
Application of CRISPR/Cas Systems in the Nucleic Acid Detection of Infectious Diseases
 , ,
, ,
Abstract
:1. Introduction
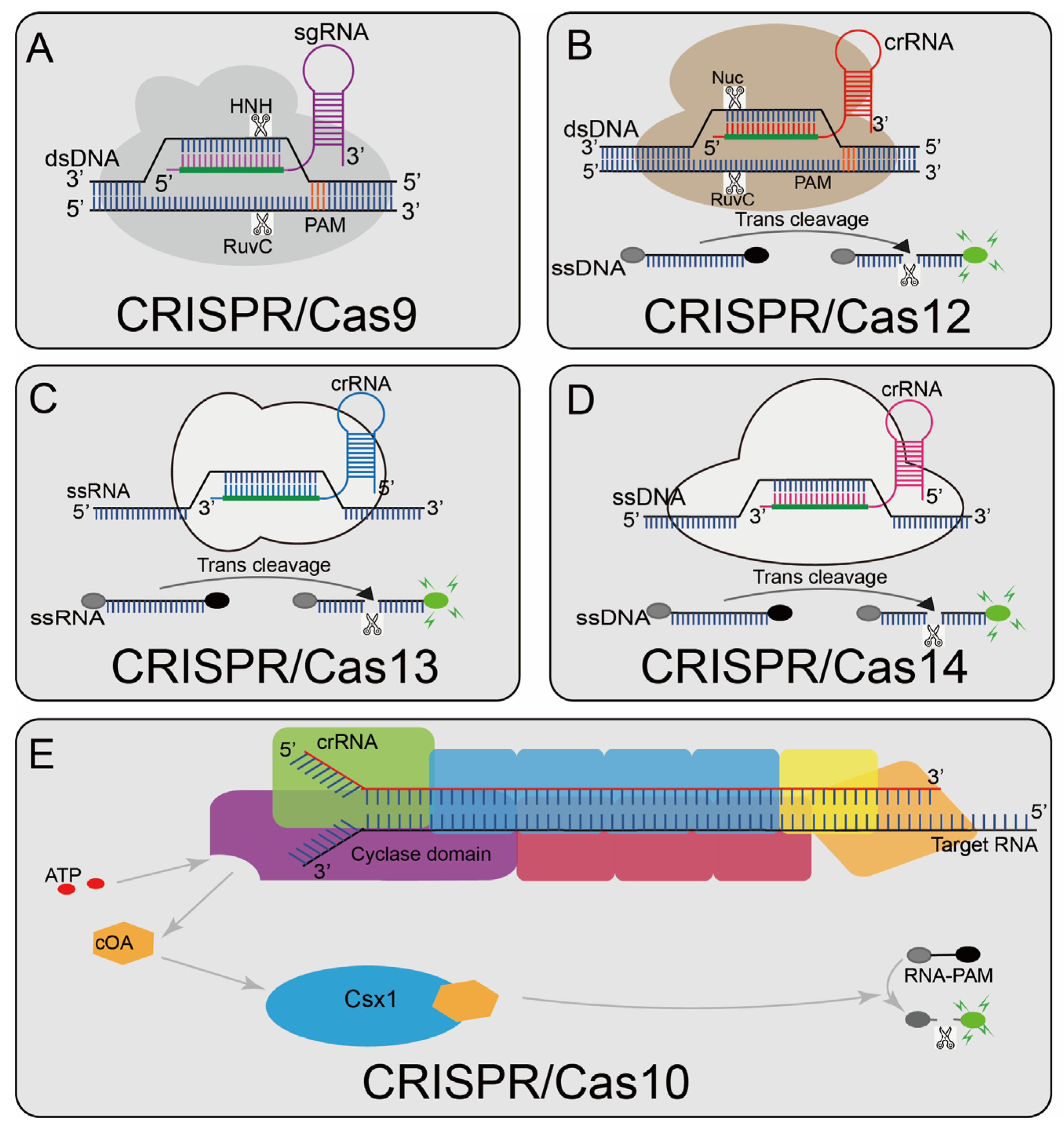
2. Classification of CRISPR Biosensing Systems
