

Anticancer Chemotherapy-Induced Atherosclerotic Cardiovascular Disease: A Comprehensive Review

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Research Question
2.2. Study Design
2.3. Search Strategies
2.4. Selection Criteria
2.5. Bias Assessment
2.6. Data Synthesis
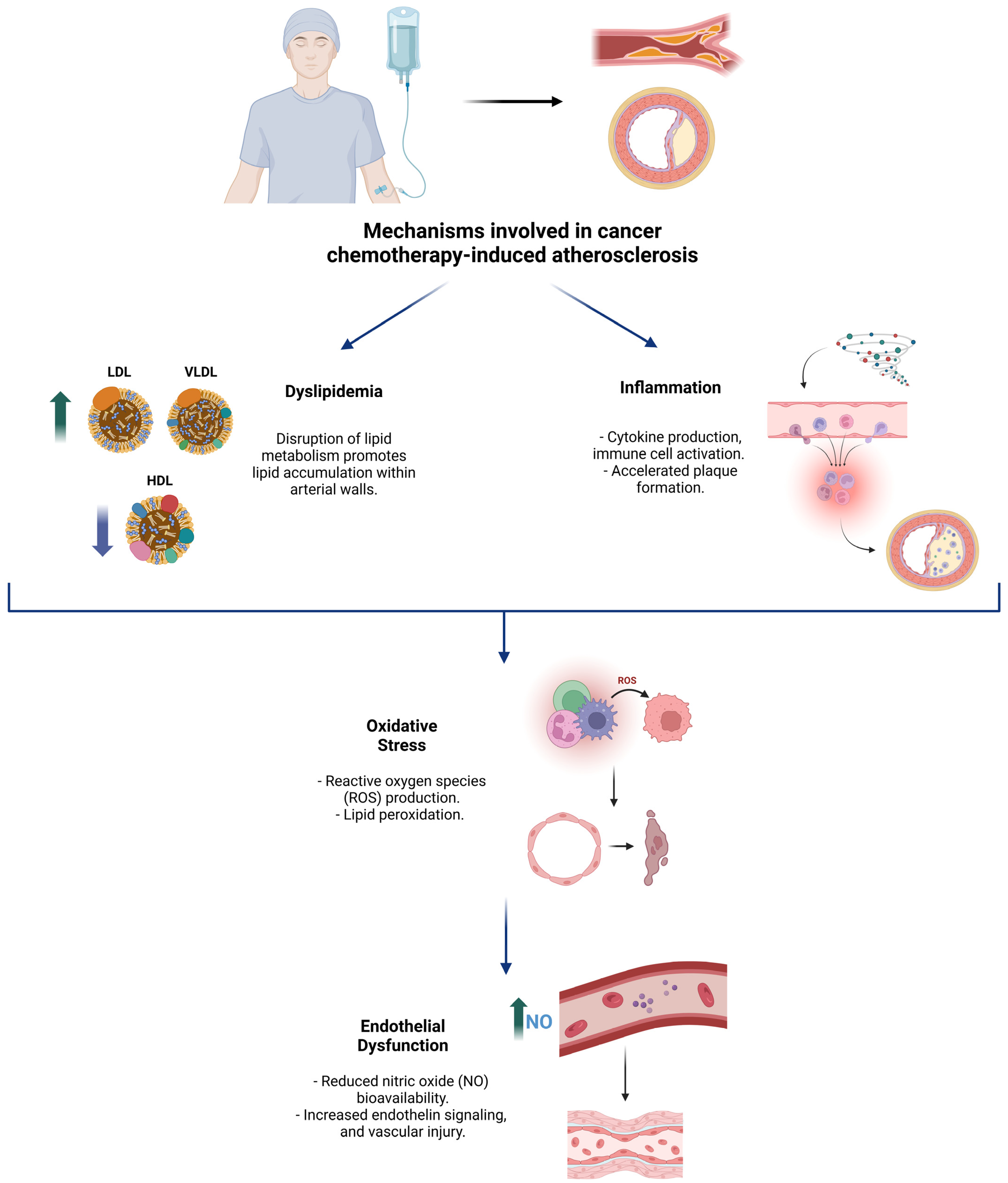
3. Mechanisms of Atherosclerosis Induction by Chemotherapy
3.1. Dyslipidemia
3.2. Inflammation
3.3. Oxidative Stress
3.4. Endothelial Dysfunction
4. Pharmacological Groups Related to Atherosclerosis and Their Effects
4.1. Anthracyclines
4.2. Taxanes
4.3. VEGF Inhibitors
4.4. Anti-Metabolites
4.5. mTOR Inhibitors
4.6. Hormonal Therapies
4.7. Tyrosine Kinase Inhibitors (TKIs)
4.8. Alkylating Agents
4.9. Platinum-Based Agents
4.10. Immune Checkpoint Inhibitors (ICIs)
4.11. Confounding Factors
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ASCVD | atherosclerovascular disease |
| LDL | low-density lipoprotein |
| HDL | high-density lipoprotein |
| oxLDL | oxidized low-density lipoprotein |
| VEGF | vascular endothelial growth factor |
| TNF-α | tumor necrosis factor-alpha |
| IL | interleukin |
| IFN-γ | interferon-gamma |
| SASP | senescence-associated secretory phenotype |
| ROS | reactive oxygen species |
| DDR | DNA damage response |
| NO | nitric oxide |
| PGI2 | prostacyclin |
| TS3 | Type III Secretion System |
| Hfq | Host Factor for RNA Binding |
| mTOR | mammalian target of rapamycin |
| HMGCR | 3-hydroxy-3-methylglutaryl-coenzyme a reductase |
| LXR-α | liver X receptor-alpha |
| PPAR-γ | peroxisome proliferator-activated receptor-gamma |
| ABCA1 | ATP-binding cassette transporter A1 |
| ApoB | apolipoprotein B |
| GDP | Guanosine Diphosphate |
| 5-FU | 5-fluorouracil |
| TG | triglyceride |
| ICAM-1 | Intercellular Adhesion Molecule 1 |
| VCAM-1 | Vascular Cell Adhesion Molecule 1 |
| ADT | androgen deprivation therapy |
| SERMs | selective estrogen receptor modulators |
| AIs | aromatase inhibitors |
