Current Update of Laboratory Molecular Diagnostics Advancement in Management of Colorectal Cancer (CRC)

 ,
,
Abstract
:1. Colorectal Cancer (CRC) and Its Treatment
2. Molecular Biomarkers
2.1. Methylated Septin9 (mSEPT9)
2.2. Microsatellite Instability (MSI)
2.3. KRAS (Kirsten Rat Sarcoma Viral Oncogene Homolog)
2.4. BRAF (v-raf Murine Sarcoma Viral Oncogene Homolog B1)
2.5. Other Potential Biomarkers
2.5.1. Programmed Death-Ligand 1 (PD-L1)
2.5.2. Phosphatidylinositol-4,5-bisphosphate 3-kinase, Catalytic Subunit Alpha (PIK3CA)
2.5.3. Phosphatase and Tensin Homolog (PTEN)
2.5.4. Human Epidermal Growth Factor Receptor 2 (HER2)
2.5.5. Micro-RNA (miRNA)
2.5.6. Consensus Molecular Subtypes (CMS)
3. Current Molecular Diagnostic Technologies
3.1. Detection of KRAS and BRAF Mutations
3.1.1. PCR-Based Detection
3.1.2. Nucleic Acid Hybridization Assay
3.1.3. Pre-PCR Isolation of Circulating Tumor Cells (CTCs)
3.1.4. Circulating Free DNA (cfDNA) Detection
3.2. Detection of Microsatellite Instability (MSI)/Microsatellite Stable (MSS) Status
3.3. miRNA Detection
3.4. Detection of Other Biomarkers
4. Challenges and Limitations
4.1. Choice of Molecular Platform
4.2. Laboratory Operations
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | Adenomatous polyposis coli |
| ARMS | Amplification refractory mutation system |
| BRAF | B-Raf protein |
| cfDNA | Circulating free DNA |
| COLD-PCR | Coamplification at lower denaturation temperature PCR |
| CRC | Colorectal cancer |
| CTC | Circulating tumor cells |
| ddPCR | Droplet digital PCR |
| DNA | Deoxyribonucleic acid |
| EGFR | Epidermal growth factor receptor |
| FDA | Food and Drug Administration |
| FFPE | Formalin-fixed paraffin embedded |
| FIT | Fecal immunochemical test |
| FOBT | Fecal occult blood test |
| HER2 | Human epidermal growth factor 2 |
| IHC | Immunohistochemistry |
| KRAS | Kirten rat sarcoma viral oncogene homolog |
| LNA | Locked nucleic acid |
| mAb | monoclonal antibody |
| mCRC | metastatic colorectal cancer |
| miRNA | microRNA |
| MMR | Mismatch repair |
| MSI | Microsatellite instability |
| NGS | Next-generation sequencing |
| NRAS | Neuroblastoma RAS viral oncogene homolog |
| ORR | Overall response rate |
| OS | Overall survival |
| PD-L1 | Programmed cell death protein ligand 1 |
| PFS | Progression-free survival |
| PNA | Peptide nucleic acid |
| RR | Response rate |
| RT-qPCR | Quantitative reverse transcription PCR |
| SEPT9 | Septin 9 |
| TKI | Tyrosine kinase inhibitor |
| TMBWNT | Tumor mutational burdenWingless and int-1 |
| WT | Wild-type |
| 5-FU | 5-fluorouracil |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Heemskerk-Gerritsen, B.A.M.; Rookus, M.A.; Aalfs, C.M.; Ausems, M.G.E.M.; Collée, J.M.; Jansen, L.; Kets, C.M.; Keymeulen, K.B.M.I.; Koppert, L.B.; Meijers-Heijboer, H.E.J.; et al. Improved overall survival after contralateral risk-reducing mastectomy in brca1/2 mutation carriers with a history of unilateral breast cancer: A prospective analysis. Int. J. Cancer 2015, 136, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyan, A.; Kircher, S.; Shah, H.; Mulcahy, M.; Benson, A. Updates on immunotherapy for colorectal cancer. J. Gastrointest. Oncol. 2018, 9, 160–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kircher, S.M.; Nimeiri, H.S.; Benson, A.B. Targeting angiogenesis in colorectal cancer tyrosine kinase inhibitors. Cancer J. 2016, 22, 182–189. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, B.H.; Wallmark, J.M.; Lorente, D.; Elez, E.; Raimbourg, J.; Gomez-Roca, C.; Ejadi, S.; Piha-Paul, S.A.; Stein, M.N.; Abdul Razak, A.R.; et al. Safety and antitumor activity of the anti–PD-1 antibody pembrolizumab in patients with advanced colorectal carcinoma. PLoS ONE 2017, 12, e0189848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA approval summary: Pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.J.; Chu, E. The adjuvant treatment of stage III colon cancer: Might less be more? Oncology 2018, 32, 437–442. [Google Scholar]
- FDA Approves Nivolumab Plus Ipilimumab Combination for Intermediate or Poor-Risk Advanced Renal Cell Carcinoma. Available online: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm604685.htm (accessed on 23 July 2019).