3. CRISPR/Cas9 Systems
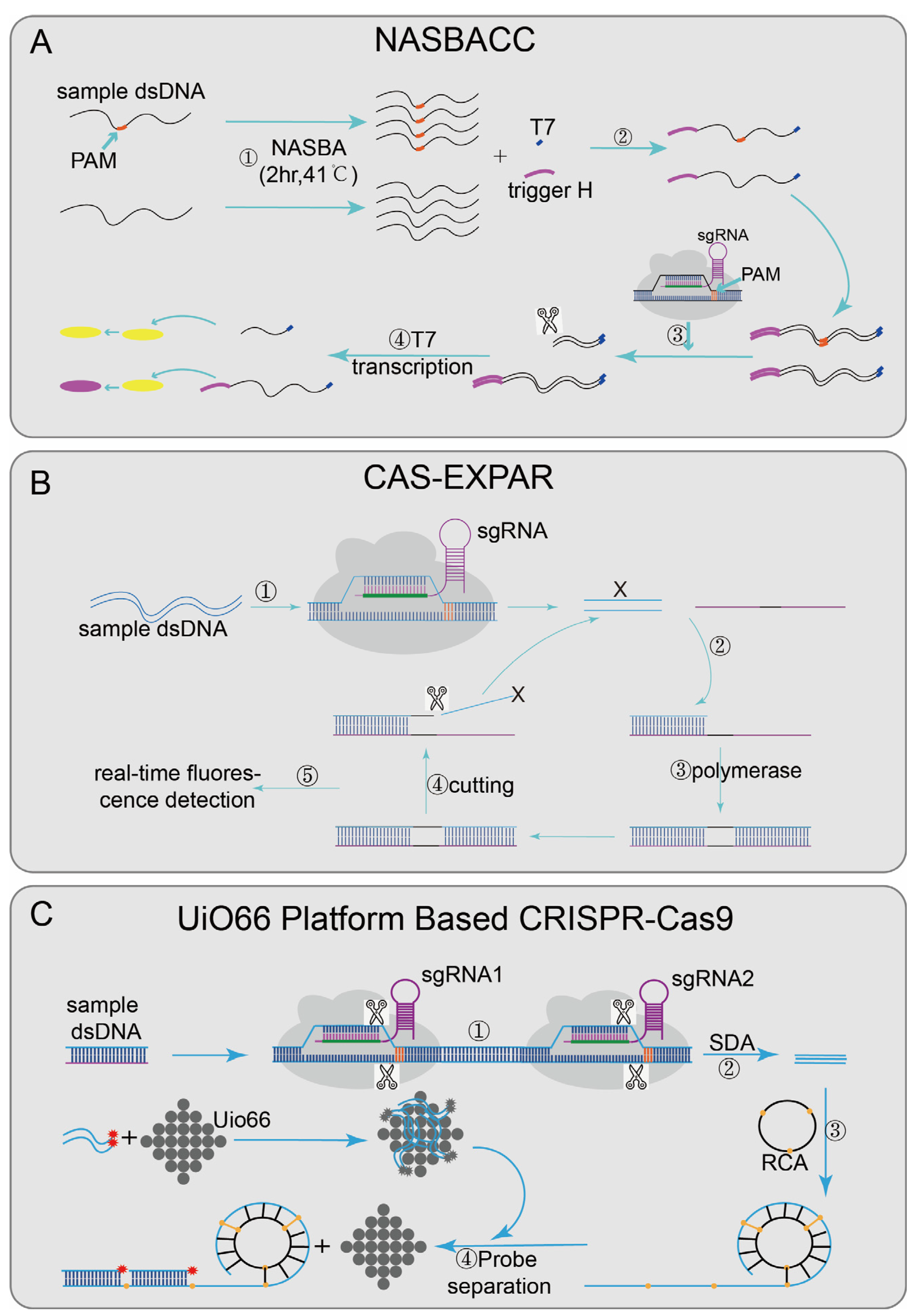
3.1. CRISPR/Cas9-Based Biosensing Systems
3.2. Evaluation of CRISPR/Cas9-Based Biosensing Systems
