
Point Mutation Specific Antibodies in B-Cell and T-Cell Lymphomas and Leukemias: Targeting IDH2, KRAS, BRAF and Other Biomarkers RHOA, IRF8, MYD88, ID3, NRAS, SF3B1 and EZH2

 , ,
, ,
Abstract
:1. Introduction
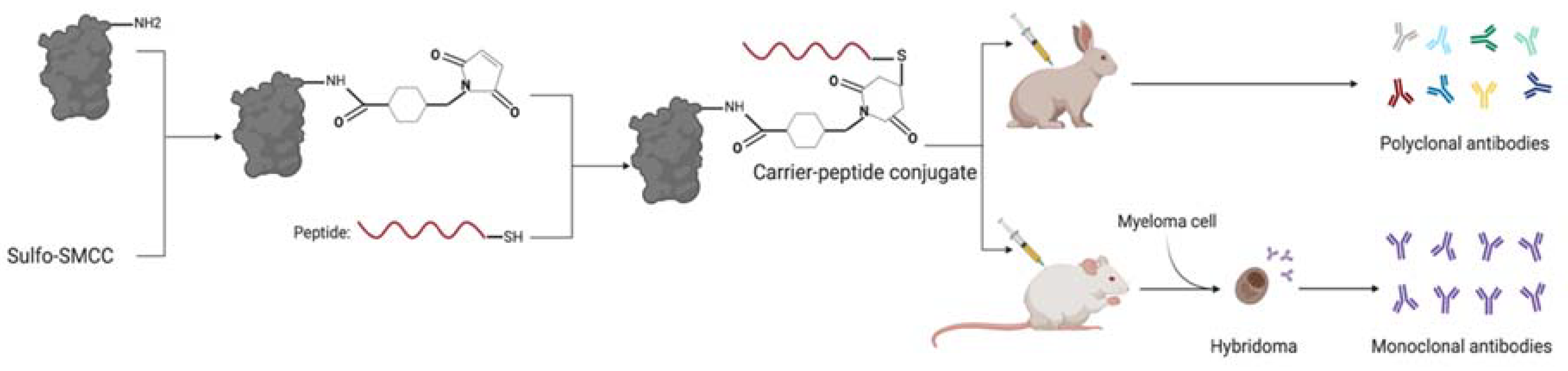
2. Materials and Methods
2.1. Target Selection
2.2. Peptide and Antibody Evaluation
3. Results
3.1. BRAF V600E
3.2. RHOA G17V
3.3. IRF8 K66R
3.4. MYD88 L265P
3.5. IDH2 R172K
3.6. DNMT3A R882H
3.7. KRAS G12D
3.8. NRAS Q61K
3.9. SF3B1 K700E
3.10. ID3 L64F
3.11. EZH2 Y646H
4. Discussion
Funding
Conflicts of Interest
References
- The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia | Blood | American Society of Hematology. Available online: https://ashpublications.org/blood/article/127/20/2391/35255/The-2016-revision-to-the-World-Health-Organization (accessed on 13 December 2020).
- Feller, J.K.; Yang, S.; Mahalingam, M. Immunohistochemistry with a mutation-specific monoclonal antibody as a screening tool for the BRAFV600E mutational status in primary cutaneous malignant melanoma. Mod. Pathol. 2013, 26, 414–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Kane, S.; Wu, J.; Benedettini, E.; Li, D.; Reeves, C.; Innocenti, G.; Wetzel, R.; Crosby, K.; Becker, A.; et al. Mutation-Specific Antibodies for the Detection of EGFR Mutations in Non–Small-Cell Lung Cancer. Clin. Cancer Res. 2009, 15, 3023–3028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Disanto, M.G.; Ambrosio, M.R.; Rocca, B.J.; Ibrahim, H.A.H.; Leoncini, L.; Naresh, K.N. Optimal Minimal Panels of Immunohistochemistry for Diagnosis of B-Cell Lymphoma for Application in Countries With Limited Resources and for Triaging Cases Before Referral to Specialist Centers. Am. J. Clin. Pathol. 2016, 145, 687–695. [Google Scholar] [CrossRef] [Green Version]
- Lear, S.; Cobb, S.L. Pep-Calc.com: A set of web utilities for the calculation of peptide and peptoid properties and automatic mass spectral peak assignment. J. Comput. Aided Mol. Des. 2016, 30, 271–277. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Khedkar, S.; Bork, P. SMART: Recent updates, new developments and status in 2020. Nucleic Acids Res. 2021, 49, D458–D460. [Google Scholar] [CrossRef] [PubMed]
- Wootton, J.C.; Federhen, S. Statistics of local complexity in amino acid sequences and sequence databases. Comput. Chem. 1993, 17, 149–163. [Google Scholar] [CrossRef]
- Porollo, A.A.; Adamczak, R.; Meller, J. POLYVIEW: A flexible visualization tool for structural and functional annotations of proteins. Bioinformatics 2004, 20, 2460–2462. [Google Scholar] [CrossRef]
- Arcaini, L.; Zibellini, S.; Boveri, E.; Riboni, R.; Rattotti, S.; Varettoni, M.; Guerrera, M.L.; Lucioni, M.; Tenore, A.; Merli, M.; et al. The BRAF V600E mutation in hairy cell leukemia and other mature B-cell neoplasms. Blood 2012, 119, 188–191. [Google Scholar] [CrossRef] [Green Version]
