Retrospective Definition of Clostridioides difficile PCR Ribotypes on the Basis of Whole Genome Polymorphisms: A Proof of Principle Study

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. In Silico PCR-Based Ribotyping
2.2. DBGWAS-Mediated Discovery New RT-Specific Markers
2.3. Validation of Markers
2.4. Statistically Reliable Ribotype Prediction
2.5. Functional Annotation of Unitigs
3. Results and Discussion
3.1. In Silico PCR
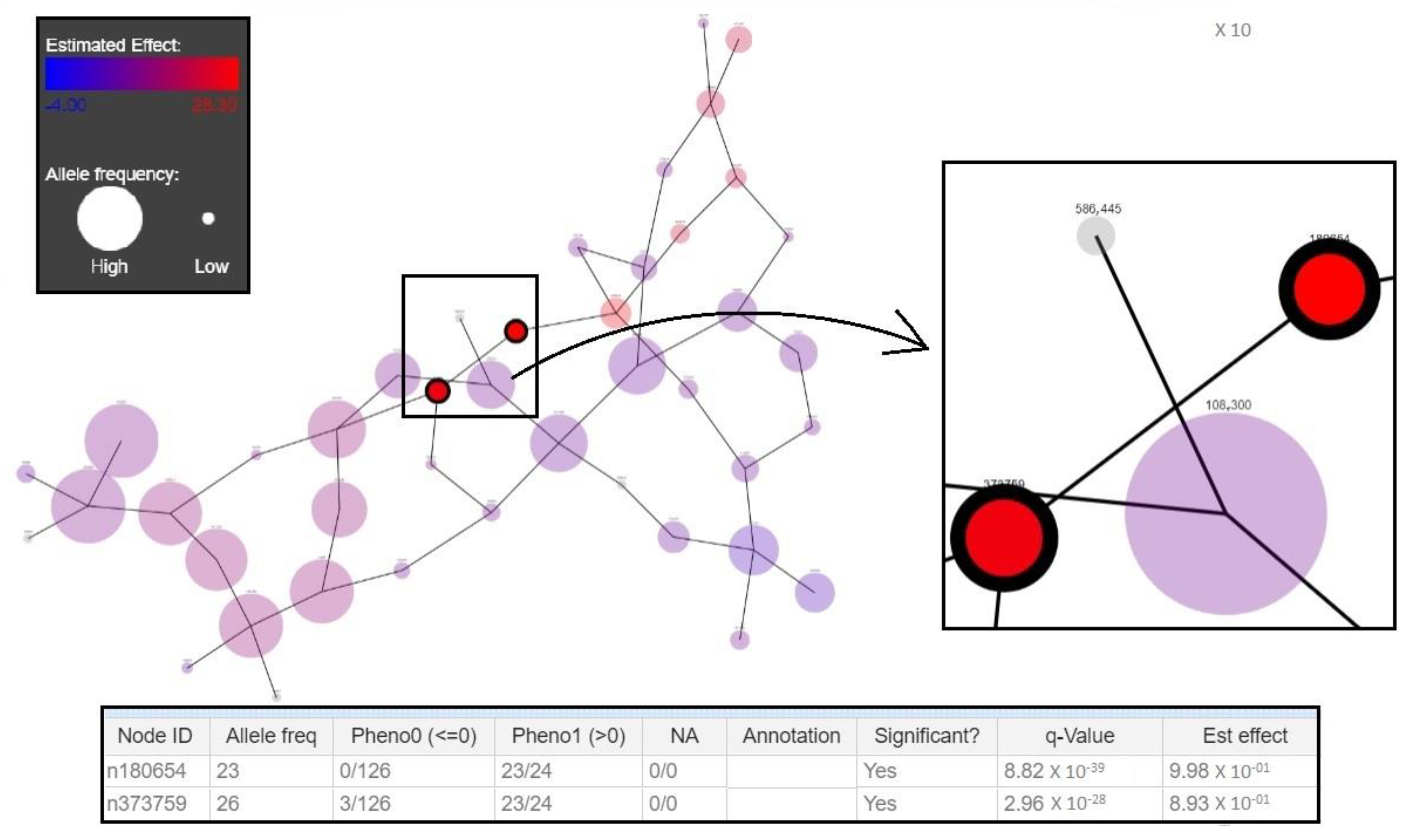
3.2. New Genotyping Markers
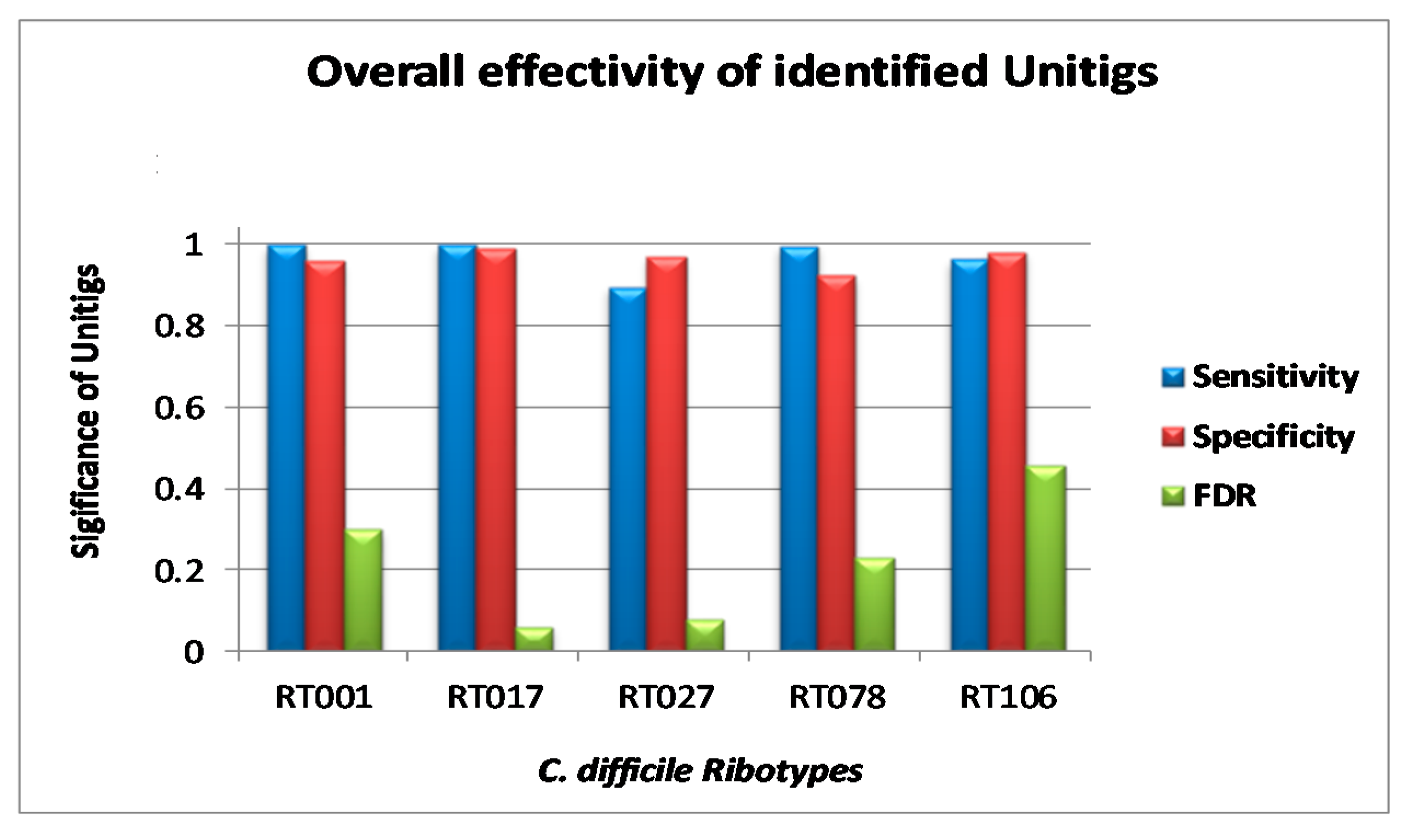
3.3. Validation of Markers
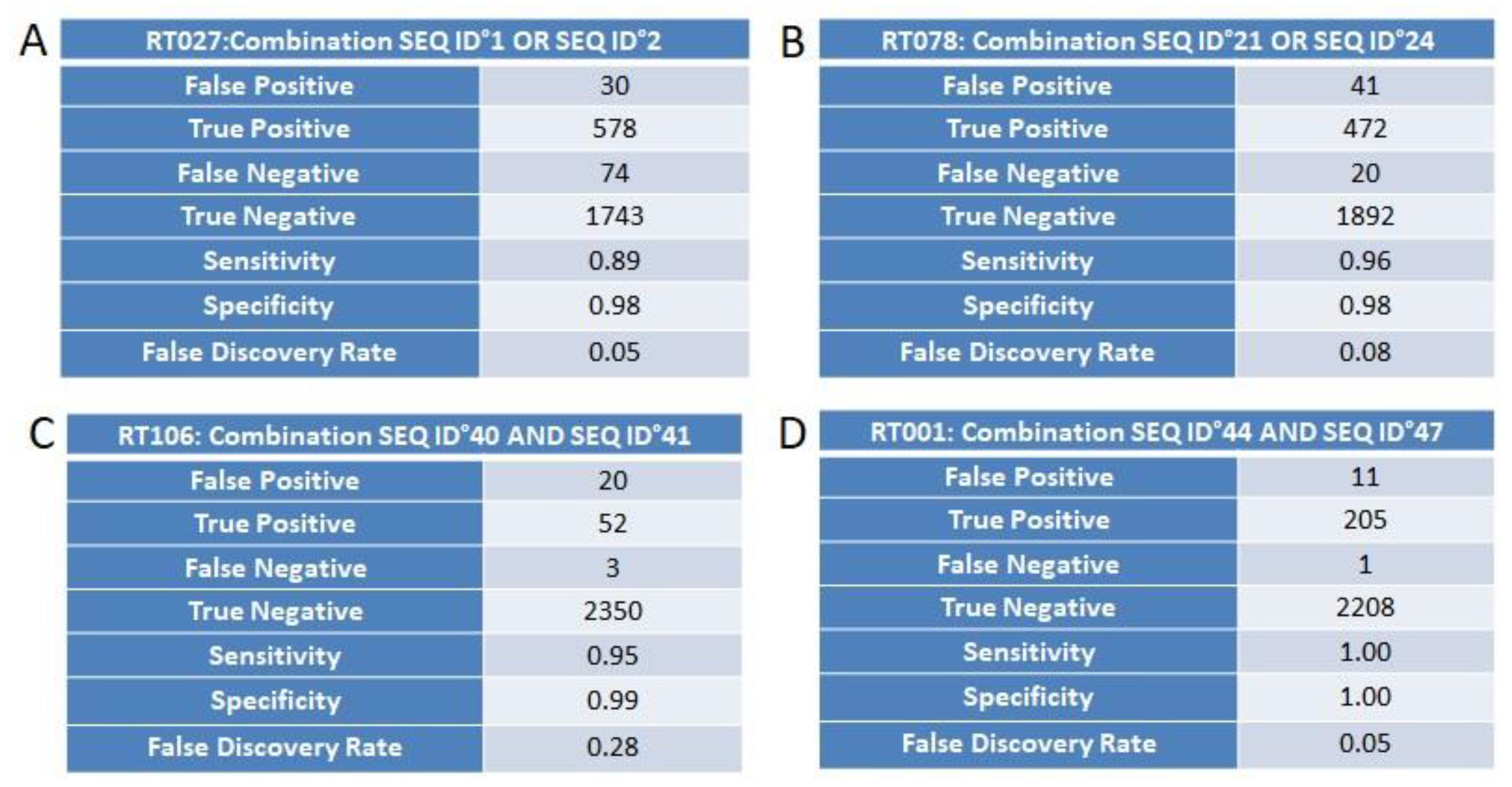
3.4. Marker Combination Study
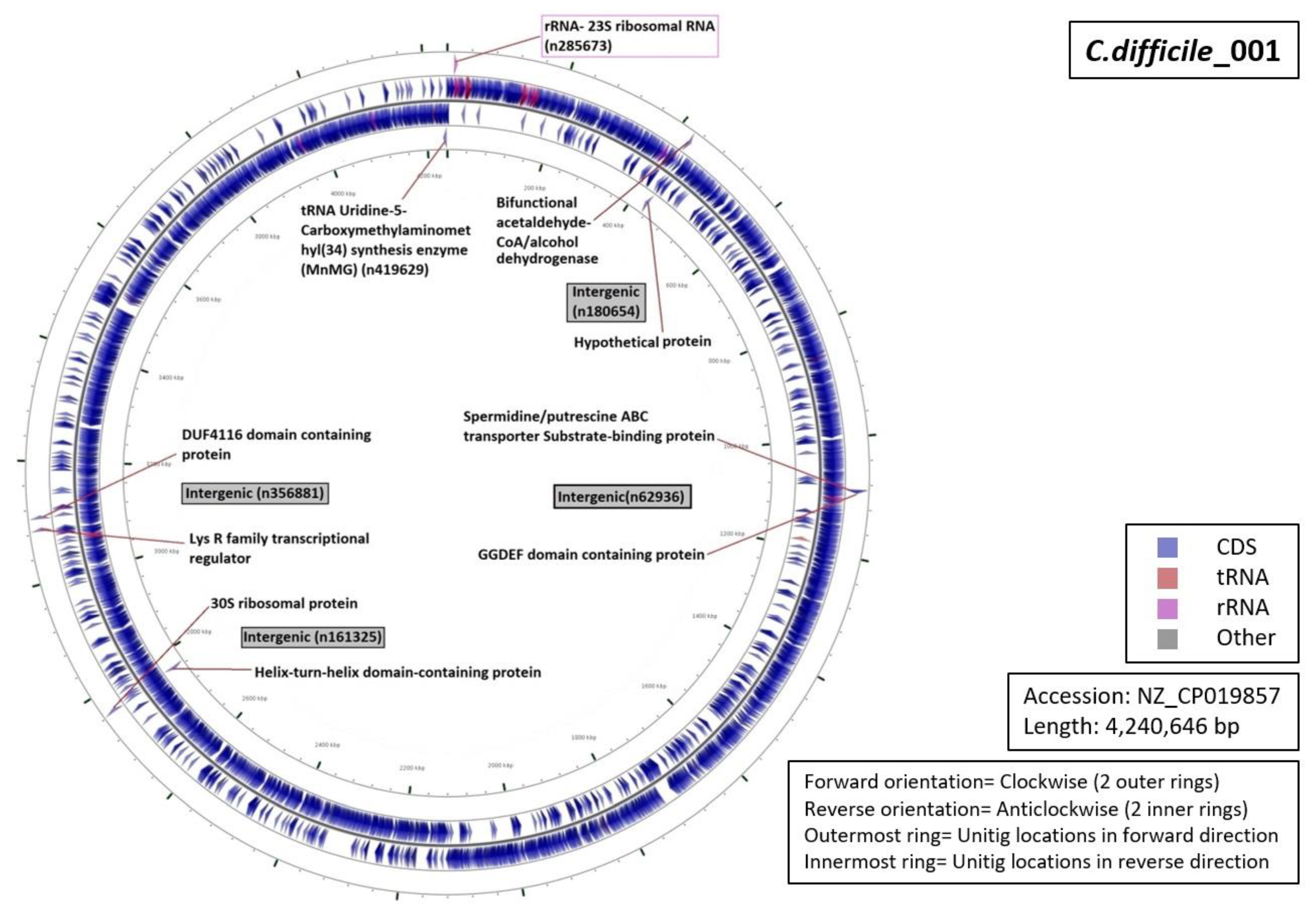
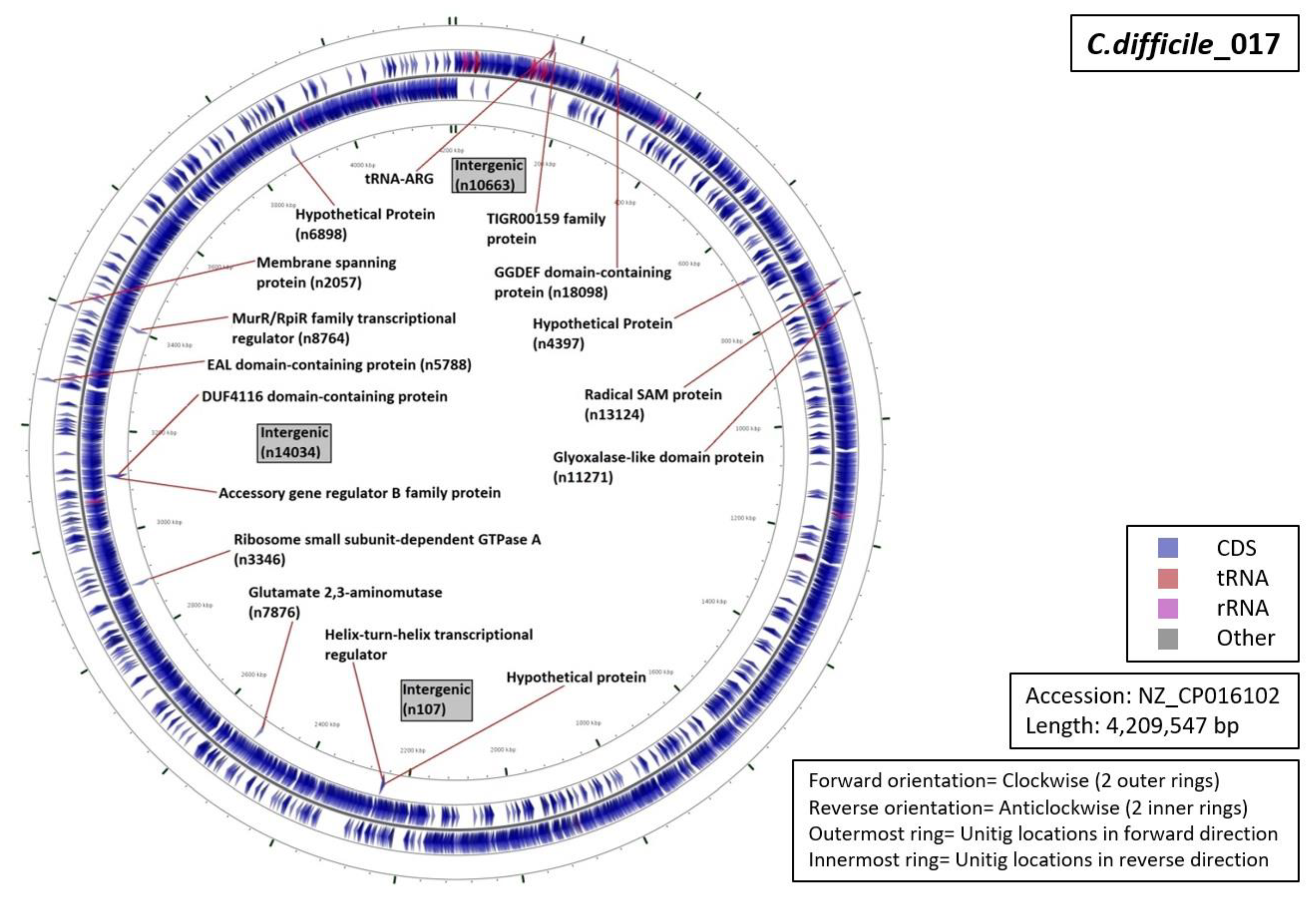
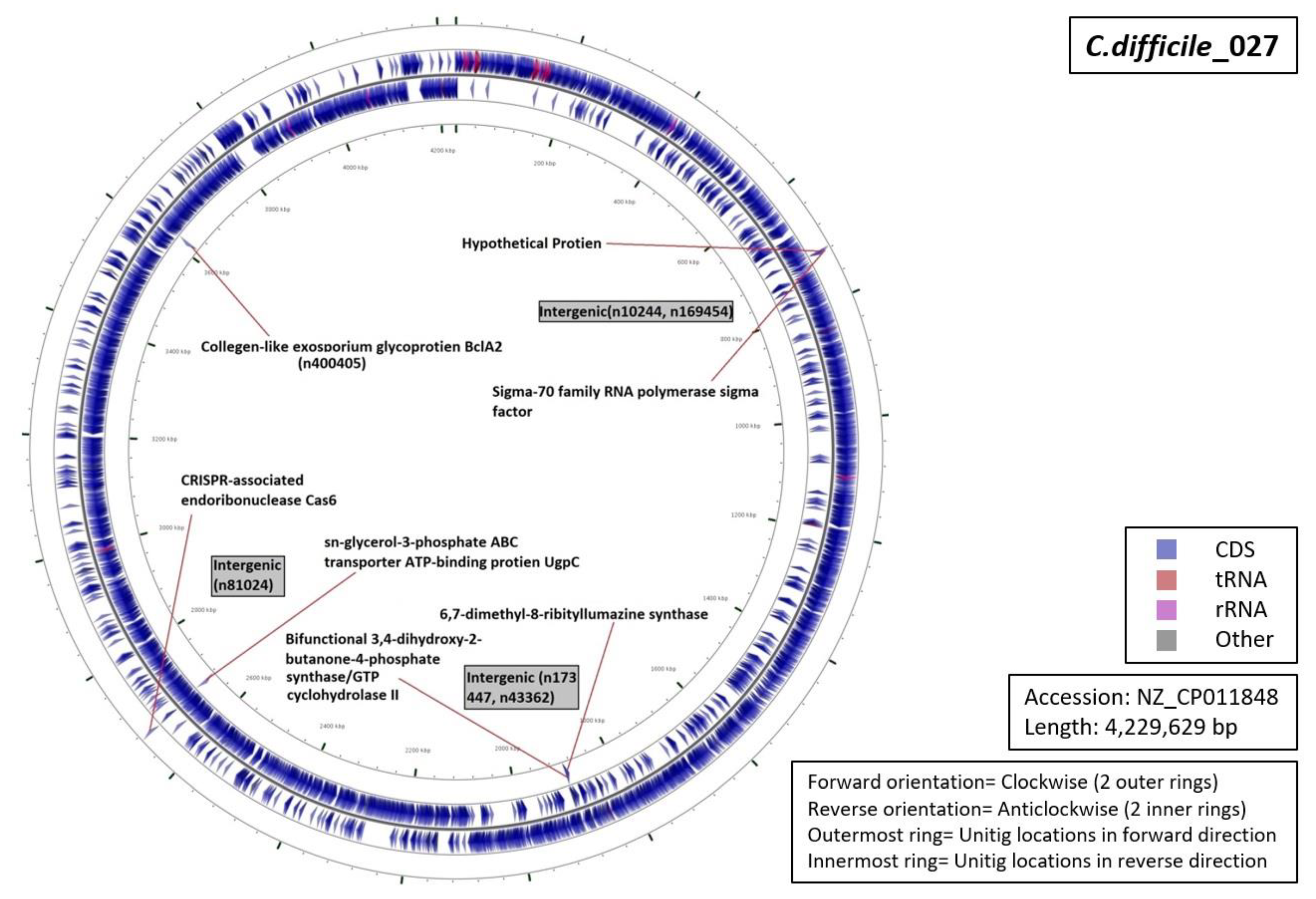
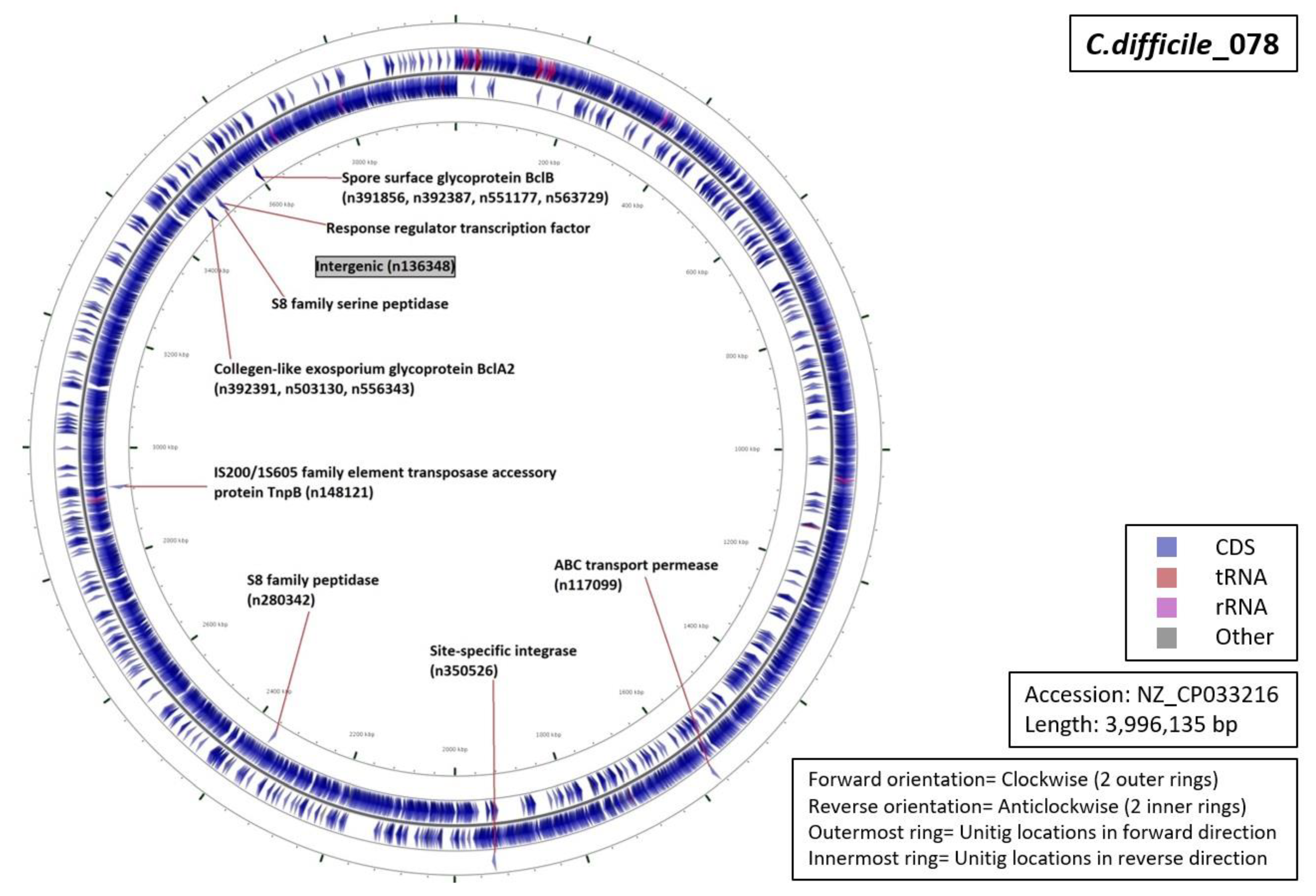
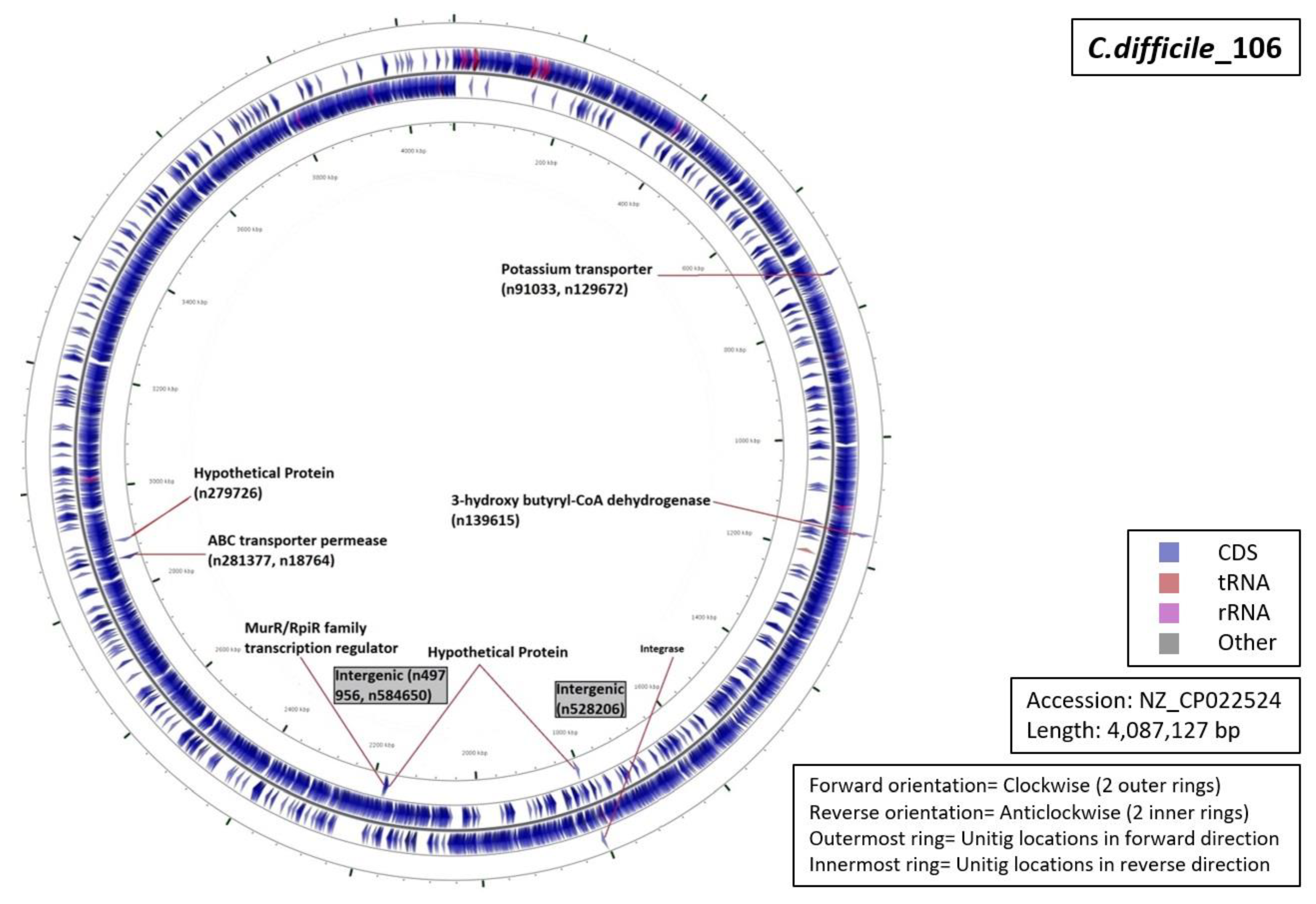
3.5. Functional Annotation of Markers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Walk, S.T.; Micic, D.; Jain, R.; Lo, E.S.; Trivedi, I.; Liu, E.W.; Almassalha, L.M.; Ewing, S.A.; Ring, C.; Galecki, A.T.; et al. Clostridium difficile Ribotype Does Not Predict Severe Infection. Clin. Infect. Dis. 2012, 55, 1661–1668. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.S.; Eyre, D.W.; Wyllie, D.H.; Dingle, K.E.; Griffiths, D.; Shine, B.; Walker, A.S.; O’Connor, L.; Finney, J.; Vaughan, A.; et al. Relationship Between Bacterial Strain Type, Host Biomarkers, and Mortality in Clostridium difficile Infection. Clin. Infect. Dis. 2013, 56, 1589–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFarland, L.V. Renewed interest in a difficult disease: Clostridium difficile infections—Epidemiology and current treatment strategies. Curr. Opin. Gastroenterol. 2009, 25, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Lessa, F.C.; Gould, C.V.; McDonald, L.C. Current Status of Clostridium difficile Infection Epidemiology. Clin. Infect. Dis. 2012, 55, S65–S70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiegand, P.N.; Nathwani, D.; Wilcox, M.H.; Stephens, J.; Shelbaya, A.; Haider, S. Clinical and economic burden of Clostridium difficile infection in Europe: A systematic review of healthcare-facility-acquired infection. J. Hosp. Infect. 