- Rogers, J.E. Patient considerations in metastatic colorectal cancer–role of panitumumab. Onco Targets Ther. 2017, 10, 2033–2044. [Google Scholar] [CrossRef] [Green Version]
- Jitawatanarat, P.; Ma, W.W. Update on antiangiogenic therapy in colorectal cancer: Aflibercept and regorafenib. J. Gastrointest. Oncol. 2013, 4, 231–238. [Google Scholar] [CrossRef]
- Kurkjian, C.; Kummar, S. Advances in the treatment of metastatic colorectal cancer. Disease-a-Month 2010, 56, 187–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothenberg, M.L. Irinotecan (CPT-11): Recent developments and future directions-colorectal cancer and beyond. Oncologist 2004, 6, 66–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, A. FDA drug approval summaries: Oxaliplatin. Oncologist 2004, 9, 8–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertino, J.R. Chemotherapy of colorectal cancer: History and new themes. Semin. Oncol. 1997, 24, S18-3–S18-7. [Google Scholar] [PubMed]
- Milsom, J.W.; Hammerhofer, K.A. Role of laparoscopic techniques in colorectal cancer surgery. Oncology 1995, 9, 393–398. [Google Scholar]
- Heald, R.J.; Husband, E.M.; Ryall, R.D.H. The mesorectum in rectal cancer surgery—The clue to pelvic recurrence? Br. J. Surg. 1982, 69, 613–616. [Google Scholar] [CrossRef]
- Warren, J.D.; Xiong, W.; Bunker, A.M.; Vaughn, C.P.; Furtado, L.V.; Roberts, W.L.; Fang, J.C.; Samowitz, W.S.; Heichman, K.A. Septin 9 methylated DNA is a sensitive and specific blood test for colorectal cancer. BMC Med. 2011, 9, 133. [Google Scholar] [CrossRef] [Green Version]
- Sepulveda, A.R.; Hamilton, S.R.; Allegra, C.J.; Grody, W.; Cushman-Vokoun, A.M.; Funkhouser, W.K.; Kopetz, S.E.; Lieu, C.; Lindor, N.M.; Minsky, B.D.; et al. Molecular biomarkers for the evaluation of colorectal cancer: Guideline from the American society for clinical pathology, college of American pathologists, association for molecular pathology, and American society of clinical oncology. Arch. Pathol. Lab. Med. 2017, 35, 1453–1486. [Google Scholar] [CrossRef] [Green Version]
- Bordeaux, J.; Welsh, A.W.; Agarwal, S.; Killiam, E.; Baquero, M.T.; Hanna, J.A.; Anagnostou, V.K.; Rimm, D.L. Antibody validation. Biotechniques 2010, 48, 197–209. [Google Scholar] [CrossRef] [Green Version]
- O’Hurley, G.; Sjöstedt, E.; Rahman, A.; Li, B.; Kampf, C.; Pontén, F.; Gallagher, W.M.; Lindskog, C. Garbage in, garbage out: A critical evaluation of strategies used for validation of immunohistochemical biomarkers. Mol. Oncol. 2014, 8, 783–798. [Google Scholar] [CrossRef]
- Allott, E.H.; Geradts, J.; Sun, X.; Cohen, S.M.; Zirpoli, G.R.; Khoury, T.; Bshara, W.; Chen, M.; Sherman, M.E.; Palmer, J.R.; et al. Intratumoral heterogeneity as a source of discordance in breast cancer biomarker classification. Breast Cancer Res. 2016, 18, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comǎnescu, M.; Arsene, D.; Ardeleanu, C.; Bussolati, G. The mandate for a proper preservation in histopathological tissues. Rom. J. Morphol. Embryol. 2012, 53, 233–242. [Google Scholar] [PubMed]
- Laterza, O.F.; Hendrickson, R.C.; Wagner, J.A. Molecular biomarkers. Drug Inf. J. 2007, 41, 573–585. [Google Scholar] [CrossRef]
- Pignone, M.; Saha, S.; Hoerger, T.; Mandelblatt, J. Cost-effectiveness analyses of colorectal cancer screening: A systematic review for the U.S. Preventive Services Task Force. Ann. Intern. Med. 2002, 137, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Smits, A.M.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberman, D.A. Clinical practice. Screening for colorectal cancer. N. Engl. J. Med. 2009, 361, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, J.; Zhou, Y.; Sheng, S.; Qian, S.Y.; Huo, X. Evaluation of serum CEA, CA19-9, CA72-4, CA125 and Ferritin as diagnostic markers and factors of clinical parameters for colorectal cancer. Sci. Rep. 2018, 8, 2732. [Google Scholar] [CrossRef] [PubMed]
- Shiratsuchi, I.; Akagi, Y.; Kawahara, A.; Kinugasa, T.; Romeo, K.; Yoshida, T.; Ryu, Y.; Gotanda, Y.; Kage, M.; Shirouzu, K. Expression of IGF-1 and IGF-1R and their relation to clinicopathological factors in colorectal cancer. Anticancer Res. 2011, 31, 2541–2545. [Google Scholar]
- Tóth, K.; Wasserkort, R.; Sipos, F.; Kalmár, A.; Wichmann, B.; Leiszter, K.; Valcz, G.; Juhász, M.; Miheller, P.; Patai, Á.V.; et al. Detection of methylated Septin 9 in tissue and plasma of colorectal patients with neoplasia and the relationship to the amount of circulating cell-free DNA. PLoS One 2014, 9, e115415. [Google Scholar] [CrossRef]
- Wang, J.Y.; Hsieh, J.S.; Chang, M.Y.; Huang, T.J.; Chen, F.M.; Cheng, T.L.; Alexandersen, K.; Huang, Y.S.; Tzou, W.S.; Lin, S.R. Molecular detection of APC, K-ras, and p53 mutations in the serum of colorectal cancer patients as circulating biomarkers. World J. Surg. 2004, 28, 721–726. [Google Scholar] [CrossRef]