- Chiba, S.; Enami, T.; Ogawa, S.; Sakata-Yanagimoto, M. G17V RHOA: Genetic evidence of GTP-unbound RHOA playing a role in tumorigenesis in T cells. Small GTPases 2015, 6, 100–103. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.; Ramis-Zaldivar, J.E.; Nadeu, F.; Gonzalez-Farre, B.; Navarro, A.; Egan, C.; Montes-Mojarro, I.A.; Marafioti, T.; Cabeçadas, J.; van der Walt, J.; et al. Mutations of MAP2K1 are frequent in pediatric-type follicular lymphoma and result in ERK pathway activation. Blood 2017, 130, 323–327. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D. Role of MYD88 in lymphoplasmacytic lymphoma diagnosis and pathogenesis. Hematology 2014, 2014, 113–118. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Iqbal, J.; Lemonnier, F.; Kucuk, C.; de Leval, L.; Jais, J.-P.; Parrens, M.; Martin, A.; Xerri, L.; Brousset, P.; et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood 2012, 119, 1901–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Rau, R.; Goodell, M.A. DNMT3A in haematological malignancies. Nat. Rev. Cancer 2015, 15, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Kindler, T.; Cornejo, M.G.; Scholl, C.; Liu, J.; Leeman, D.S.; Haydu, J.E.; Fröhling, S.; Lee, B.H.; Gilliland, D.G. K-RasG12D–induced T-cell lymphoblastic lymphoma/leukemias harbor Notch1 mutations and are sensitive to γ-secretase inhibitors. Blood 2008, 112, 3373–3382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- KieΔling, M.K.; Nicolay, J.P.; Schlör, T.; Klemke, C.-D.; Süss, D.; Krammer, P.H.; Gülow, K. NRAS mutations in cutaneous T cell lymphoma (CTCL) sensitize tumors towards treatment with the multikinase inhibitor Sorafenib. Oncotarget 2017, 8, 45687–45697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.; Wu, C.J. SF3B1 mutations in chronic lymphocytic leukemia. Blood 2013, 121, 4627–4634. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.; Zhang, M.; Wright, G.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt Lymphoma Pathogenesis and Therapeutic Targets from Structural and Functional Genomics. Nature 2012, 490, 116–120. [Google Scholar] [CrossRef]
- Bödör, C.; O’Riain, C.; Wrench, D.; Matthews, J.; Iyengar, S.; Tayyib, H.; Calaminici, M.; Clear, A.; Iqbal, S.; Quentmeier, H.; et al. EZH2 Y641 mutations in follicular lymphoma. Leukemia 2011, 25, 726–729. [Google Scholar] [CrossRef]
- Ritterhouse, L.L.; Barletta, J.A. BRAF V600E mutation-specific antibody: A review. Semin. Diagn. Pathol. 2015, 32, 400–408. [Google Scholar] [CrossRef]
- Pillonel, V.; Juskevicius, D.; Ng, C.K.Y.; Bodmer, A.; Zettl, A.; Jucker, D.; Dirnhofer, S.; Tzankov, A. High-throughput sequencing of nodal marginal zone lymphomas identifies recurrent BRAF mutations. Leukemia 2018, 32, 2412–2426. [Google Scholar] [CrossRef] [Green Version]
- Tadmor, T.; Tiacci, E.; Falini, B.; Polliack, A. The BRAF-V600E mutation in hematological malignancies: A new player in hairy cell leukemia and Langerhans cell histiocytosis. Leuk. Lymphoma 2012, 53, 2339–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anti-BRAF (V600E) Antibody Produced in Rabbit SAB4200772. Sigma-Aldrich. Available online: https://www.sigmaaldrich.com/catalog/product/sigma/sab4200772 (accessed on 6 December 2020).