| TKIs | tyrosine kinase inhibitors |
| PD-1 | Programmed Death-1 |
| CTLA4 | Cytotoxic T-Lymphocyte-Associated Protein 4 |
| CVD | cardiovascular disease |
| CRP | C-reactive protein |
| ARB | angiotensin receptor blocker |
| CVAD | cardiovascular adverse disease |
| TMA | trimethylamine |
| TMAO | trimethylamine-N-oxide |
| SCFAs | short-chain fatty acids |
References
- Michaeli, D.T.; Michaeli, J.C.; Michaeli, T. Advances in Cancer Therapy: Clinical Benefit of New Cancer Drugs. Aging 2023, 15, 5232–5234. [Google Scholar] [CrossRef] [PubMed]
- Rosa, G.M.; Gigli, L.; Tagliasacchi, M.I.; Di Iorio, C.; Carbone, F.; Nencioni, A.; Montecucco, F.; Brunelli, C. Update on Cardiotoxicity of Anti-Cancer Treatments. Eur. J. Clin. Investig. 2016, 46, 264–284. [Google Scholar] [CrossRef] [PubMed]
- Boerma, M.; Sridharan, V.; Mao, X.-W.; Nelson, G.A.; Cheema, A.K.; Koturbash, I.; Singh, S.P.; Tackett, A.J.; Hauer-Jensen, M. Effects of Ionizing Radiation on the Heart. Mutat. Res. Mutat. Res. 2016, 770, 319–327. [Google Scholar] [CrossRef]
- Henson, K.E.; Reulen, R.C.; Winter, D.L.; Bright, C.J.; Fidler, M.M.; Frobisher, C.; Guha, J.; Wong, K.F.; Kelly, J.; Edgar, A.B.; et al. Cardiac Mortality Among 200 000 Five-Year Survivors of Cancer Diagnosed at 15 to 39 Years of Age. Circulation 2016, 134, 1519–1531. [Google Scholar] [CrossRef] [PubMed]
- Muhandiramge, J.; Zalcberg, J.R.; Van Londen, G.J.; Warner, E.T.; Carr, P.R.; Haydon, A.; Orchard, S.G. Cardiovascular Disease in Adult Cancer Survivors: A Review of Current Evidence, Strategies for Prevention and Management, and Future Directions for Cardio-Oncology. Curr. Oncol. Rep. 2022, 24, 1579–1592. [Google Scholar] [CrossRef]
- Raposeiras Roubín, S.; Cordero, A. The Two-Way Relationship Between Cancer and Atherosclerosis. Rev. Espanola Cardiol. Engl. Ed. 2019, 72, 487–494. [Google Scholar] [CrossRef]
- ASCVD Risk Stratification Among Cancer Survivors; American College of Cardiology: Washington, DC, USA, 2021.