2012, 81, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.; Pasupuleti, V.; Thota, P.; Pant, C.; Rolston, D.D.K.; Sferra, T.J.; Hernandez, A.V.; Donskey, C.J. Community-associated Clostridium difficile infection and antibiotics: A meta-analysis. J. Antimicrob. Chemother. 2013, 68, 1951–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namiki, H.; Kobayashi, T. Long-term, low-dose of clarithromycin as a cause of community-acquired Clostridium difficile infection in a 5-year-old boy. Oxf. Med. Case Rep. 2018, 2018, omx106. [Google Scholar] [CrossRef] [Green Version]
- Lessa, F.C.; Mu, Y.; Bamberg, W.M.; Beldavs, Z.G.; Dumyati, G.K.; Dunn, J.R.; Farley, M.M.; Holzbauer, S.M.; Meek, J.I.; Phipps, E.C.; et al. Burden of Clostridium difficile infection in the United States. N. Engl. J. Med. 2015, 372, 825–834. [Google Scholar] [CrossRef] [Green Version]
- Davies, K.G.; Longshaw, C.M.; Davis, G.L.; Bouza, E.; Barbut, F.; Barna, Z.; Delmée, M.; Fitzpatrick, F.; Ivanova, K.; Kuijper, E.; et al. Underdiagnosis of Clostridium difficile across Europe: The European, multicentre, prospective, biannual, point-prevalence study of Clostridium difficile infection in hospitalised patients with diarrhoea (EUCLID). Lancet Infect. Dis. 2014, 14, 1208–1219. [Google Scholar] [CrossRef]
- Borren, N.Z.; Ghadermarzi, S.; Hutfless, S.; Ananthakrishnan, A.N. The emergence of Clostridium difficile infection in Asia: A systematic review and meta-analysis of incidence and impact. PLoS ONE 2017, 12, e0176797. [Google Scholar] [CrossRef] [Green Version]
- Balsells, E.; Shi, T.; Leese, C.; Lyell, I.; Burrows, J.; Wiuff, C.; Campbell, H.; Kyaw, M.H.; Nair, H. Global burden of Clostridium difficile infections: A systematic review and meta-analysis. J. Glob. Health 2018, 9, 010407. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.P.; Rao, K.; Young, V.B. Probiotics for prevention of Clostridium difficile infection. Curr. Opin. Gastroenterol. 2018, 34, 3–10. [Google Scholar] [CrossRef] [PubMed]
- CDC. Biggest Threats Antibiotic/Antimicrobial Resistance; CDC: Atlanta, GA, USA, 2017. [Google Scholar]