- Church, T.R.; Wandell, M.; Lofton-Day, C.; Mongin, S.J.; Burger, M.; Payne, S.R.; Castaños-Vélez, E.; Blumenstein, B.A.; Rösch, T.; Osborn, N.; et al. Prospective evaluation of methylated SEPT9 in plasma for detection of asymptomatic colorectal cancer. Gut 2014, 63, 317–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Zhou, G.; Jin, P.; Zhu, J.; Li, S.; Wu, Q.; Wang, G.; Sheng, J.; Wang, J.; Song, L.; et al. Detection of colorectal cancer using a simplified SEPT9 gene methylation assay is a reliable method for opportunistic screening. J. Mol. Diagn. 2016, 18, 535–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, P.; Kang, Q.; Wang, X.; Yang, L.; Yu, Y.; Li, N.; He, Y.Q.; Han, X.; Hang, J.; Zhang, J.; et al. Performance of a second-generation methylated SEPT9 test in detecting colorectal neoplasm. J. Gastroenterol. Hepatol. 2015, 30, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Su, X.L.; Wang, Y.F.; Li, S.J.; Zhang, F.; Cui, H.W. High methylation of the SEPT9 gene in Chinese colorectal cancer patients. Genet. Mol. Res. 2014, 13, 2513–2520. [Google Scholar] [CrossRef] [PubMed]
- Tóth, K.; Sipos, F.; Kalmár, A.; Patai, Á.V.; Wichmann, B.; Stoehr, R.; Golcher, H.; Schellerer, V.; Tulassay, Z.; Molnár, B. Detection of methylated SEPT9 in plasma is a reliable screening method for both left- and right-sided colon cancers. PLoS One 2012, 7, e46000. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, P.-M.; Liu, R.-B. Advance in plasma SEPT9 gene methylation assay for colorectal cancer early detection. World J. Gastrointest. Oncol. 2018, 10, 15–22. [Google Scholar] [CrossRef]
- Kojima, K.; Sakai, I.; Hasegawa, A.; Niiya, H.; Azuma, T.; Matsuo, Y.; Fujii, N.; Tanimoto, M.; Fujita, S. FLJ10849, a septin family gene, fuses MLL in a novel leukemia cell line CNLBC1 derived from chronic neutrophilic leukemia in transformation with t(4;11)(q21;q23). Leukemia 2004, 18, 998–1005. [Google Scholar] [CrossRef]
- Montagna, C.; Lyu, M.S.; Hunter, K.; Lukes, L.; Lowther, W.; Reppert, T.; Hissong, B.; Weaver, Z.; Ried, T. The Septin 9 (MSF) gene is amplified and overexpressed in mouse mammary gland adenocarcinomas and human breast cancer cell lines. Cancer Res. 2003, 63, 2179–2187. [Google Scholar]
- Burrows, J.F.; Chanduloy, S.; McIlhatton, M.A.; Nagar, H.; Yeates, K.; Donaghy, P.; Price, J.; Godwin, A.K.; Johnston, P.G.; Russell, S.E.H. Altered expression of the septin gene, SEPT9, in ovarian neoplasia. J. Pathol. 2003, 201, 581–588. [Google Scholar] [CrossRef]
- Amir, S.; Golan, M.; Mabjeesh, N.J. Targeted knockdown of SEPT9_v1 inhibits tumor growth and angiogenesis of human prostate cancer cells concomitant with disruption of hypoxia-inducible factor-1 pathway. Mol. Cancer Res. 2010, 8, 643–652. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-S.; Hubbard, S.-L.; Peraud, A.; Salhia, B.; Sakai, K.; Rutka, J.T. Analysis of mammalian Septin expression in human malignant brain tumors. Neoplasia 2004, 6, 168–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.Y.; Law, W.L.; Ng, E.K.O.; Chan, C.S.Y.; Lau, K.S.; Cheng, Y.Y.; Shin, V.Y.; Kwong, A.; Leung, W.K. Methylated Septin 9 and carcinoembryonic antigen for serological diagnosis and monitoring of patients with colorectal cancer after surgery. Sci. Rep. 2019, 9, 10326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.; Williams, M.; Cheng, Y.Y.; Leung, W.K. Roles of methylated DNA biomarkers in patients with colorectal cancer. Dis. Mark. 2019, 2019, 2673543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, M.T.; Weinberg, D.S. Biomarkers in colorectal cancer screening. JNCCN J. Natl. Compr. Cancer Netw. 2016, 14, 1033–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vymetalkova, V.; Cervena, K.; Bartu, L.; Vodicka, P. Circulating cell-free DNA and colorectal cancer: A systematic review. Int. J. Mol. Sci. 2018, 19, 3356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, A.H.F.; Cheng, K.H.; Wong, A.S.P.; Ng, S.S.M.; Ma, B.B.Y.; Chan, C.M.L.; Tsui, N.B.Y.; Chan, L.W.C.; Yung, B.Y.M.; Wong, S.C.C. Current and future molecular diagnostics in colorectal cancer and colorectal adenoma. World J. Gastroenterol. 2014, 20, 3847–3857. [Google Scholar] [CrossRef]
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer-the stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Legolvan, M.P.; Taliano, R.J.; Resnick, M.B. Application of molecular techniques in the diagnosis, prognosis and management of patients with colorectal cancer: A practical approach. Hum. Pathol. 2012, 43, 1157–1168. [Google Scholar] [CrossRef]
- Huth, L.; Jäkel, J.; Dahl, E. Molecular diagnostic applications in colorectal cancer. Microarrays 2014, 3, 168–179. [Google Scholar] [CrossRef] [Green Version]
- Jover, R.; Zapater, P.; Castells, A.; Llor, X.; Andreu, M.; Cubiella, J.; Balaguer, F.; Sempere, L.; Xicola, R.M.; Bujanda, L.; et al. The efficacy of adjuvant chemotherapy with 5-fluorouracil in colorectal cancer depends on the mismatch repair status. Eur. J. Cancer 2009, 45, 365–373. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Gong, J.; Wang, C.; Lee, P.P.; Chu, P.; Fakih, M. Response to PD-1 blockade in microsatellite stable metastatic colorectal cancer harboring a POLE mutation. JNCCN J. Natl. Compr. Cancer Netw. 2017, 15, 142–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorich, M.J.; Wiese, M.D.; Rowland, A.; Kichenadasse, G.; McKinnon, R.A.; Karapetis, C.S. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: A meta-analysis of randomized, controlled trials. Ann. Oncol. 2015, 26, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Lenz, H.J.; Köhne, C.H.; Heinemann, V.; Tejpar, S.; Melezínek, I.; Beier, F.; Stroh, C.; Rougier, P.; Han Van Krieken, J.; et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J. Clin. Oncol. 