- Mohammed, T.; Parekh, T.; Desai, A. Cardiovascular Risk Management in Cancer Survivors: Are We Doing It Right? World J. Clin. Oncol. 2021, 12, 144–149. [Google Scholar] [CrossRef]
- Zhang, X.; Pawlikowski, M.; Olivo-Marston, S.; Williams, K.P.; Bower, J.K.; Felix, A.S. Ten-Year Cardiovascular Risk among Cancer Survivors: The National Health and Nutrition Examination Survey. PLoS ONE 2021, 16, e0247919. [Google Scholar] [CrossRef]
- Caller, T.; Fardman, A.; Gerber, Y.; Moshkovits, Y.; Tiosano, S.; Kaplan, A.; Kalstein, M.; Bayshtok, G.; Itkin, T.; Avigdor, A.; et al. Atherosclerotic Cardiovascular Diseases Are Associated With Incident Metastatic and Nonmetastatic Cancer. JACC CardioOncol. 2024, 6, 949–961. [Google Scholar] [CrossRef]
- Gallucci, G.; Turazza, F.M.; Inno, A.; Canale, M.L.; Silvestris, N.; Farì, R.; Navazio, A.; Pinto, C.; Tarantini, L. Atherosclerosis and the Bidirectional Relationship between Cancer and Cardiovascular Disease: From Bench to Bedside—Part 1. Int. J. Mol. Sci. 2024, 25, 4232. [Google Scholar] [CrossRef]
- Minhas, A.S.; Goerlich, E.; Corretti, M.C.; Arbab-Zadeh, A.; Kelle, S.; Leucker, T.; Lerman, A.; Hays, A.G. Imaging Assessment of Endothelial Function: An Index of Cardiovascular Health. Front. Cardiovasc. Med. 2022, 9, 778762. [Google Scholar] [CrossRef] [PubMed]
- Mukai, M.; Komori, K.; Oka, T. Mechanism and Management of Cancer Chemotherapy-Induced Atherosclerosis. J. Atheroscler. Thromb. 2018, 25, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Gusev, E.; Sarapultsev, A. Atherosclerosis and Inflammation: Insights from the Theory of General Pathological Processes. Int. J. Mol. Sci. 2023, 24, 7910. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, R.; Dixit, N.M.; Yang, E.H.; Sallam, T. Cancer Therapy’s Impact on Lipid Metabolism: Mechanisms and Future Avenues. Front. Cardiovasc. Med. 2022, 9, 925816. [Google Scholar] [CrossRef]
- Singh, S.; Singh, K. Atherosclerosis, Ischemia, and Anticancer Drugs. Heart Views 2021, 22, 127–133. [Google Scholar] [CrossRef]
- Yue, Z.; Nie, L.; Zhao, P.; Ji, N.; Liao, G.; Wang, Q. Senescence-Associated Secretory Phenotype and Its Impact on Oral Immune Homeostasis. Front. Immunol. 2022, 13, 1019313. [Google Scholar] [CrossRef]
- Satoh, K.; Nigro, P.; Berk, B.C. Oxidative Stress and Vascular Smooth Muscle Cell Growth: A Mechanistic Linkage by Cyclophilin A. Antioxid. Redox Signal. 2010, 12, 675–682. [Google Scholar] [CrossRef]
- Terwoord, J.D.; Beyer, A.M.; Gutterman, D.D. Endothelial Dysfunction as a Complication of Anti-Cancer Therapy. Pharmacol. Ther. 2022, 237, 108116. [Google Scholar] [CrossRef]
- Gao, F.; Lucke-Wold, B.P.; Li, X.; Logsdon, A.F.; Xu, L.-C.; Xu, S.; LaPenna, K.B.; Wang, H.; Talukder, M.A.H.; Siedlecki, C.A.; et al. Reduction of Endothelial Nitric Oxide Increases the Adhesiveness of Constitutive Endothelial Membrane ICAM-1 through Src-Mediated Phosphorylation. Front. Physiol. 2018, 8, 1124. [Google Scholar] [CrossRef]
- Kciuk, M.; Gielecińska, A.; Mujwar, S.; Kołat, D.; Kałuzińska-Kołat, Ż.; Celik, I.; Kontek, R. Doxorubicin—An Agent with Multiple Mechanisms of Anticancer Activity. Cells 2023, 12, 659. [Google Scholar] [CrossRef]
- Li, H.; Wang, M.; Huang, Y. Anthracycline-Induced Cardiotoxicity: An Overview from Cellular Structural Perspective. Biomed. Pharmacother. 2024, 179, 117312. [Google Scholar] [CrossRef] [PubMed]
- Kidani, Y.; Bensinger, S.J. Liver X Receptor and Peroxisome Proliferator-activated Receptor as Integrators of Lipid Homeostasis and Immunity. Immunol. Rev. 2012, 249, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Narezkina, A.; Narayan, H.K.; Zemljic-Harpf, A.E. Molecular Mechanisms of Anthracycline Cardiovascular Toxicity. Clin. Sci. Lond. Engl. 1979 2021, 135, 1311–1332. [Google Scholar] [CrossRef] [PubMed]