- Burnham, C.-A.D.; Carroll, K.C. Diagnosis of Clostridium difficile Infection: An Ongoing Conundrum for Clinicians and for Clinical Laboratories. Clin. Microbiol. Rev. 2013, 26, 604–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planche, T.; Wilcox, M. Reference assays for Clostridium difficile infection: One or two gold standards? J. Clin. Pathol. 2011, 64, 1–5. [Google Scholar] [CrossRef] [Green Version]
- She, R.C.; Durrant, R.J.; Petti, C.A. Evaluation of Enzyme Immunoassays to Detect Clostridium difficile Toxin from Anaerobic Stool Culture. Am. J. Clin. Pathol. 2009, 131, 81–84. [Google Scholar] [CrossRef]
- Shetty, N.; Wren, M.; Coen, P. The role of glutamate dehydrogenase for the detection of Clostridium difficile in faecal samples: A meta-analysis. J. Hosp. Infect. 2011, 77, 1–6. [Google Scholar] [CrossRef]
- Eckert, C.; Jones, G.; Barbut, F. Diagnosis of Clostridium difficile infection: The molecular approach. Future Microbiol. 2013, 8, 1587–1598. [Google Scholar] [CrossRef]
- Krutova, M.; Wilcox, M.; Kuijper, E. A two-step approach for the investigation of a Clostridium difficile outbreak by molecular methods. Clin. Microbiol. Infect. 2019, 25, 1300–1301. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Raval, I.H. Chapter 32—Pathogenic Microbial Genetic Diversity with Reference to Health. In Microbial Diversity in the Genomic Era; Academic Press: Cambridge, MA, USA, 2019; pp. 559–577. [Google Scholar]
- Dingle, T.C.; MacCannell, D.R. Chapter 9—Molecular Strain Typing and Characterisation of Toxigenic Clostridium difficile. In Methods in Microbiology; Elsevier: Amsterdam, The Netherlands, 2015; pp. 329–357. [Google Scholar]
- Krutova, M.; Kinross, P.; Barbut, F.; Hajdu, A.; Wilcox, M.; Kuijper, E.; Allerberger, F.; Delmée, M.; Van Broeck, J.; Vatcheva-Dobrevska, R.; et al. How to: Surveillance of Clostridium difficile infections. Clin. Microbiol. Infect. 2018, 24, 469–475. [Google Scholar] [CrossRef] [Green Version]
- Collins, D.A.; Elliott, B.; Riley, T.V. Molecular methods for detecting and typing of Clostridium difficile. Pathology 2015, 47, 211–218. [Google Scholar] [CrossRef]