- Dietrich, S.; Pircher, A.; Endris, V.; Peyrade, F.; Wendtner, C.-M.; Follows, G.A.; Hüllein, J.; Jethwa, A.; Ellert, E.; Walther, T.; et al. BRAF inhibition in hairy cell leukemia with low-dose vemurafenib. Blood 2016, 127, 2847–2855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- RHOA ras Homolog Family Member A [Homo Sapiens (Human)]-Gene-NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/387 (accessed on 6 December 2020).
- Schaefer, A.; Reinhard, N.R.; Hordijk, P.L. Toward understanding RhoGTPase specificity: Structure, function and local activation. Small GTPases 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watatani, Y.; Sato, Y.; Miyoshi, H.; Sakamoto, K.; Nishida, K.; Gion, Y.; Nagata, Y.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; et al. Molecular heterogeneity in peripheral T-cell lymphoma, not otherwise specified revealed by comprehensive genetic profiling. Leukemia 2019, 33, 2867–2883. [Google Scholar] [CrossRef] [PubMed]
- Blood Atlas—IRF8—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000140968-IRF8/blood (accessed on 13 December 2020).
- Tamura, T.; Ozato, K. Review: ICSBP/IRF-8: Its Regulatory Roles in the Development of Myeloid Cells. J. Interferon Cytokine Res. 2002, 22, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Liu, J.; Cao, X. Regulation of type I interferon signaling in immunity and inflammation: A comprehensive review. J. Autoimmun. 2017, 83, 1–11. [Google Scholar] [CrossRef]
- Tamura, T.; Thotakura, P.; Tanaka, T.S.; Ko, M.S.H.; Ozato, K. Identification of target genes and a unique cis element regulated by IRF-8 in developing macrophages. Blood 2005, 106, 1938–1947. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Hu, X.; Zimmerman, M.; Torres, C.M.; Yang, D.; Smith, S.B.; Liu, K. Cutting Edge: IRF8 Regulates Bax Transcription In Vivo in Primary Myeloid Cells. J. Immunol. 2011, 187, 4426–4430. [Google Scholar] [CrossRef] [Green Version]
- IRF8 Regulates Transcription of Naips for NLRC4 Inflammasome Activation: Cell. Available online: https://www.cell.com/cell/fulltext/S0092-8674(18)30232-0?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS0092867418302320%3Fshowall%3Dtrue (accessed on 13 December 2020).
- Ozawa, M.G.; Bhaduri, A.; Chisholm, K.M.; Baker, S.A.; Ma, L.; Zehnder, J.L.; Luna-Fineman, S.; Link, M.P.; Merker, J.D.; Arber, D.A.; et al. A study of the mutational landscape of pediatric-type follicular lymphoma and pediatric nodal marginal zone lymphoma. Mod. Pathol. 2016, 29, 1212–1220. [Google Scholar] [CrossRef] [Green Version]
- Deguine, J.; Barton, G.M. MyD88: A central player in innate immune signaling. F1000Prime Rep. 2014, 6, 97. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Hoshino, K. Myeloid Differentiation Factor 88–Dependent and –Independent Pathways in Toll-Like Receptor Signaling. J. Infect. Dis. 2003, 187 (Suppl 2), S356–S363. [Google Scholar] [CrossRef]
- Kawai, T.; Sato, S.; Ishii, K.J.; Coban, C.; Hemmi, H.; Yamamoto, M.; Terai, K.; Matsuda, M.; Inoue, J.; Uematsu, S.; et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 2004, 5, 1061–1068. [Google Scholar] [CrossRef]
- Gilmore, T.D. The Rel/NF-κB signal transduction pathway: Introduction. Oncogene 1999, 18, 6842–6844. [Google Scholar] [CrossRef] [Green Version]
- Kramer, I.M. Chapter 13—Activation of the Innate Immune System: The Toll-Like Receptor-4 and Signaling through Ubiquitinylation. In Signal Transduction, 3rd ed.; Kramer, I.M., Ed.; Academic Press: Boston, MA, USA, 2016; pp. 741–775. [Google Scholar] [CrossRef]