- Borowiec, A.; Ozdowska, P.; Rosinska, M.; Jagiello-Gruszfeld, A.; Jasek, S.; Waniewska, J.; Kotowicz, B.; Kosela-Paterczyk, H.; Lampka, E.; Makowka, A.; et al. Prognostic Value of Coronary Atherosclerosis and CAC Score for the Risk of Chemotherapy-Related Cardiac Dysfunction (CTRCD): The Protocol of ANTEC Study. PLoS ONE 2023, 18, e0288146. [Google Scholar] [CrossRef]
- Lyon, A.R.; López-Fernández, T.; Couch, L.S.; Asteggiano, R.; Aznar, M.C.; Bergler-Klein, J.; Boriani, G.; Cardinale, D.; Cordoba, R.; Cosyns, B.; et al. 2022 ESC Guidelines on Cardio-Oncology Developed in Collaboration with the European Hematology Association (EHA), the European Society for Therapeutic Radiology and Oncology (ESTRO) and the International Cardio-Oncology Society (IC-OS). Eur. Heart J. 2022, 43, 4229–4361. [Google Scholar] [CrossRef]
- Marsh, S. Taxane Pharmacogenetics. Pers. Med. 2006, 3, 33–43. [Google Scholar] [CrossRef]
- Sharma, M.; Tuaine, J.; McLaren, B.; Waters, D.L.; Black, K.; Jones, L.M.; McCormick, S.P.A. Chemotherapy Agents Alter Plasma Lipids in Breast Cancer Patients and Show Differential Effects on Lipid Metabolism Genes in Liver Cells. PLoS ONE 2016, 11, e0148049. [Google Scholar] [CrossRef]
- Ghalehbandi, S.; Yuzugulen, J.; Pranjol, M.Z.I.; Pourgholami, M.H. The Role of VEGF in Cancer-Induced Angiogenesis and Research Progress of Drugs Targeting VEGF. Eur. J. Pharmacol. 2023, 949, 175586. [Google Scholar] [CrossRef]
- Versmissen, J.; Mirabito Colafella, K.M.; Koolen, S.L.W.; Danser, A.H.J. Vascular Cardio-Oncology: Vascular Endothelial Growth Factor Inhibitors and Hypertension. Cardiovasc. Res. 2019, 115, 904–914. [Google Scholar] [CrossRef]
- Mihalcea, D.; Memis, H.; Mihaila, S.; Vinereanu, D. Cardiovascular Toxicity Induced by Vascular Endothelial Growth Factor Inhibitors. Life 2023, 13, 366. [Google Scholar] [CrossRef]
- Sommer, J.; Mahli, A.; Freese, K.; Schiergens, T.S.; Kuecuekoktay, F.S.; Teufel, A.; Thasler, W.E.; Müller, M.; Bosserhoff, A.K.; Hellerbrand, C. Analysis of Molecular Mechanisms of 5-Fluorouracil-Induced Steatosis and Inflammation in Vitro and in Mice. Oncotarget 2016, 8, 13059–13072. [Google Scholar] [CrossRef] [PubMed]
- Coomes, E.; Chan, E.S.L.; Reiss, A.B. Methotrexate in Atherogenesis and Cholesterol Metabolism. Cholesterol 2011, 2011, 503028. [Google Scholar] [CrossRef] [PubMed]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and Mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef] [PubMed]
- Zaza, G.; Granata, S.; Caletti, C.; Signorini, L.; Stallone, G.; Lupo, A. mTOR Inhibition Role in Cellular Mechanisms. Transplantation 2018, 102, S3–S16. [Google Scholar] [CrossRef] [PubMed]
- Szwed, A.; Kim, E.; Jacinto, E. Regulation and Metabolic Functions of mTORC1 and mTORC2. Physiol. Rev. 2021, 101, 1371–1426. [Google Scholar] [CrossRef]
- Liu, X.; Guo, B.; Li, Q.; Nie, J. mTOR in Metabolic Homeostasis and Disease. Exp. Cell Res. 2024, 441, 114173. [Google Scholar] [CrossRef]
- Chaput, G.; Sumar, N. Endocrine Therapies for Breast and Prostate Cancers. Can. Fam. Physician 2022, 68, 271–276. [Google Scholar] [CrossRef]
- Joensuu, H.; Holli, K.; Oksanen, H.; Valavaara, R. Serum Lipid Levels during and after Adjuvant Toremifene or Tamoxifen Therapy for Breast Cancer. Breast Cancer Res. Treat. 2000, 63, 225–234. [Google Scholar] [CrossRef]
- Liu, C.-L.; Yang, T.-L. Sequential Changes in Serum Triglyceride Levels During Adjuvant Tamoxifen Therapy in Breast Cancer Patients and the Effect of Dose Reduction. Breast Cancer Res. Treat. 2003, 79, 11–16. [Google Scholar] [CrossRef]
- Hozumi, Y.; Kawano, M.; Saito, T.; Miyata, M. Effect of Tamoxifen on Serum Lipid Metabolism. J. Clin. Endocrinol. Metab. 1998, 83, 1633–1635. [Google Scholar] [CrossRef]
- Love, R.R.; Wiebe, D.A.; Feyzi, J.M.; Newcomb, P.A.; Chappell, R.J. Effects of Tamoxifen on Cardiovascular Risk Factors in Postmenopausal Women After 5 Years of Treatment. JNCI J. Natl. Cancer Inst. 1994, 86, 1534–1539. [Google Scholar] [CrossRef] [PubMed]