- Bidet, P.; Barbut, F.; Lalande, V.; Burghoffer, B.; Petit, J.-C. Development of a new PCR-ribotyping method for Clostridium difficile based on ribosomal RNA gene sequencing. FEMS Microbiol. Lett. 1999, 175, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Indra, A.; Blaschitz, M.; Kernbichler, S.; Reischl, U.; Wewalka, G.; Allerberger, F. Mechanisms behind variation in the Clostridium difficile 16S–23S rRNA intergenic spacer region. J. Med. Microbiol. 2010, 59, 1317–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indra, A.; Huhulescu, S.; Schneeweis, M.; Hasenberger, P.; Kernbichler, S.; Fiedler, A.; Wewalka, G.; Allerberger, F.; Kuijper, E.J. Characterization of Clostridium difficile isolates using capillary gel electrophoresis-based PCR ribotyping. J. Med. Microbiol. 2008, 57, 1377–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waslawski, S.; Lo, E.S.; Ewing, S.A.; Young, V.B.; Aronoff, D.M.; Sharp, S.E.; Novak-Weekley, S.M.; Crist, A.E.; Dunne, W.M.; Hoppe-Bauer, J.; et al. Clostridium difficile Ribotype Diversity at Six Health Care Institutions in the United States. J. Clin. Microbiol. 2013, 51, 1938–1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janezic, S. Direct PCR-Ribotyping of Clostridium difficile. In Clostridium difficile Methods in Molecular Biology; Humana Press: New York, NY, USA, 2016; Volume 1476. [Google Scholar]
- Martinez-Melendez, A.; Camacho-Ortiz, A.; Morfin-Otero, R.; Maldonado-Garza, H.J.; Villarreal-Trevino, L.; Garza-Gonzalez, E. Current knowledge on the laboratory diagnosis of Clostridium difficile infection. World J. Gastroenterol. 2017, 23, 1552–1567. [Google Scholar] [CrossRef] [PubMed]
- Polage, C.R.; Gyorke, C.E.; Kennedy, M.A.; Leslie, J.L.; Chin, D.L.; Wang, S.; Nguyen, H.H.; Huang, B.; Tang, Y.W.; Lee, L.W.; et al. Overdiagnosis of Clostridium difficile Infection in the Molecular Test Era. JAMA Intern. Med. 2015, 175, 1792–1801. [Google Scholar] [CrossRef] [Green Version]
- Janezic, S.; Rupnik, M. Development and Implementation of Whole Genome Sequencing-Based Typing Schemes for Clostridioides difficile. Front. Public Health 2019, 7. [Google Scholar] [CrossRef]