- Lin, S.-C.; Lo, Y.-C.; Wu, H. Helical assembly in the MyD88–IRAK4–IRAK2 complex in TLR/IL-1R signalling. Nat. Cell Biol. 2010, 465, 885–890. [Google Scholar] [CrossRef] [Green Version]
- Dolcet, X.; Llobet, D.; Pallares, J.; Matias-Guiu, X. NF-kB in development and progression of human cancer. Virchows Archiv. 2005, 446, 475–482. [Google Scholar] [CrossRef]
- Pahl, H.L. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef] [Green Version]
- May, M.J.; Ghosh, S. Signal transduction through NF-κB. Immunol. Today 1998, 19, 80–88. [Google Scholar] [CrossRef]
- Sims, J.E.; March, C.J.; Cosman, D.; Widmer, M.B.; MacDonald, H.R.; McMahan, C.J.; Grubin, C.E.; Wignall, J.M.; Jackson, J.L.; Call, S.M.; et al. cDNA expression cloning of the IL-1 receptor, a member of the immunoglobulin superfamily. Science 1988, 241, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.-H.; Kohlhammer, H.; Xu. W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, M.T.; Asthana, S.; Gao, S.P.; Lee, B.H.; Chapman, J.S.; Kandoth, C.; Gao, J.; Socci, N.D.; Solit, D.B.; Olshen, A.B.; et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 2016, 34, 155–163. [Google Scholar] [CrossRef]
- Vlahopoulos, S.A. Aberrant control of NF-κB in cancer permits transcriptional and phenotypic plasticity, to curtail dependence on host tissue: Molecular mode. Cancer Biol. Med. 2017, 14, 254–270. [Google Scholar] [CrossRef] [Green Version]
- Munshi, M.; Liu, X.; Chen, J.G.; Xu, L.; Tsakmaklis, N.; Demos, M.G.; Kofides, A.; Guerrera, M.L.; Jimenez, C.; Chan, G.G.; et al. SYK is activated by mutated MYD88 and drives pro-survival signaling in MYD88 driven B-cell lymphomas. Blood Cancer J. 2020, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zou, Y.; Liu, W.; Guan, P.; Tao, Q.; Xiang, C.; Zhang, W.; Ye, Y.; Yan, J.; Zhao, S.; et al. Morphologic Patterns and the Correlation with MYD88 L265P, CD79B Mutations in Primary Adrenal Diffuse Large B-Cell Lymphoma. Am. J. Surg. Pathol. 2020, 44, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.-T.; Wang, R.C.; Kuo, C.-C.; Hsieh, Y.-C.; Su, Y.-Z.; Chuang, S.-S. MYD88 L265P mutation analysis is a useful diagnostic adjunct for lymphoplasmacytic lymphoma with pleural effusion. Pathol. Int. 2019, 69, 601–607. [Google Scholar] [CrossRef]
- Wu, Y.-Y.; Jia, M.-N.; Cai, H.; Qiu, Y.; Zhou, D.-B.; Li, J.; Cao, X.-X. Detection of the MYD88L265P and CXCR4S338X mutations by cell-free DNA in Waldenström macroglobulinemia. Ann. Hematol. 2020, 99, 1763–1769. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.W.; Kim, S.-M.; Kim, J.-A.; Park, H.S.; Hwang, S.M.; Im, K.; Kim, S.; Kim, J.; Kwon, S.; Yoon, S.S.; et al. Characteristics of Waldenström Macroglobulinemia in Korean Patients According to Mutational Status of MYD88 and CXCR4: Analysis Using Ultra-Deep Sequencing. Clin. Lymphoma Myeloma Leuk. 2019, 19, e496–e505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauw, M.I.S.; Lucas, C.-H.G.; Ohgami, R.S.; Wen, K.W. Primary Central Nervous System Lymphomas: A Diagnostic Overview of Key Histomorphologic, Immunophenotypic, and Genetic Features. Diagnostics 2020, 10, 1076. [Google Scholar] [CrossRef] [PubMed]
- Treon, S.P.; Xu, L.; Yang, G.; Zhou, Y.; Liu, X.; Cao, Y.; Sheehy, P.; Manning, R.J.; Patterson, C.J.; Tripsas, C.; et al. MYD88 L265P Somatic Mutation in Waldenström’s Macroglobulinemia. N. Engl. J. Med. 2012, 367, 826–833. [Google Scholar] [CrossRef] [Green Version]