- Miller, W. Aromatase Inhibitors: Mechanism of Action and Role in the Treatment of Breast Cancer. Semin. Oncol. 2003, 30, 3–11. [Google Scholar] [CrossRef]
- Chang, W.-T.; Chen, P.-W.; Lin, H.-W.; Kuo, Y.-H.; Lin, S.-H.; Li, Y.-H. Risks of Aromatase Inhibitor-Related Cardiotoxicity in Patients with Breast Cancer in Asia. Cancers 2022, 14, 508. [Google Scholar] [CrossRef] [PubMed]
- Goldvaser, H.; Barnes, T.A.; Šeruga, B.; Cescon, D.W.; Ocaña, A.; Ribnikar, D.; Amir, E. Toxicity of Extended Adjuvant Therapy With Aromatase Inhibitors in Early Breast Cancer: A Systematic Review and Meta-Analysis. JNCI J. Natl. Cancer Inst. 2018, 110, 31–39. [Google Scholar] [CrossRef] [PubMed]
- De Haas, E.C.; Altena, R.; Boezen, H.M.; Zwart, N.; Smit, A.J.; Bakker, S.J.L.; Van Roon, A.M.; Postma, A.; Wolffenbuttel, B.H.R.; Hoekstra, H.J.; et al. Early Development of the Metabolic Syndrome after Chemotherapy for Testicular Cancer. Ann. Oncol. 2013, 24, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Gotink, K.J.; Verheul, H.M.W. Anti-Angiogenic Tyrosine Kinase Inhibitors: What Is Their Mechanism of Action? Angiogenesis 2010, 13, 1–14. [Google Scholar] [CrossRef]
- Ashry, N.A.; Abdelaziz, R.R.; Suddek, G.M. The Potential Effect of Imatinib against Hypercholesterolemia Induced Atherosclerosis, Endothelial Dysfunction and Hepatic Injury in Rabbits. Life Sci. 2020, 243, 117275. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, H.; Dai, J.; Liu, Z.; Zhu, X.; Ni, Y.; Yin, X.; Sun, G.; Zhu, S.; Chen, J.; et al. The Influence of Dynamic Changes in Lipid Metabolism on Survival Outcomes in Patients with Metastatic Renal Cell Carcinoma Treated with Tyrosine Kinase Inhibitors. Jpn. J. Clin. Oncol. 2020, 50, 1454–1463. [Google Scholar] [CrossRef]
- Shyam Sunder, S.; Sharma, U.C.; Pokharel, S. Adverse Effects of Tyrosine Kinase Inhibitors in Cancer Therapy: Pathophysiology, Mechanisms and Clinical Management. Signal Transduct. Target. Ther. 2023, 8, 262. [Google Scholar] [CrossRef]
- Curieses Andrés, C.M.; Pérez de la Lastra, J.M.; Munguira, E.B.; Andrés Juan, C.; Pérez-Lebeña, E. Dual-Action Therapeutics: DNA Alkylation and Antimicrobial Peptides for Cancer Therapy. Cancers 2024, 16, 3123. [Google Scholar] [CrossRef]
- Bhat, N.; Kalthur, S.G.; Padmashali, S.; Monappa, V. Toxic Effects of Different Doses of Cyclophosphamide on Liver and Kidney Tissue in Swiss Albino Mice: A Histopathological Study. Ethiop. J. Health Sci. 2018, 28, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Mythili, Y.; Sudharsan, P.T.; Sudhahar, V.; Varalakshmi, P. Protective Effect of Dl-α-Lipoic Acid on Cyclophosphamide Induced Hyperlipidemic Cardiomyopathy. Eur. J. Pharmacol. 2006, 543, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Zeng, K.; Cao, J.; Zhou, T.; Lin, S.; Ma, W.; Yang, Y.; Zhang, Z.; Lu, F.; Huang, Y.; et al. Predictive Value of a Reduction in the Level of High-Density Lipoprotein-Cholesterol in Patients with Non-Small-Cell Lung Cancer Undergoing Radical Resection and Adjuvant Chemotherapy: A Retrospective Observational Study. Lipids Health Dis. 2021, 20, 109. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef]
- Lutgens, E.; Seijkens, T.T.P. Cancer Patients Receiving Immune Checkpoint Inhibitor Therapy Are at an Increased Risk for Atherosclerotic Cardiovascular Disease. J. Immunother. Cancer 2020, 8, e000300. [Google Scholar] [CrossRef]
- Vuong, J.T.; Stein-Merlob, A.F.; Nayeri, A.; Sallam, T.; Neilan, T.G.; Yang, E.H. Immune Checkpoint Therapies and Atherosclerosis: Mechanisms and Clinical Implications. J. Am. Coll. Cardiol. 2022, 79, 577–593. [Google Scholar] [CrossRef]
- Drobni, Z.D.; Alvi, R.M.; Taron, J.; Zafar, A.; Murphy, S.P.; Rambarat, P.K.; Mosarla, R.C.; Lee, C.; Zlotoff, D.A.; Raghu, V.K.; et al. Association Between Immune Checkpoint Inhibitors with Cardiovascular Events and Atherosclerotic Plaque. Circulation 2020, 142, 2299–2311. [Google Scholar] [CrossRef]