- Fawley, W.N.; Knetsch, C.W.; MacCannell, D.R.; Harmanus, C.; Du, T.; Mulvey, M.R.; Paulick, A.; Anderson, L.; Kuijper, E.J.; Wilcox, M.H. Development and Validation of an Internationally-Standardized, High-Resolution Capillary Gel-Based Electrophoresis PCR-Ribotyping Protocol for Clostridium difficile. PLoS ONE 2015, 10, e0118150. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.P.; Notermans, D.W.; Van Benthem, B.H.; Brazier, J.S.; Wilcox, M.H.; Rupnik, M.; Monnet, D.L.; Van Dissel, J.T.; Kuijper, E.J. Clostridium difficile infection in Europe: A hospital-based survey. Lancet Infect. Dis. 2011, 377, 63–73. [Google Scholar] [CrossRef]
- Arvand, M.; Hauri, A.M.; Zaiss, N.H.; Witte, W.; Bettge-Weller, G. Clostridium difficile ribotypes 001, 017, and 027 are associated with lethal C. difficile infection in Hesse, Germany. Eurosurveillance 2009, 14, 19403. [Google Scholar] [CrossRef] [Green Version]
- Vanek, J.; Hill, K.; Collins, J.; Berrington, A.; Perry, J.; Inns, T.; Gorton, R.; Magee, J.; Sails, A.; Mullan, A.; et al. Epidemiological survey of Clostridium difficile ribotypes in the North East of England during an 18-month period. J. Hosp. Infect. 2012, 81, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Giancola, S.E.; Williams, R.J.; Gentry, C.A. Prevalence of the Clostridium difficile BI/NAP1/027 strain across the United States Veterans Health Administration. Clin. Microbiol. Infect. 2018, 24, 877–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howell, M.D.; Novack, V.; Grgurich, P.; Soulliard, D.; Novack, L.; Pencina, M.; Talmor, D. Latrogenic gastric acid suppression and the risk of nosocomial Clostridium difficile infection. Arch. Intern. Med. 2010, 170, 784–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imwattana, K.; Knight, D.R.; Kullin, B.; Collins, D.A.; Putsathit, P.; Kiratisin, P.; Riley, T.V. Clostridium difficile ribotype 017—Characterization, evolution and epidemiology of the dominant strain in Asia. Emerg. Microbes Infect. 2019, 8, 796–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kim, Y.; Pai, H. Clinical Characteristics and Treatment Outcomes of Clostridium difficile Infections by PCR Ribotype 017 and 018 Strains. PLoS ONE 2016, 11, e0168849. [Google Scholar] [CrossRef] [Green Version]
