration of dendritic cells through CD74 and the activation of the Src/PI3K/myosin II pathway. FASEB J. 2021, 35, e21418. [Google Scholar] [CrossRef]
- Bloom, B.R.; Bennett, B. Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science 1966, 153, 80–82. [Google Scholar] [CrossRef]
- David, J.R. Delayed hypersensitivity in vitro: Its mediation by cell-free substances formed by lymphoid cell-antigen interaction. Proc. Natl. Acad. Sci. USA 1966, 56, 72–77. [Google Scholar] [CrossRef]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage M1–M2 polarization balance. Front. Immunol. 2014, 5, 113523. [Google Scholar] [CrossRef]
- Yaddanapudi, K.; Putty, K.; Rendon, B.E.; Lamont, G.J.; Faughn, J.D.; Satoskar, A.; Lasnik, A.; Eaton, J.W.; Mitchell, R.A. Control of tumor-associated macrophage alternative activation by macrophage migration inhibitory factor. J. Immunol. 2013, 190, 2984–2993. [Google Scholar] [CrossRef]
- Bernhagen, J.; Krohn, R.; Lue, H.; Gregory, J.L.; Zernecke, A.; Koenen, R.R.; Dewor, M.; Georgiev, I.; Schober, A.; Leng, L. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 2007, 13, 587–596. [Google Scholar] [CrossRef]
- Pan, J.H.; Sukhova, G.K.; Yang, J.T.; Wang, B.; Xie, T.; Fu, H.; Zhang, Y.; Satoskar, A.R.; David, J.R.; Metz, C.N.; et al. Macrophage migration inhibitory factor deficiency impairs atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 2004, 109, 3149–3153. [Google Scholar] [CrossRef]
- Schindler, L.; Smyth, L.C.D.; Bernhagen, J.; Hampton, M.B.; Dickerhof, N. Macrophage migration inhibitory factor (MIF) enhances hypochlorous acid production in phagocytic neutrophils. Redox Biol. 2021, 41, 101946. [Google Scholar] [CrossRef] [PubMed]
- Alampour-Rajabi, S.; El Bounkari, O.; Rot, A.; Muller-Newen, G.; Bachelerie, F.; Gawaz, M.; Weber, C.; Schober, A.; Bernhagen, J. MIF interacts with CXCR7 to promote receptor internalization, ERK1/2 and ZAP-70 signaling, and lymphocyte chemotaxis. FASEB J. 2015, 29, 4497–4511. [Google Scholar] [CrossRef] [PubMed]
- Leng, L.; Metz, C.N.; Fang, Y.; Xu, J.; Donnelly, S.; Baugh, J.; Delohery, T.; Chen, Y.; Mitchell, R.A.; Bucala, R. MIF signal transduction initiated by binding to CD74. J. Exp. Med. 2003, 197, 1467–1476. [Google Scholar] [CrossRef]
- Schindler, L.; Zwissler, L.; Krammer, C.; Hendgen-Cotta, U.; Rassaf, T.; Hampton, M.B.; Dickerhof, N.; Bernhagen, J. Macrophage migration inhibitory factor inhibits neutrophil apoptosis by inducing cytokine release from mononuclear cells. J. Leukoc. Biol. 2021, 110, 893–905. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Rouhi, N.; Slotabec, L.A.; Seale, B.C.; Wen, C.; Filho, F.; Adenawoola, M.I.; Li, J. Myeloid Cells in Myocardial Ischemic Injury: The Role of the Macrophage Migration Inhibitory Factor. Life 2024, 14, 981. https://doi.org/10.3390/life14080981
Wang H, Rouhi N, Slotabec LA, Seale BC, Wen C, Filho F, Adenawoola MI, Li J. Myeloid Cells in Myocardial Ischemic Injury: The Role of the Macrophage Migration Inhibitory Factor. Life. 2024; 14(8):981. https://doi.org/10.3390/life14080981
Chicago/Turabian StyleWang, Hao, Nadiyeh Rouhi, Lily A. Slotabec, Blaise C. Seale, Changhong Wen, Fernanda Filho, Michael I. Adenawoola, and Ji Li. 2024. "Myeloid Cells in Myocardial Ischemic Injury: The Role of the Macrophage Migration Inhibitory Factor" Life 14, no. 8: 981. https://doi.org/10.3390/life14080981