- Jiménez, C.; Sebastián, E.; Chillón, M.C.; Giraldo, P.; Mariano Hernández, J.; Escalante, F.; González-López, T.J.; Aguilera, C.; de Coca, A.G.; Murillo, I.; et al. MYD88 L265P is a marker highly characteristic of, but not restricted to, Waldenström’s macroglobulinemia. Leukemia 2013, 27, 1722–1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varettoni, M.; Arcaini, L.; Zibellini, S.; Boveri, E.; Rattotti, S.; Riboni, R.; Corso, A.; Orlandi, E.; Bonfichi, M.; Gotti, M.; et al. Prevalence and clinical significance of the MYD88 (L265P) somatic mutation in Waldenström’s macroglobulinemia and related lymphoid neoplasms. Blood 2013, 121, 2522–2528. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Zhu, H.; Fu, L.; Li, Y.; Bao, X.; Fu, H.; Quan, H.; Wang, L.; Lou, L. Pharmacological characterization of TQ 05310, a potent inhibitor of isocitrate dehydrogenase 2 R140Q and R172K mutants. Cancer Sci. 2019, 110, 3306–3314. [Google Scholar] [CrossRef] [Green Version]
- Mutation Overview Page IDH2-p.R172K (Substitution-Missense). Available online: https://cancer.sanger.ac.uk/cosmic/mutation/overview?id=173874954 (accessed on 13 December 2020).
- Mondesir, J.; Willekens, C.; Touat, M.; de Botton, S. IDH1 and IDH2 mutations as novel therapeutic targets: Current perspectives. J. Blood Med. 2016, 7, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Anti-IDH2-R172K (Human) mAb (Monoclonal Antibody). Available online: https://www.mblintl.com/products/d328-3/ (accessed on 6 December 2020).
- Sakata-Yanagimoto, M.; Nakamoto-Matsubara, R.; Komori, D.; Nguyen, T.B.; Hattori, K.; Nanmoku, T.; Kato, T.; Kurita, N.; Yokoyama, Y.; Obara, N.; et al. Detection of the circulating tumor DNAs in angioimmunoblastic T- cell lymphoma. Ann. Hematol. 2017, 96, 1471–1475. [Google Scholar] [CrossRef]
- Gowher, H.; Loutchanwoot, P.; Vorobjeva, O.; Handa, V.; Jurkowska, R.Z.; Jurkowski, T.P.; Jeltsch, A. Mutational Analysis of the Catalytic Domain of the Murine Dnmt3a DNA-(cytosine C5)-methyltransferase. J. Mol. Biol. 2006, 357, 928–941. [Google Scholar] [CrossRef]
- Jančík, S.; Drábek, J.; Radzioch, D.; Hajdúch, M. Clinical Relevance of KRAS in Human Cancers. J. Biomed. Biotechnol. 2010, 2010, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gideon, P.; John, J.; Frech, M.; Lautwein, A.; Clark, R.; Scheffler, J.; Wittinghofer, A. Mutational and kinetic analyses of the GTPase-activating protein (GAP)-p21 interaction: The C-terminal domain of GAP is not sufficient for full activity. Mol. Cell. Biol. 1992, 12, 2050–2056. [Google Scholar] [CrossRef] [Green Version]
- Giglione, C.; Parrini, M.; Baouz, S.; Bernardi, A.; Parmeggiani, A. A new function of p120-GTPase-activating protein—Prevention of the guanine nucleotide exchange factor-stimulated nucleotide exchange on the active form of Ha-Ras p21. J. Biol. Chem. 1997, 272, 25128–25134. [Google Scholar] [CrossRef] [Green Version]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [Green Version]
- Kong, G.; Du, J.; Liu, Y.; Meline, B.; Chang, Y.-I.; Ranheim, E.A.; Wang, J.; Zhang, J. Notch1 Gene Mutations Target KRAS G12D-expressing CD8+ Cells and Contribute to Their Leukemogenic Transformation. J. Biol. Chem. 2013, 288, 18219–18227. [Google Scholar] [CrossRef] [Green Version]
- Anti-Ras (Mutated G12D) Antibody (ab221163)|Abcam. Available online: https://www.abcam.com/ras-mutated-g12d-antibody-ab221163.html (accessed on 6 December 2020).