- Jaillard, M.; Lima, L.; Tournoud, M.; Mahé, P.; Van Belkum, A.; Lacroix, V.; Jacob, L. A fast and agnostic method for bacterial genome-wide association studies: Bridging the gap between k-mers and genetic events. PLoS Genet. 2018, 14, e1007758. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.T.; Petit, R.A.; Crispell, E.K.; Thornton, T.A.; Conneely, K.N.; Jiang, Y.; Satola, S.W.; Read, T.D. Dissecting vancomycin-intermediate resistance in staphylococcus aureus using genome-wide association. Genome Biol. Evol. 2014, 6, 1174–1185. [Google Scholar] [CrossRef]
- Chewapreecha, C.; Marttinen, P.; Croucher, N.J.; Salter, S.J.; Harris, S.R.; Mather, A.E.; Hanage, W.P.; Goldblatt, D.; Nosten, F.H.; Turner, C.; et al. Comprehensive Identification of Single Nucleotide Polymorphisms Associated with Beta-lactam Resistance within Pneumococcal Mosaic Genes. PLoS Genet. 2014, 10, e1004547. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Frentrup, M.; Zhou, Z.; Steglich, M.; Meier-Kolthoff, J.P.; Göker, M.; Riedel, T.; Bunk, B.; Spröer, C.; Overmann, J.; Blaschitz, M.; et al. A publicly accessible database for Clostridioides difficile genome sequences supports tracing of transmission chains and epidemics. Microb. Genom. 2020, 6, 410. [Google Scholar] [CrossRef]
- Baratloo, A.; Hosseini, M.; Negida, A.; El Ashal, G. Simple Definition and Calculation of Accuracy, Sensitivity and Specificity. Emergency 2015, 3, 48–49. [Google Scholar] [PubMed]
- Bradbury, P.; Parker, T.; Hamblin, M.T.; Jannink, J. Assessment of Power and False Discovery Rate in Genome-Wide Association Studies using the BarleyCAP Germplasm. Crop. Sci. 2011, 51, 52–59. [Google Scholar] [CrossRef]
- Xiao, M.; Kong, F.; Jin, P.; Wang, Q.; Xiao, K.; Jeoffreys, N.; James, G.; Gilbert, G.L. Comparison of Two Capillary Gel Electrophoresis Systems for Clostridium difficile Ribotyping, Using a Panel of Ribotype 027 Isolates and Whole-Genome Sequences as a Reference Standard. J. Clin. Microbiol. 2012, 50, 2755–2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Perez, S.; Blanco, J.L.; Harmanus, C.; Kuijper, E.; Garcia, M.E. Subtyping and antimicrobial susceptibility of Clostridium difficile PCR ribotype 078/126 isolates of human and animal origin. Vet. Microbiol. 2017, 199, 15–22. [Google Scholar] [CrossRef]