2015, 33, 692–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, D.; Longatto-Filho, A.; Martins, S.F. Predictive biomarkers in colorectal cancer: From the single therapeutic target to a plethora of options. Biomed Res. Int. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Douillard, J.Y.; Siena, S.; Cassidy, J.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Randomized, Phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) Versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: The PRIME study. J. Clin. Oncol. 2010, 28, 4697–4705. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Köhne, C.-H.; Hitre, E.; Zaluski, J.; Chang Chien, C.-R.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [Green Version]
- Douillard, J.-Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab–FOLFOX4 Treatment and RAS Mutations in Colorectal Cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Ursem, C.; Atreya, C.E.; Van Loon, K. Emerging treatment options for BRAF-mutant colorectal cancer. Gastrointest. Cancer Targets Ther. 2018, 8, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Di Nicolantonio, F.; Martini, M.; Molinari, F.; Sartore-Bianchi, A.; Arena, S.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; Frattini, M.; Siena, S.; et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 5705–5712. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Voigt, A.; Bozukova, G.; Ahadova, A.; Kickingereder, P.; Von Deimling, A.; Von Knebel Doeberitz, M.; Kloor, M. BRAF V600E-specific immunohistochemistry for the exclusion of Lynch syndrome in MSI-H colorectal cancer. Int. J. Cancer 2013, 133, 1624–1630. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Swanson, B.J.; Frankel, W.L. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn. Pathol. 2017, 12, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, S.; Roepman, P.; Popovici, V.; Michaut, M.; Majewski, I.; Salazar, R.; Santos, C.; Rosenberg, R.; Nitsche, U.; Mesker, W.E.; et al. A robust genomic signature for the detection of colorectal cancer patients with microsatellite instability phenotype and high mutation frequency. J. Pathol. 2012, 228, 586–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, A.D.; Tejpar, S.; Delorenzi, M.; Yan, P.; Fiocca, R.; Klingbiel, D.; Dietrich, D.; Biesmans, B.; Bodoky, G.; Barone, C.; et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: Results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J. Clin. Oncol. 2010, 28, 466–474. [Google Scholar] [CrossRef]
- Manthravadi, S.; Sun, W.; Saeed, A. Prognostic impact of BRAF V600E mutation in patients with non-metastatic colorectal cancer with microsatellite instability: A systematic review and meta-analysis. J. Clin. Oncol. 2018, 15, 3597. [Google Scholar] [CrossRef]
- Rosenbaum, M.W.; Bledsoe, J.R.; Morales-Oyarvide, V.; Huynh, T.G.; Mino-Kenudson, M. PD-L1 expression in colorectal cancer is associated with microsatellite instability, BRAF mutation, medullary morphology and cytotoxic tumor-infiltrating lymphocytes. Proc. Mod. Pathol. 2016, 29, 1104–1112. [Google Scholar] [CrossRef]
- Lee, L.H.; Cavalcanti, M.S.; Segal, N.H.; Hechtman, J.F.; Weiser, M.R.; Smith, J.J.; Garcia-Aguilar, J.; Sadot, E.; Ntiamoah, P.; Markowitz, A.J.; et al. Patterns and prognostic relevance of PD-1 and PD-L1 expression in colorectal carcinoma. Mod. Pathol. 2016, 29, 1433–1442. [Google Scholar] [CrossRef] [Green Version]
- Droeser, R.A.; Hirt, C.; Viehl, C.T.; Frey, D.M.; Nebiker, C.; Huber, X.; Zlobec, I.; Eppenberger-Castori, S.; Tzankov, A.; Rosso, R.; et al. Clinical impact of programmed cell death ligand 1 expression in colorectal cancer. Eur. J. Cancer 2013, 49, 2233–2242. [Google Scholar] [CrossRef]
- Wang, H.B.; Yao, H.; Li, C.S.; Liang, L.X.; Zhang, Y.; Chen, Y.X.; Fang, J.Y.; Xu, J. Rise of PD-L1 expression during metastasis of colorectal cancer: Implications for immunotherapy. J. Dig. Dis. 2017, 18, 574–581. [Google Scholar] [CrossRef]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.F.; Yu, B.H.; Li, D.L.; Ke, H.L.; Guo, X.Z.; Xiao, X.Y. PI3K expression and PIK3CA mutations are related to colorectal cancer metastases. World J. Gastroenterol. 2012, 18, 3745–3751. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Yang, Z.Y.; Hu, X.F.; Chen, Q.; Tang, J.L. PIK3CA exon 20 mutations as a potential biomarker for resistance to anti-EGFR monoclonal antibodies in KRAS wild-type metastatic colorectal cancer: A systematic review and meta-analysis. Ann. Oncol. 2012, 23, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Prenen, H.; De Schutter, J.; Jacobs, B.; De Roock, W.; Biesmans, B.; Claes, B.; Lambrechts, D.; Van Cutsem, E.; Tejpar, S. PIK3CA mutations are not a major determinant of resistance to the epidermal growth factor receptor inhibitor cetuximab in metastatic colorectal cancer. Clin. Cancer Res. 2009, 15, 3184–3188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Garcia, V.; Bartos, C.; Keraite, I.; Trivedi, U.; Brennan, P.M.; Kersaudy-Kerhoas, M.; Gharbi, K.; Oikonomidou, O.; Leslie, N.R. A simple and robust real-time qPCR method for the detection of PIK3CA mutations. Sci. Rep. 2018, 8, 4290. [Google Scholar] [CrossRef] [Green Version]