- Kurihara, O.; Takano, M.; Miyauchi, Y.; Mizuno, K.; Shimizu, W. Vulnerable Atherosclerotic Plaque Features: Findings from Coronary Imaging. J. Geriatr. Cardiol. JGC 2021, 18, 577–584. [Google Scholar] [CrossRef]
- Jo, W.; Won, T.; Daoud, A.; Čiháková, D. Immune Checkpoint Inhibitors Associated Cardiovascular Immune-Related Adverse Events. Front. Immunol. 2024, 15, 1340373. [Google Scholar] [CrossRef]
- Bays, H.E.; Taub, P.R.; Epstein, E.; Michos, E.D.; Ferraro, R.A.; Bailey, A.L.; Kelli, H.M.; Ferdinand, K.C.; Echols, M.R.; Weintraub, H.; et al. Ten Things to Know about Ten Cardiovascular Disease Risk Factors. Am. J. Prev. Cardiol. 2021, 5, 100149. [Google Scholar] [CrossRef]
- Björkegren, J.L.M.; Lusis, A.J. Atherosclerosis: Recent Developments. Cell 2022, 185, 1630–1645. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Trucksäss, A.; Lichtenstein, A.H.; von Känel, R. Lifestyle Factors as Determinants of Atherosclerotic Cardiovascular Health. Atherosclerosis 2024, 395, 117577. [Google Scholar] [CrossRef]
- Peng, Q.; Zhu, J.; Zhang, Y.; Jing, Y. Blood Hypercoagulability and Thrombosis Mechanisms in Cancer Patients—A Brief Review. Heliyon 2024, 10, e38831. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Yu, W.; Liu, J.; Tang, D.; Yang, L.; Chen, X. Oxidative Cell Death in Cancer: Mechanisms and Therapeutic Opportunities. Cell Death Dis. 2024, 15, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Pursnani, S.; Merchant, M. South Asian Ethnicity as a Risk Factor for Coronary Heart Disease. Atherosclerosis 2020, 315, 126–130. [Google Scholar] [CrossRef]
- Santos Volgman, A.; Palaniappan, L.S.; Aggarwal, N.T.; Gupta, M.; Khandelwal, A.; Krishnan, A.V.; Lichtman, J.H.; Mehta, L.S.; Patel, H.N.; Shah, K.S.; et al. Atherosclerotic Cardiovascular Disease in South Asians in the United States: Epidemiology, Risk Factors, and Treatments: A Scientific Statement From the American Heart Association. Circulation 2018, 138, e1–e34. [Google Scholar] [CrossRef]
- Abdel-Qadir, H.; Austin, P.C.; Lee, D.S.; Amir, E.; Tu, J.V.; Thavendiranathan, P.; Fung, K.; Anderson, G.M. A Population-Based Study of Cardiovascular Mortality Following Early-Stage Breast Cancer. JAMA Cardiol. 2017, 2, 88–93. [Google Scholar] [CrossRef]
- Guo, J.; Fang, P.; Shi, W.; Luo, P.; Huo, S.; Yan, D.; Wang, M.; Peng, D.; Men, L.; Li, S.; et al. Preexisting Cardiovascular Risk Factors and Coronary Artery Atherosclerosis in Patients with and without Cancer. Cardiol. Res. Pract. 2022, 2022, 4570926. [Google Scholar] [CrossRef]
- Sekijima, T.; Tanabe, A.; Maruoka, R.; Fujishiro, N.; Yu, S.; Fujiwara, S.; Yuguchi, H.; Yamashita, Y.; Terai, Y.; Ohmichi, M. Impact of Platinum-Based Chemotherapy on the Progression of Atherosclerosis. Climacteric J. Int. Menopause Soc. 2011, 14, 31–40. [Google Scholar] [CrossRef]
- le Noble, F.A.C.; Mourad, J.-J.; Levy, B.I.; Struijker-Boudier, H.A.J. VEGF (Vascular Endothelial Growth Factor) Inhibition and Hypertension: Does Microvascular Rarefaction Play a Role? Hypertension 2023, 80, 901–911. [Google Scholar] [CrossRef]
- Bravo-Jaimes, K.; Marcellon, R.; Varanitskaya, L.; Kim, P.Y.; Iliescu, C.; Gilchrist, S.C.; Baldassarre, L.A.; Manisty, C.; Ghosh, A.K.; Guha, A.; et al. Opportunities for Improved Cardiovascular Disease Prevention in Oncology Patients. Curr. Opin. Cardiol. 2020, 35, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Al Samarraie, A.; Pichette, M.; Rousseau, G. Role of the Gut Microbiome in the Development of Atherosclerotic Cardiovascular Disease. Int. J. Mol. Sci. 2023, 24, 5420. [Google Scholar] [CrossRef] [PubMed]