- Li, A.; Ma, Y.; Jin, M.; Mason, S.; Mort, R.L.; Blyth, K.; Larue, L.; Sansom, O.J.; Machesky, L.M. Activated Mutant NRasQ61K Drives Aberrant Melanocyte Signaling, Survival, and Invasiveness via a Rac1-Dependent Mechanism. J. Investig. Dermatol. 2012, 132, 2610–2621. [Google Scholar] [CrossRef] [Green Version]
- SF3B1 Splicing Factor 3b Subunit 1 [Homo Sapiens (Human)]-Gene-NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/23451 (accessed on 13 December 2020).
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium. SomaticSF3B1Mutation in Myelodysplasia with Ring Sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395. [Google Scholar] [CrossRef] [Green Version]
- Cazzola, M.; Della Porta, M.G.; Malcovati, L. The genetic basis of myelodysplasia and its clinical relevance. Blood 2013, 122, 4021–4034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ali, A.M.; Lieu, Y.K.; Liu, Z.; Gao, J.; Rabadan, R.; Raza, A.; Mukherjee, S.; Manley, J.L. Disease-Causing Mutations in SF3B1 Alter Splicing by Disrupting Interaction with SUGP1. Mol. Cell 2019, 76, 82–95.e7. [Google Scholar] [CrossRef]
- Zhou, Z.; Gong, Q.; Wang, Y.; Li, M.; Wang, L.; Ding, H.; Li, P. The biological function and clinical significance of SF3B1 mutations in cancer. Biomark. Res. 2020, 8, 38. [Google Scholar] [CrossRef]
- Ebert, B. Targeting SF3B1 for the Treatment of MDS. Available online: https://grantome.com/grant/NIH/P50-CA206963-01A1-5029 (accessed on 3 February 2021).
- Love, C.; Sun, Z.; Jima, D.; Li, G.; Zhang, J.; Miles, R.; Richards, K.L.; Dunphy, C.H.; Choi, W.W.; Srivastava, G.; et al. The genetic landscape of mutations in Burkitt lymphoma. Nat. Genet. 2012, 44, 1321–1325. [Google Scholar] [CrossRef] [Green Version]
- Lasorella, A.; Benezra, R.; Iavarone, A. The ID proteins: Master regulators of cancer stem cells and tumour aggressiveness. Nat. Rev. Cancer 2014, 14, 77–91. [Google Scholar] [CrossRef]
- Deed, R.W.; Hirose, T.; Mitchell, E.L.; Santibanez-Koref, M.F.; Norton, J.D. Structural organisation and chromosomal mapping of the human Id-3 gene. Gene 1994, 151, 309–314. [Google Scholar] [CrossRef]
- Lim, R.W.-S.; Wu, J.-M. Molecular mechanisms regulating expression and function of transcription regulator “inhibitor of differentiation 3”. Acta Pharmacol. Sin. 2005, 26, 1409–1420. [Google Scholar] [CrossRef] [Green Version]
- Lyden, D.; Young, A.Z.; Zagzag, D.; Yan, W.; Gerald, W.; O’Reilly, R.; Bader, B.L.; Hynes, R.O.; Zhuang, Y.; Manova, K.; et al. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nat. Cell Biol. 1999, 401, 670–677. [Google Scholar] [CrossRef]
- Pan, L.; Sato, S.; Frederick, J.P.; Sun, X.H.; Zhuang, Y. Impaired Immune Responses and B-Cell Proliferation in Mice Lacking the Id3 Gene. Mol. Cell. Biol. 1999, 19, 5969–5980. [Google Scholar] [CrossRef] [Green Version]
- Müller, J.; Hart, C.M.; Francis, N.J.; Vargas, M.L.; Sengupta, A.; Wild, B.; Miller, E.L.; O’Connor, M.B.; Kingston, R.E.; Simon, J.A. Histone Methyltransferase Activity of a Drosophila Polycomb Group Repressor Complex. Cell 2002, 111, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Czermin, B.; Melfi, R.; McCabe, D.; Seitz, V.; Imhof, A.; Pirrotta, V. Drosophila Enhancer of Zeste/ESC Complexes Have a Histone H3 Methyltransferase Activity that Marks Chromosomal Polycomb Sites. Cell 2002, 111, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nat. Cell Biol. 2011, 469, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Di Croce, L.; Helin, K. Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 2013, 20, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef]