- Yazdani, Y.; Farazmandfar, T.; Azadeh, H.; Zekavatian, Z. The prognostic effect of PTEN expression status in colorectal cancer development and evaluation of factors affecting it: MiR-21 and promoter methylation. J. Biomed. Sci. 2016, 23, 9. [Google Scholar] [CrossRef] [Green Version]
- Negri, F.V.; Bozzetti, C.; Lagrasta, C.A.; Crafa, P.; Bonasoni, M.P.; Camisa, R.; Pedrazzi, G.; Ardizzoni, A. PTEN status in advanced colorectal cancer treated with cetuximab. Br. J. Cancer 2010, 102, 162–164. [Google Scholar] [CrossRef] [Green Version]
- Laurent-Puig, P.; Cayre, A.; Manceau, G.; Buc, E.; Bachet, J.B.; Lecomte, T.; Rougier, P.; Lievre, A.; Landi, B.; Boige, V.; et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J. Clin. Oncol. 2009, 27, 5924–5930. [Google Scholar] [CrossRef]
- Loupakis, F.; Pollina, L.; Stasi, I.; Ruzzo, A.; Scartozzi, M.; Santini, D.; Masi, G.; Graziano, F.; Cremolini, C.; Rulli, E.; et al. PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J. Clin. Oncol. 2009, 27, 2622–2629. [Google Scholar] [CrossRef]
- Bohn, B.A.; Mina, S.; Krohn, A.; Simon, R.; Kluth, M.; Harasimowicz, S.; Quaas, A.; Bockhorn, M.; Izbicki, J.R.; Sauter, G.; et al. Altered PTEN function caused by deletion or gene disruption is associated with poor prognosis in rectal but not in colon cancer. Hum. Pathol. 2013, 44, 1524–1533. [Google Scholar] [CrossRef]
- Atreya, C.E.; Sangale, Z.; Xu, N.; Matli, M.R.; Tikishvili, E.; Welbourn, W.; Stone, S.; Shokat, K.M.; Warren, R.S. PTEN expression is consistent in colorectal cancer primaries and metastases and associates with patient survival. Cancer Med. 2013, 2, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Eklöf, V.; Wikberg, M.L.; Edin, S.; Dahlin, A.M.; Jonsson, B.A.; Öberg, Å.; Rutegård, J.; Palmqvist, R. The prognostic role of KRAS, BRAF, PIK3CA and PTEN in colorectal cancer. Br. J. Cancer 2013, 108, 2153–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, F.L.; Jorissen, R.N.; Lipton, L.; Mouradov, D.; Sakthianandeswaren, A.; Christie, M.; Li, S.; Tsui, C.; Tie, J.; Desai, J.; et al. PIK3CA and PTEN gene and exon mutation-specific clinicopathologic and molecular associations in colorectal cancer. Clin. Cancer Res. 2013, 19, 3285–3296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pazhoomand, R.; Keyhani, E.; Banan, M.; Najmabadi, H.; Khodadadi, F.; Iraniparast, A.; Feiz, F.; Majidzadeh, K.; Bahman, I.; Moghadam, F.A.; et al. Detection of HER2 status in breast cancer: Comparison of current methods with MLPA and real-time RT-PCR. Asian Pac. J. Cancer Prev. 2013, 14, 7621–7628. [Google Scholar] [CrossRef] [Green Version]
- Valtorta, E.; Martino, C.; Sartore-Bianchi, A.; Penaullt-Llorca, F.; Viale, G.; Risio, M.; Rugge, M.; Grigioni, W.; Bencardino, K.; Lonardi, S.; et al. Assessment of a HER2 scoring system for colorectal cancer: Results from a validation study. Mod. Pathol. 2015, 28, 1481–1491. [Google Scholar] [CrossRef] [Green Version]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Schepeler, T.; Reinert, J.T.; Ostenfeld, M.S.; Christensen, L.L.; Silahtaroglu, A.N.; Dyrskjøt, L.; Wiuf, C.; Sørensen, F.J.; Kruhøffer, M.; Laurberg, S.; et al. Diagnostic and prognostic microRNAs in stage II colon cancer. Cancer Res. 2008, 68, 6416–6424. [Google Scholar] [CrossRef] [Green Version]
- Nagel, R.; Le Sage, C.; Diosdado, B.; Van Der Waal, M.; Oude Vrielink, J.A.F.; Bolijn, A.; Meijer, G.A.; Agami, R. Regulation of the adenomatous polyposis coli gene by the miR-135 family in colorectal cancer. Cancer Res. 2008, 68, 5795–5802. [Google Scholar] [CrossRef] [Green Version]
- Kan, S.F.; Yang, J.S.; Sun, G.X.; Sun, J.J. MicroRNA-135b is associated with tumor progression in colorectal cancer. Int. J. Clin. Exp. Med. 2016, 9, 6533–6538. [Google Scholar]
- Zhou, W.; Li, X.; Liu, F.; Xiao, Z.; He, M.; Shen, S.; Liu, S. MiR-135a promotes growth and invasion of colorectal cancer via metastasis suppressor 1 in vitro. Acta Biochim. Biophys. Sin. 2012, 44, 838–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, D.; Tang, L.; Zhuang, Y.; Zhao, P. MiR-17-3P regulates the proliferation and survival of colon cancer cells by targeting Par4. Mol. Med. Rep. 2018, 17, 618–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.; Li, Q.; Wang, H.; Yang, F.; Min, L.; Yang, J. MiR-92a promotes tumorigenesis of colorectal cancer, a transcriptomic and functional based study. Biomed. Pharmacother. 2018, 106, 1370–1377. [Google Scholar] [CrossRef]
- Yang, X.; Zeng, Z.; Hou, Y.; Yuan, T.; Gao, C.; Jia, W.; Yi, X.; Liu, M. MicroRNA-92a as a potential biomarker in diagnosis of colorectal cancer: A systematic review and meta-analysis. PLoS One 2014, 9, e88745. [Google Scholar] [CrossRef] [Green Version]
- Pu, X.X.; Huang, G.L.; Guo, H.Q.; Guo, C.C.; Li, H.; Ye, S.; Ling, S.; Jiang, L.; Tian, Y.; Lin, T.Y. Circulating miR-221 directly amplified from plasma is a potential diagnostic and prognostic marker of colorectal cancer and is correlated with p53 expression. J. Gastroenterol. Hepatol. 2010, 25, 1674–1680. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zhao, J.; Wu, C.-W.; Zhang, L.; Liu, X.; Kang, W.; Leung, W.-W.; Zhang, N.; Chan, F.K.L.; Sung, J.J.Y.; et al. Tumor suppressor functions of miR-133a in colorectal cancer. Mol. Cancer Res. 2013, 11, 1051–1060. [Google Scholar] [CrossRef] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Williams, D.S.; Mouradov, D.; Jorissen, R.N.; Newman, M.R.; Amini, E.; Nickless, D.K.; Teague, J.A.; Fang, C.G.; Palmieri, M.; Parsons, M.J.; et al. Lymphocytic response to tumour and deficient DNA mismatch repair identify subtypes of stage II/III colorectal cancer associated with patient outcomes. Gut 2019, 68, 465–474. [Google Scholar] [CrossRef]