- Schneeberg, A.; Neubauer, H.; Schmoock, G.; Baier, S.; Harlizius, J.; Nienhoff, H.; Brase, K.; Zimmermann, S.; Seyboldt, C. Clostridium difficile Genotypes in Piglet Populations in Germany. J. Clin. Microbiol. 2013, 51, 3796–3803. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Gene Target | GenBank Accession No. | Sequence (5’–3’) | Tm (°C) | Reference |
|---|---|---|---|---|---|
| 16S-USA (Forward) | 16S rRNA gene | FN545816 | (12293)GTGCGGCTGGATCACCTCCT (12312) | 71.0 | Xiao et al., 2012 (46) |
| 23S-USA (Reverse) | 23S rRNA gene | FN545816 | (12621)CCCTGCACCCTTAATAACTTGACC (12598) | 67.1 |
| C. difficile Ribotype | Number of Genomes | Source |
|---|---|---|
| RT001 | 24 | Enterobase, NCBI, Creighton University |
| RT002 | 2 | NCBI, Creighton University |
| RT003 | 19 | NCBI, Creighton University |
| RT005 | 19 | NCBI, Creighton University |
| RT010 | 3 | NCBI, Creighton University |
| RT014 | 11 | NCBI, Creighton University |
| RT015 | 2 | NCBI, Creighton University |
| RT017 | 15 | NCBI, Creighton University |
| RT023 | 3 | NCBI, Creighton University |
| RT027 | 15 | NCBI, Creighton University |
| RT046 | 4 | NCBI, Creighton University |
| RT078 | 15 | NCBI, Creighton University |
| RT106 | 22 | Enterobase, NCBI, Creighton University |
| RT126 | 6 | NCBI, Creighton University |
| TOTAL | 160 | |
| Ribotype | Count | Ribotype | Count | Ribotype | Count | Ribotype | Count |
|---|---|---|---|---|---|---|---|
| RT001 | 206 | RT046 | 3 | RT127 | 1 | RT375 | 1 |
| RT002 | 53 | RT049 | 9 | RT129 | 1 | RT404 | 15 |
| RT003 | 11 | RT050 | 4 | RT137 | 1 | RT413 | 13 |
| RT005 | 14 | RT051 | 1 | RT138 | 1 | RT446 | 2 |
| RT006 | 1 | RT053 | 5 | RT149 | 1 | RT449 | 2 |
| RT009 | 2 | RT054 | 2 | RT150 | 1 | RT451 | 1 |
| RT010 | 7 | RT056 | 5 | RT153 | 1 | RT453 | 1 |
| RT011 | 3 | RT058 | 1 | RT156 | 1 | RT454 | 1 |
| RT012 | 45 | RT060 | 1 | RT157 | 1 | RT456 | 1 |
| RT013 | 1 | RT062 | 2 | RT158 | 1 | RT470 | 1 |
| RT014 | 113 | RT063 | 1 | RT176 | 13 | RT500 | 21 |
| RT015 | 36 | RT066 | 3 | RT193 | 1 | RT534 | 1 |
| RT017 | 272 | RT067 | 1 | RT194 | 1 | RT547 | 1 |
| RT018 | 55 | RT069 | 1 | RT212 | 1 | RT559 | 1 |