- Stanton, K.M.; Liu, H.; Kienzle, V.; Bursill, C.; Bao, S.; Celermajer, D.S. The Effects of Exercise on Plaque Volume and Composition in a Mouse Model of Early and Late Life Atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 837371. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Liao, J.K. Statins and Cardiovascular Diseases: From Cholesterol Lowering to Pleiotropy. Curr. Pharm. Des. 2009, 15, 467–478. [Google Scholar] [CrossRef]
- GBD 2015 Risk Factors Collaborators. Global, Regional, and National Comparative Risk Assessment of 79 Behavioural, Environmental and Occupational, and Metabolic Risks or Clusters of Risks, 1990–2015: A Systematic Analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1659–1724. [Google Scholar] [CrossRef]
- Bosch, X.; Rovira, M.; Sitges, M.; Domènech, A.; Ortiz-Pérez, J.T.; de Caralt, T.M.; Morales-Ruiz, M.; Perea, R.J.; Monzó, M.; Esteve, J. Enalapril and Carvedilol for Preventing Chemotherapy-Induced Left Ventricular Systolic Dysfunction in Patients with Malignant Hemopathies: The OVERCOME Trial (preventiOn of Left Ventricular Dysfunction with Enalapril and caRvedilol in Patients Submitted to Intensive ChemOtherapy for the Treatment of Malignant hEmopathies). J. Am. Coll. Cardiol. 2013, 61, 2355–2362. [Google Scholar] [CrossRef]
- Tajiri, K.; Aonuma, K.; Sekine, I. Cardio-Oncology: A Multidisciplinary Approach for Detection, Prevention and Management of Cardiac Dysfunction in Cancer Patients. Jpn. J. Clin. Oncol. 2017, 47, 678–682. [Google Scholar] [CrossRef]
- Ng, C.T.; Bonilla, H.M.G.; Bryce, A.H.; Singh, P.; Herrmann, J. Approaches to Prevent and Manage Cardiovascular Disease in Patients Receiving Therapy for Prostate Cancer. Curr. Cardiol. Rep. 2023, 25, 889–899. [Google Scholar] [CrossRef]
- Cheng, A.; Krauter, M.; Mullen, K.-A.; Liu, P. The Continuing Scourge of Atherosclerotic Cardiovascular Disease: Importance of Multidisciplinary and Innovative Person-Centred Approaches. Can. J. Cardiol. 2024, 40, S43–S52. [Google Scholar] [CrossRef]
- Alizadehasl, A.; Amin, A.; Maleki, M.; Noohi, F.; Ghavamzadeh, A.; Farrashi, M. Cardio-oncology Discipline: Focus on the Necessities in Developing Countries. ESC Heart Fail. 2020, 7, 2175–2183. [Google Scholar] [CrossRef]
- de Jesus, M.; Mohammed, T.; Singh, M.; Tiu, J.G.; Kim, A.S. Etiology and Management of Dyslipidemia in Patients with Cancer. Front. Cardiovasc. Med. 2022, 9, 892335. [Google Scholar] [CrossRef]
- Lee, Y.R.; Oh, S.S.; Jang, S.-I.; Park, E.-C. Statin Adherence and Risk of All-Cause, Cancer, and Cardiovascular Mortality among Dyslipidemia Patients: A Time-Dependent Analysis. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 2207–2214. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.L.; Roddick, A.J. Association of Aspirin Use for Primary Prevention with Cardiovascular Events and Bleeding Events. JAMA 2019, 321, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Cvetković, R.S.; Scott, L.J. Dexrazoxane: A Review of Its Use for Cardioprotection during Anthracycline Chemotherapy. Drugs 2005, 65, 1005–1024. [Google Scholar] [CrossRef] [PubMed]
- Brånén, L.; Hovgaard, L.; Nitulescu, M.; Bengtsson, E.; Nilsson, J.; Jovinge, S. Inhibition of Tumor Necrosis Factor-Alpha Reduces Atherosclerosis in Apolipoprotein E Knockout Mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2137–2142. [Google Scholar] [CrossRef]
- Gallucci, G.; Larocca, M.; Navazio, A.; Turazza, F.M.; Inno, A.; Canale, M.L.; Oliva, S.; Besutti, G.; Tedeschi, A.; Aschieri, D.; et al. Atherosclerosis and the Bidirectional Relationship Between Cancer and Cardiovascular Disease: From Bench to Bedside, Part 2 Management. Int. J. Mol. Sci. 2025, 26, 334. [Google Scholar] [CrossRef]
- Mitchell, J.D.; Laurie, M.; Xia, Q.; Dreyfus, B.; Jain, N.; Jain, A.; Lane, D.; Lenihan, D.J. Risk Profiles and Incidence of Cardiovascular Events across Different Cancer Types. ESMO Open 2023, 8, 101830. [Google Scholar] [CrossRef]

{kind=link}
| Anticancer Agent Class | Mechanisms of Action | Effects on ASCVD | Clinical Implications |
|---|---|---|---|