- Marisa, L.; Ayadi, M.; Balogoun, R.; Pilati, C.; Le Malicot, K.; Lepage, C.; Emile, J.-F.; Salazar, R.; Aust, D.E.; Duval, A.; et al. Clinical utility of colon cancer molecular subtypes: Validation of two main colorectal molecular classifications on the PETACC-8 phase III trial cohort. J. Clin. Oncol. 2017, 15, 3509. [Google Scholar] [CrossRef]
- Mooi, J.K.; Wirapati, P.; Asher, R.; Lee, C.K.; Savas, P.; Price, T.J.; Townsend, A.; Hardingham, J.; Buchanan, D.; Williams, D.; et al. The prognostic impact of consensus molecular subtypes (CMS) and its predictive effects for bevacizumab benefit in metastatic colorectal cancer: Molecular analysis of the AGITG MAX clinical trial. Ann. Oncol. 2018, 29, 2240–2246. [Google Scholar] [CrossRef]
- Lenz, H.-J.; Ou, F.-S.; Venook, A.P.; Hochster, H.S.; Niedzwiecki, D.; Goldberg, R.M.; Mayer, R.J.; Bertagnolli, M.M.; Blanke, C.D.; Zemla, T.; et al. Impact of consensus molecular subtype on survival in patients with metastatic colorectal cancer: results from CALGB/SWOG 80405 (Alliance). J. Clin. Oncol. 2019, 37, 1876–1885. [Google Scholar] [CrossRef] [PubMed]
- Okita, A.; Takahashi, S.; Ouchi, K.; Inoue, M.; Watanabe, M.; Endo, M.; Honda, H.; Yamada, Y.; Ishioka, C. Consensus molecular subtypes classification of colorectal cancer as a predictive factor for chemotherapeutic efficacy against metastatic colorectal cancer. Oncotarget 2018, 9, 18698–18711. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Andrews, K.S.; Brooks, D.; Fedewa, S.A.; Manassaram-Baptiste, D.; Saslow, D.; Brawley, O.W.; Wender, R.C. Cancer screening in the United States, 2018: A review of current American Cancer Society guidelines and current issues in cancer screening. CA Cancer J. Clin. 2018, 68, 297–316. [Google Scholar] [CrossRef] [PubMed]
- Sargent, D.J.; Marsoni, S.; Monges, G.; Thibodeau, S.N.; Labianca, R.; Hamilton, S.R.; French, A.J.; Kabat, B.; Foster, N.R.; Torri, V.; et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J. Clin. Oncol. 2010, 28, 3219–3226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Yang, Z.; Eshleman, J.R.; Netto, G.J.; Lin, M.T. Molecular diagnostics for precision medicine in colorectal cancer: Current status and future perspective. BioMed Res. Int. 2016, 2016, 9850690. [Google Scholar] [CrossRef]
- Cree, I.A. Diagnostic RAS mutation analysis by polymerase chain reaction (PCR). Biomol. Detect. Quantif. 2016, 8, 29–32. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.; Forestier, J.; Dusserre, E.; Wozny, A.-S.; Geiguer, F.; Merle, P.; Tissot, C.; Ferraro-Peyret, C.; Jones, F.S.; Edelstein, D.L.; et al. Cross-platform comparison for the detection of RAS mutations in cfDNA (ddPCR Biorad detection assay, BEAMing assay, and NGS strategy). Oncotarget 2018, 9, 21122–21131. [Google Scholar] [CrossRef]
- Neumann, J.; Zeindl-Eberhart, E.; Kirchner, T.; Jung, A. Frequency and type of KRAS mutations in routine diagnostic analysis of metastatic colorectal cancer. Pathol. Res. Pract. 2009, 205, 858–862. [Google Scholar] [CrossRef]
- Tsiatis, A.C.; Norris-Kirby, A.; Rich, R.G.; Hafez, M.J.; Gocke, C.D.; Eshleman, J.R.; Murphy, K.M. Comparison of Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations: Diagnostic and clinical implications. J. Mol. Diagn. 2010, 12, 425–432. [Google Scholar] [CrossRef]
- Szankasi, P.; Reading, N.S.; Vaughn, C.P.; Prchal, J.T.; Bahler, D.W.; Kelley, T.W. A quantitative allele-specific PCR test for the BRAF V600E mutation using a single heterozygous control plasmid for quantitation: A model for qPCR testing without standard curves. J. Mol. Diagn. 2013, 15, 248–254. [Google Scholar] [CrossRef]
- Do, H.; Krypuy, M.; Mitchell, P.L.; Fox, S.B.; Dobrovic, A. High resolution melting analysis for rapid and sensitive EGFR and KRAS mutation detection in formalin fixed paraffin embedded biopsies. BMC Cancer 2008, 8, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliner, K.; Juan, T.; Suggs, S.; Wolf, M.; Sarosi, I.; Freeman, D.J.; Gyuris, T.; Baron, W.; Bakker, A.; Parker, A.; et al. A comparability study of 5 commercial KRAS tests. Diagn. Pathol. 2010, 5, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobunai, T.; Watanabe, T.; Yamamoto, Y.; Eshima, K. The frequency of KRAS mutation detection in human colon carcinoma is influenced by the sensitivity of assay methodology: A comparison between direct sequencing and real-time PCR. Biochem. Biophys. Res. Commun. 2010, 395, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Sarasqueta, A.F.; Moerland, E.; De Bruyne, H.; De Graaf, H.; Vrancken, T.; Van Lijnschoten, G.; Van Den Brule, A.J.C. SNaPshot and StripAssay as valuable alternatives to direct sequencing for KRAS mutation detection in colon cancer routine diagnostics. J. Mol. Diagn. 2011, 13, 199–205. [Google Scholar] [CrossRef]