- Yap, D.B.; Chu, J.; Berg, T.; Schapira, M.; Cheng, S.-W.G.; Moradian, A.; Morin, R.D.; Mungall, A.J.; Meissner, B.; Boyle, M.; et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 2011, 117, 2451–2459. [Google Scholar] [CrossRef] [Green Version]
- Sneeringer, C.J.; Scott, M.P.; Kuntz, K.W.; Knutson, S.K.; Pollock, R.M.; Richon, V.M.; Copeland, R.A. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc. Natl. Acad. Sci. USA 2010, 107, 20980–20985. [Google Scholar] [CrossRef] [Green Version]
- Lund, K.; Adams, P.D.; Copland, M. EZH2 in normal and malignant hematopoiesis. Leukemia 2014, 28, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. Histone-writer cancer drugs enter center stage. Nat. Biotechnol. 2020, 38, 909–912. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Protein | Mutation | Disease Prevalence | Commercial Antibody Available | Predicted Solubility | Conjugation Chemistry Likelihood | AA Sequence Complexity | Surface Epitope Exposure 0–9; 9 Most Accessible (PDB File Code) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| MBS | EDC | aEDC | Wild-Type | Mutant | ||||||
| BRAF | V600E | ~100% of HCL [9] | Yes | Likely | Yes | No | No | High | 8 (3Q4C) | 5 (4MNF) |
| RHOA | G17V | Up to 70% of AITL [10] | No | Unlikely | No | No | No | High | 2 (1FTN) | NA |
| IRF8 | K66R | ~15% of PTFL [11] | No | Likely | Yes | No | No | High | NA ** | NA ** |
| MYD88 | L265P | >90% of LPL [12] | No | Likely | No | No | No | High | NA | 0 (4EO7) |
| IDH2 | R172K | up to 45% of AITL [13] | Yes | Likely | Yes | No | No | High | 2 (5SVO) | 0 (5SVN) |
| DNMT3A | R882H | ~30% of nodal TCL [14] | No | Likely | Yes | No | Yes | High | 6 (4U7P) | 2 (6W89) |
| KRAS | G12D | ~10–15% of B and T ALL [15] | Yes | Unlikely | Yes | No | No | Low | 9 (4EPT) | 5 (6GJ7) |
| NRAS | Q61K | up to 11% of CTCL [16] | No | Likely | Yes * | No | No | High | 2 (5UHV) | NA |
| SF3B1 | K700E | up to 18% of CLL [17] | No | Likely | Yes | No | Yes | High | 2 (5IFE) | NA |
| ID3 | L64F | Up to 58% of BL [18] | No | Unlikely | Yes | No | Yes | High | 1 (2LFH) | NA |
| EZH2 | Y646H | ~9% of FL [19] | No | Likely | No | No | No | High | 0 (4MI5) | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, K.; Gollapudi, S.; Mittal, S.; Small, C.; Kumar, J.; Ohgami, R.S. Point Mutation Specific Antibodies in B-Cell and T-Cell Lymphomas and Leukemias: Targeting IDH2, KRAS, BRAF and Other Biomarkers RHOA, IRF8, MYD88, ID3, NRAS, SF3B1 and EZH2. Diagnostics 2021, 11, 600. https://doi.org/10.3390/diagnostics11040600
Singh K, Gollapudi S, Mittal S, Small C, Kumar J, Ohgami RS. Point Mutation Specific Antibodies in B-Cell and T-Cell Lymphomas and Leukemias: Targeting IDH2, KRAS, BRAF and Other Biomarkers RHOA, IRF8, MYD88, ID3, NRAS, SF3B1 and EZH2. Diagnostics. 2021; 11(4):600. https://doi.org/10.3390/diagnostics11040600
Chicago/Turabian StyleSingh, Kunwar, Sumanth Gollapudi, Sasha Mittal, Corinn Small, Jyoti Kumar, and Robert S. Ohgami. 2021. "Point Mutation Specific Antibodies in B-Cell and T-Cell Lymphomas and Leukemias: Targeting IDH2, KRAS, BRAF and Other Biomarkers RHOA, IRF8, MYD88, ID3, NRAS, SF3B1 and EZH2" Diagnostics 11, no. 4: 600. https://doi.org/10.3390/diagnostics11040600