| Anthracyclines | Inhibit topoisomerase II, induce mitochondrial dysfunction, generate ROS | Impair endothelial function, promote oxidative stress, disrupt cholesterol efflux, and elevate ApoB levels | Promote plaque formation and instability by reducing HMGCR activity despite elevated LDL levels |
| Taxanes | Stabilize GDP-bound tubulin, disrupting microtubule function | Alter lipid metabolism: increase HMGCR activity, reduce ApoB activity, and lower LDL receptor expression | Associated with dyslipidemia and proteomic changes in lipid metabolic pathways |
| VEGF Inhibitors | Block tumor angiogenesis, impair endothelial homeostasis | Reduce NO and PGI2 levels; promote vasoconstriction, platelet aggregation, microvascular thrombosis | Accelerate plaque progression, increase hypertension risk, and raise overall ASCVD incidence |
| Anti-Metabolites | Incorporated into DNA/RNA, disrupting cell proliferation | Increase ROS, pro-inflammatory cytokines, adhesion molecules, and endothelial stress | Contribute to endothelial injury, plaque vulnerability, and coronary vasospasm in some cases |
| mTOR Inhibitors | Inhibit mTOR signaling, affecting cell proliferation and metabolism | Downregulate lipogenic enzymes and LDL receptors; impair autophagy and endothelial repair | Induce dyslipidemia (elevated LDL/TG) and potentially impact ASCVD outcomes, though evidence is limited |
| Hormonal Therapies | Modulate estrogen/testosterone via SERMs, ADT, or AIs | Affect cholesterol synthesis and lipid transport; SERMs modulate lipid profiles, while AIs increase risk | ADT raises ASCVD risk (~20%), while SERMs offer some lipid protection; AIs increase dyslipidemia and CVD events |
| Tyrosine Kinase Inhibitors (TKIs) | Inhibit kinases involved in endothelial repair and NO production | Increase ROS, lipid peroxidation, and impair endothelial function | Imatinib reduces cholesterol and atherosclerosis; newer TKIs show mixed effects on lipids |
| Alkylating Agents | Cross-link DNA and RNA, halting cell division | High doses induce dyslipidemia in animals; dyslipidemia association unclear in humans | High doses induce dyslipidemia in animals; low-dose cyclophosphamide reduces atherosclerosis; human data limited |
| Platinum-Based Agents | Cross-link DNA, disrupting transcription | Promote endothelial dysfunction, increase cytokines, and induce fibrinogen | Associated with thrombotic and inflammatory states; little data on cholesterol or lipid changes in humans |
| Immune Checkpoint Inhibitors (ICIs) | Block PD-1 and CTLA4, enhancing T-cell activity | Exacerbate plaque inflammation, increase cytokines, promote vulnerable plaques | Linked to a 3-fold higher ASCVD risk, including plaque rupture and thrombotic complications |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izquierdo-Condoy, J.S.; Arias-Intriago, M.; Becerra Cardona, D.A.; García-Cañarte, S.; Vinueza-Moreano, P. Anticancer Chemotherapy-Induced Atherosclerotic Cardiovascular Disease: A Comprehensive Review. Life 2025, 15, 245. https://doi.org/10.3390/life15020245
Izquierdo-Condoy JS, Arias-Intriago M, Becerra Cardona DA, García-Cañarte S, Vinueza-Moreano P. Anticancer Chemotherapy-Induced Atherosclerotic Cardiovascular Disease: A Comprehensive Review. Life. 2025; 15(2):245. https://doi.org/10.3390/life15020245
Chicago/Turabian StyleIzquierdo-Condoy, Juan S., Marlon Arias-Intriago, Diego Alexander Becerra Cardona, Susana García-Cañarte, and Paul Vinueza-Moreano. 2025. "Anticancer Chemotherapy-Induced Atherosclerotic Cardiovascular Disease: A Comprehensive Review" Life 15, no. 2: 245. https://doi.org/10.3390/life15020245
APA StyleIzquierdo-Condoy, J. S., Arias-Intriago, M., Becerra Cardona, D. A., García-Cañarte, S., & Vinueza-Moreano, P. (2025). Anticancer Chemotherapy-Induced Atherosclerotic Cardiovascular Disease: A Comprehensive Review. Life, 15(2), 245. https://doi.org/10.3390/life15020245