- Halait, H.; Demartin, K.; Shah, S.; Soviero, S.; Langland, R.; Cheng, S.; Hillman, G.; Wu, L.; Lawrence, H.J. Analytical performance of a real-time PCR-based assay for V600 mutations in the BRAF gene, used as the companion diagnostic test for the novel BRAF inhibitor vemurafenib in metastatic melanoma. Diagn. Mol. Pathol. 2012, 21, 1–8. [Google Scholar] [CrossRef]
- Harbison, C.T.; Horak, C.E.; Ledeine, J.M.; Mukhopadhyay, P.; Malone, D.P.; O’Callaghan, C.; Jonker, D.J.; Karapetis, C.S.; Khambata-Ford, S.; Gustafson, N.; et al. Validation of companion diagnostic for detection of mutations in Codons 12 and 13 of the KRAS gene in patients with metastatic colorectal Cancer: Analysis of the NCIC CTG CO.17 trial. Arch. Pathol. Lab. Med. 2013, 137, 820–827. [Google Scholar] [CrossRef]
- Harlé, A.; Busser, B.; Rouyer, M.; Harter, V.; Genin, P.; Leroux, A.; Merlin, J.L. Comparison of COBAS 4800 KRAS, TaqMan PCR and High Resolution Melting PCR assays for the detection of KRAS somatic mutations in formalin-fixed paraffin embedded colorectal carcinomas. Virchows Arch. 2013, 462, 329–335. [Google Scholar] [CrossRef]
- Nordgård, O.; Oltedal, S.; Janssen, E.A.M.; Gilje, B.; Kørner, H.; Tjensvoll, K.; Smaaland, R. Comparison of a PNA clamp PCR and an ARMS/Scorpion PCR assay for the detection of K-ras mutations. Diagn. Mol. Pathol. 2012, 21, 9–13. [Google Scholar] [CrossRef]
- Orum, H. PCR clamping. Curr. Issues Mol. Biol. 2000, 2, 27–30. [Google Scholar] [CrossRef]
- Mancini, I.; Santucci, C.; Sestini, R.; Simi, L.; Pratesi, N.; Cianchi, F.; Valanzano, R.; Pinzani, P.; Orlando, C. The use of COLD-PCR and high-resolution melting analysis improves the limit of detection of KRAS and BRAF mutations in colorectal cancer. J. Mol. Diagnostics 2010, 12, 705–711. [Google Scholar] [CrossRef]
- Solassol, J.; Vendrell, J.; Märkl, B.; Haas, C.; Bellosillo, B.; Montagut, C.; Smith, M.; O’Sullivan, B.; D’Haene, N.; Le Mercier, M.; et al. Multi-center evaluation of the fully automated PCR-based IdyllaTM KRAS mutation assay for rapid KRAS mutation status determination on formalin-fixed paraffin-embedded tissue of human colorectal cancer. PLoS One 2016, 11, e0163444. [Google Scholar] [CrossRef] [PubMed]
- Johnston, L.; Power, M.; Sloan, P.; Long, A.; Silmon, A.; Chaffey, B.; Lisgo, A.J.; Little, L.; Vercauteren, E.; Steiniche, T.; et al. Clinical performance evaluation of the Idylla NRAS-BRAF mutation test on retrospectively collected formalin-fixed paraffin-embedded colorectal cancer tissue. J. Clin. Pathol. 2018, 71, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, M.A.; Jóåwicki, W.; Żurawski, B. KRAS and BRAF mutation analysis in colorectal adenocarcinoma specimens with a low percentage of tumor cells. Mol. Diagn. Ther. 2013, 17, 193–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnin, S.; Viel, E.; Baraquin, A.; Valmary-Degano, S.; Kantelip, B.; Pretet, J.L.; Mougin, C.; Bigand, M.; Girardo, B.; Borg, C.; et al. A multiplex SNaPshot assay as a rapid method for detecting KRAS and BRAF mutations in advanced colorectal cancers. J. Mol. Diagn. 2011, 13, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Denis, J.A.; Patroni, A.; Guillerm, E.; Pépin, D.; Benali-Furet, N.; Wechsler, J.; Manceau, G.; Bernard, M.; Coulet, F.; Larsen, A.K.; et al. Droplet digital PCR of circulating tumor cells from colorectal cancer patients can predict KRAS mutations before surgery. Mol. Oncol. 2016, 10, 1221–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, M.H.D.; Bender, S.; Krahn, T.; Schlange, T. ctDNA and CTCs in liquid biopsy–Current status and where we need to progress. Comput. Struct. Biotechnol. J. 2018, 16, 190–195. [Google Scholar] [CrossRef]
- Baudrin, L.G.; Deleuze, J.F.; How-Kit, A. Molecular and computational methods for the detection of microsatellite instability in cancer. Front. Oncol. 2018, 8, 621. [Google Scholar] [CrossRef]
- Redford, L.; Alhilal, G.; Needham, S.; O’Brien, O.; Coaker, J.; Tyson, J.; Amorim, L.M.; Middleton, I.; Izuogu, O.; Arends, M.; et al. A novel panel of short mononucleotide repeats linked to informative polymorphisms enabling effective high volume low cost discrimination between mismatch repair deficient and proficient tumours. PLoS One 2018, 13, e0203052. [Google Scholar] [CrossRef] [Green Version]
- Murphy, K.M.; Zhang, S.; Geiger, T.; Hafez, M.J.; Bacher, J.; Berg, K.D.; Eshleman, J.R. Comparison of the microsatellite instability analysis system and the Bethesda panel for the determination of microsatellite instability in colorectal cancers. J. Mol. Diagn. 2006, 8, 305–311. [Google Scholar] [CrossRef] [Green Version]
- Babaei, H.; Zeinalian, M.; Emami, M.H.; Hashemzadeh, M.; Farahani, N.; Salehi, R. Simplified microsatellite instability detection protocol provides equivalent sensitivity to robust detection strategies in Lynch syndrome patients. Cancer Biol. Med. 2017, 14, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005, 33, e179. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ruan, K. MicroRNA detection by microarray. Anal. Bioanal. Chem. 2009, 394, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.Y.; Hsiao, Y.L.; Lin, M.S.; Yen, C.C.; Chang, C.S. MicroRNA detection using lateral flow nucleic acid strips with gold nanoparticles. Talanta 2012, 99, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Xu, H.; Zeng, Q.; Zeng, L.; Liu, G. Molecular beacon-functionalized gold nanoparticles as probes in dry-reagent strip biosensor for DNA analysis. Chem. Commun. 2009, 21, 3065–3067. [Google Scholar] [CrossRef]