| RT019 | 1 | RT070 | 4 | RT220 | 4 | RT563 | 1 |
| RT020 | 44 | RT072 | 1 | RT225 | 1 | RT569 | 1 |
| RT022 | 1 | RT073 | 2 | RT226 | 1 | RT581 | 1 |
| RT023 | 16 | RT075 | 1 | RT236 | 3 | RT585 | 1 |
| RT024 | 1 | RT076 | 2 | RT238 | 1 | RT586 | 1 |
| RT026 | 6 | RT077 | 1 | RT239 | 2 | RT591 | 1 |
| RT027 | 652 | RT078 | 492 | RT241 | 5 | RT598 | 8 |
| RT029 | 3 | RT081 | 2 | RT244 | 9 | RT614 | 1 |
| RT031 | 2 | RT083 | 1 | RT251 | 1 | RT620 | 2 |
| RT032 | 1 | RT084 | 2 | RT262 | 1 | RT629 | 1 |
| RT033 | 5 | RT087 | 8 | RT284 | 1 | RT651 | 1 |
| RT035 | 2 | RT090 | 1 | RT289 | 1 | RT666 | 1 |
| RT036 | 1 | RT094 | 1 | RT290 | 1 | RT668 | 1 |
| RT037 | 1 | RT102 | 1 | RT305 | 1 | RT678 | 1 |
| RT039 | 5 | RT103 | 2 | RT316 | 1 | RT708 | 1 |
| RT042 | 2 | RT106 | 55 | RT321 | 1 | RT719 | 1 |
| RT043 | 2 | RT117 | 2 | RT328 | 2 | RT720 | 1 |
| RT044 | 2 | RT125 | 1 | RT336 | 1 | RT721 | 1 |
| RT045 | 2 | RT126 | 79 | RT356 | 8 | RT722 | 1 |
| Ribotype | Number of Markers | Average Length (Base Pairs) | Annotation (Number of Unitigs) |
|---|---|---|---|
| RT001 | 06 | 59 |
|
| RT017 | 13 | 69 |
|
| RT027 | 07 | 53 |
|
| RT078 | 12 | 42 |
|
| RT106 | 09 | 55 |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goyal, M.; Hauben, L.; Pouseele, H.; Jaillard, M.; De Bruyne, K.; van Belkum, A.; Goering, R. Retrospective Definition of Clostridioides difficile PCR Ribotypes on the Basis of Whole Genome Polymorphisms: A Proof of Principle Study. Diagnostics 2020, 10, 1078. https://doi.org/10.3390/diagnostics10121078
Goyal M, Hauben L, Pouseele H, Jaillard M, De Bruyne K, van Belkum A, Goering R. Retrospective Definition of Clostridioides difficile PCR Ribotypes on the Basis of Whole Genome Polymorphisms: A Proof of Principle Study. Diagnostics. 2020; 10(12):1078. https://doi.org/10.3390/diagnostics10121078
Chicago/Turabian StyleGoyal, Manisha, Lysiane Hauben, Hannes Pouseele, Magali Jaillard, Katrien De Bruyne, Alex van Belkum, and Richard Goering. 2020. "Retrospective Definition of Clostridioides difficile PCR Ribotypes on the Basis of Whole Genome Polymorphisms: A Proof of Principle Study" Diagnostics 10, no. 12: 1078. https://doi.org/10.3390/diagnostics10121078