- He, Y.; Zhang, S.; Zhang, X.; Baloda, M.; Gurung, A.S.; Xu, H.; Zhang, X.; Liu, G. Ultrasensitive nucleic acid biosensor based on enzyme-gold nanoparticle dual label and lateral flow strip biosensor. Biosens. Bioelectron. 2011, 26, 2018–2024. [Google Scholar] [CrossRef]
- Aravanis, A.M.; Lee, M.; Klausner, R.D. Next-generation sequencing of circulating tumor DNA for early cancer detection. Cell 2017, 168, 571–574. [Google Scholar] [CrossRef] [Green Version]
- Marcuello, M.; Vymetalkova, V.; Neves, R.P.L.; Duran-Sanchon, S.; Vedeld, H.M.; Tham, E.; van Dalum, G.; Flügen, G.; Garcia-Barberan, V.; Fijneman, R.J.; et al. Circulating biomarkers for early detection and clinical management of colorectal cancer. Mol. Asp. Med. 2019, 69, 107–122. [Google Scholar] [CrossRef]
- Andrews, D.; Chetty, Y.; Cooper, B.S.; Virk, M.; Glass, S.K.; Letters, A.; Kelly, P.A.; Sudhanva, M.; Jeyaratnam, D. Multiplex PCR point of care testing versus routine, laboratory-based testing in the treatment of adults with respiratory tract infections: A quasi-randomised study assessing impact on length of stay and antimicrobial use. BMC Infect. Dis. 2017, 17, 671. [Google Scholar] [CrossRef] [Green Version]
- Lindeman, N.I.; Cagle, P.T.; Beasley, M.B.; Chitale, D.A.; Dacic, S.; Giaccone, G.; Jenkins, R.B.; Kwiatkowski, D.J.; Saldivar, J.S.; Squire, J.; et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: Guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Patho. J. Thorac. Oncol. 2013, 8, 823–859. [Google Scholar] [CrossRef] [Green Version]
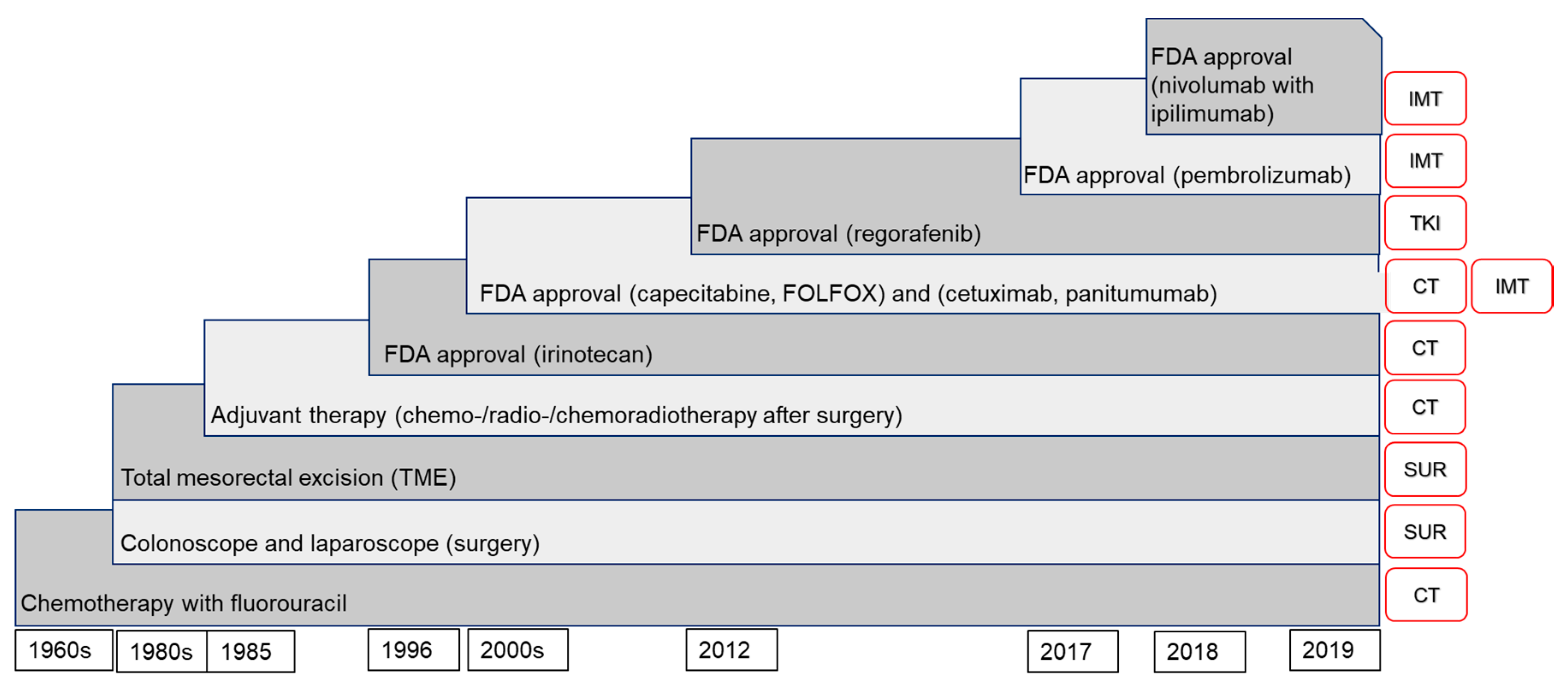
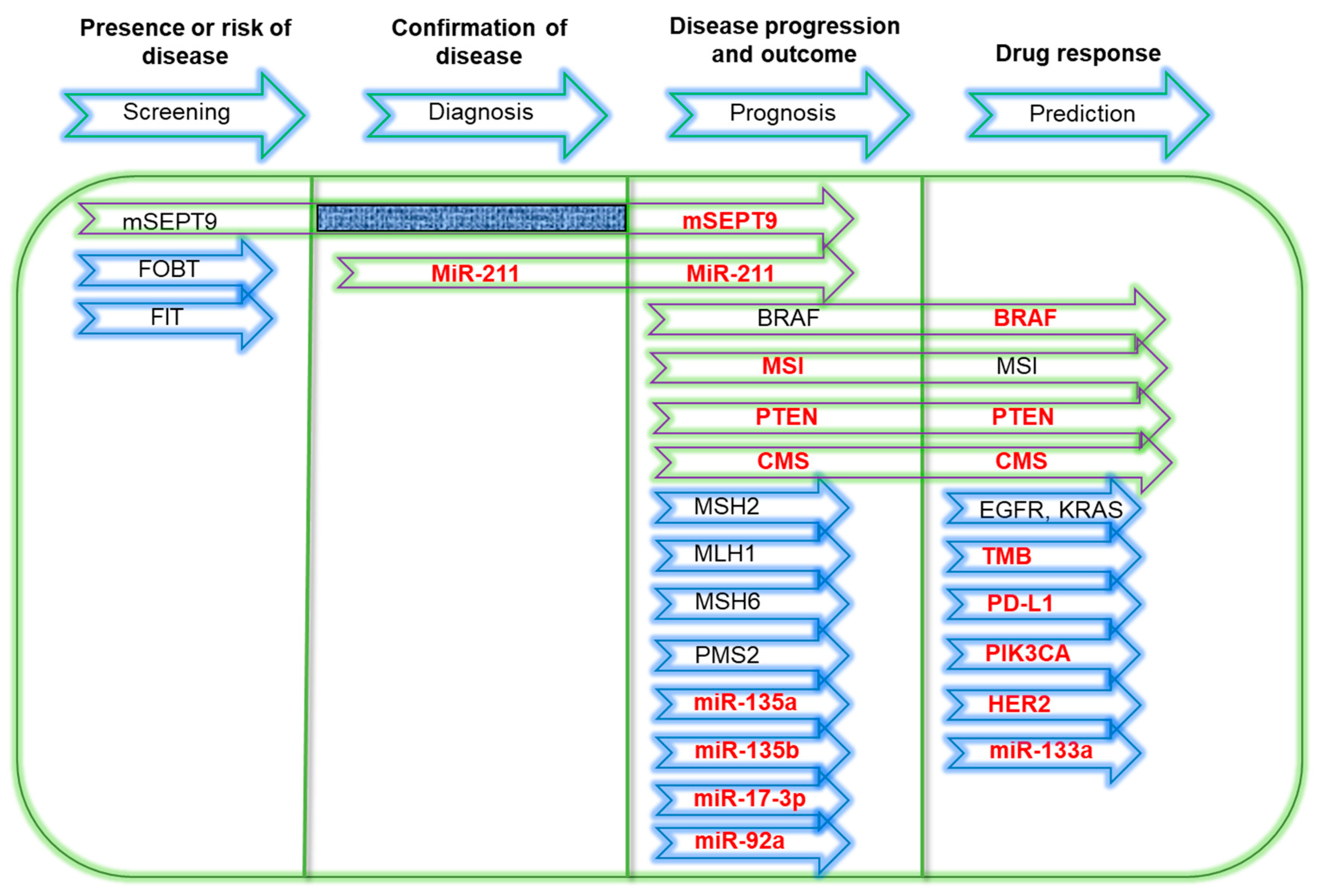

{kind=link}
{kind=link}
| Platform | Sensitivity (%) | Specificity | PPV (%) | NPV (%) |
|---|---|---|---|---|
| ddPCR | 47 | 77 | 70 | 55 |
| BEAM | 93 | 69 | 78 | 90 |
| NGS | 73 | 77 | 79 | 71 |
| Biomarker | Diagnostic Platform | References |
|---|---|---|
| mSEPT9 | • Methylation-specific real-time PCR | [18] |
| MSI | • Immunohistochemistry | [64] |
| • Next-generation sequencing (NGS) | [89] | |
| • Fragment analysis | [128] | |
| • Gene expression assay | [130] | |
| • miRNA microarray | [89] | |
| TMB | • Whole exome sequencing | [54] |
| KRAS | • qPCR (Roche Cobas 4800 KRAS mutation, Qiagen therascreen KRAS, Biocartis KRAS mutation assay) | [117,122] |
| • Direct sequencing | [106] | |
| • Pyrosequencing | [111] | |
| • Next-generation sequencing (NGS) | [108] | |
| • High-resolution melt curve (HRM) | [110] | |
| • Amplification refractory mutation system (ARMS) PCR | [119] | |
| • Peptic nucleic acid (PNA) clamp PCR | [119] | |
| • COLD-PCR | [121] | |
| • Single nucleotide primer extension (SNaPshot) | [115] | |
| • Reverse hybridization KRAS StripAssay | [115] | |
| • Digital PCR (Bio-Rad droplet digital PCR, Sysmex BEAMing) | [108] | |
| BRAF | • qPCR (Roche Cobas 4800 BRAF V600, Biocartis Idylla NRAS-BRAF mutation assay) | [117,122] |
| • Direct sequencing | [106] | |
| • Pyrosequencing | [111] | |
| • Next-generation sequencing (NGS) | [108] | |
| • High-resolution melt curve (HRM) | [110] | |
| • COLD-PCR | [121] | |
| • Immunohistochemistry | [106] | |
| • Single nucleotide primer extension (SNaPshot) | [125] | |
| • Reverse hybridization BRAF StripAssay | [124] | |
| miRNA | • Microarray—spotted locked nucleic acid (LNA) | [89] |
| • RT-qPCR | [132] | |
| • Lateral flow nucleic acid strip assay using gold nanoparticles | [47] | |
| PD-L1 | • Immunohistochemistry | [68] |
| PIK3CA | • Real-time PCR | [76] |
| • Immunohistochemistry | [73] | |
| • Gene sequencing | [73] | |
| HER2 | • Immunohistochemistry | [86] |
| • Quantitative reverse transcription PCR (RT-qPCR) | [85] | |
| PTEN | • Indirect immunofluorescence | [78] |
| • Immunohistochemistry | [77] | |
| CMS | • Gene expression microarray | [99] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, S.-W.; Awi, N.J.; Armon, S.; Lim, W.W.-D.; Low, J.S.-H.; Peh, K.-B.; Peh, S.-C.; Teow, S.-Y. Current Update of Laboratory Molecular Diagnostics Advancement in Management of Colorectal Cancer (CRC). Diagnostics 2020, 10, 9. https://doi.org/10.3390/diagnostics10010009
Pang S-W, Awi NJ, Armon S, Lim WW-D, Low JS-H, Peh K-B, Peh S-C, Teow S-Y. Current Update of Laboratory Molecular Diagnostics Advancement in Management of Colorectal Cancer (CRC). Diagnostics. 2020; 10(1):9. https://doi.org/10.3390/diagnostics10010009
Chicago/Turabian StylePang, Siew-Wai, Noel Jacques Awi, Subasri Armon, Wendy Wan-Dee Lim, John Seng-Hooi Low, Kaik-Boo Peh, Suat-Cheng Peh, and Sin-Yeang Teow. 2020. "Current Update of Laboratory Molecular Diagnostics Advancement in Management of Colorectal Cancer (CRC)" Diagnostics 10, no. 1: 9. https://doi.org/10.3390/diagnostics10010009
APA StylePang, S.-W., Awi, N. J., Armon, S., Lim, W. W.-D., Low, J. S.-H., Peh, K.-B., Peh, S.-C., & Teow, S.-Y. (2020). Current Update of Laboratory Molecular Diagnostics Advancement in Management of Colorectal Cancer (CRC). Diagnostics, 10(1), 9. https://doi.org/10.3390/diagnostics10010009