4. CRISPR/Cas12 Systems
4.1. CRISPR/Cas12-Based Biosensing Systems
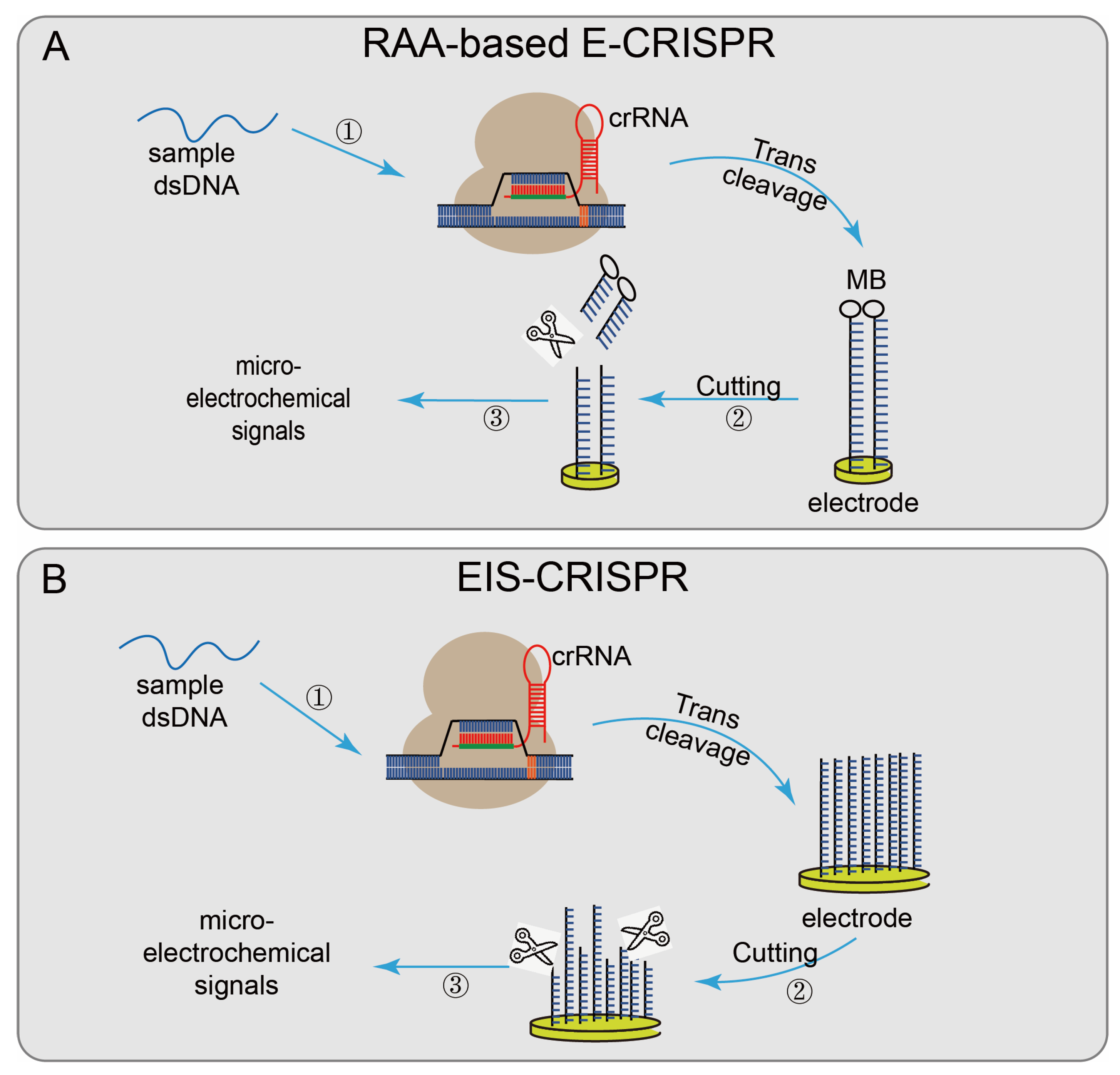
4.2. Evaluation of CRISPR/Cas12-Based Biosensing Systems
5. CRISPR/Cas 13 Systems
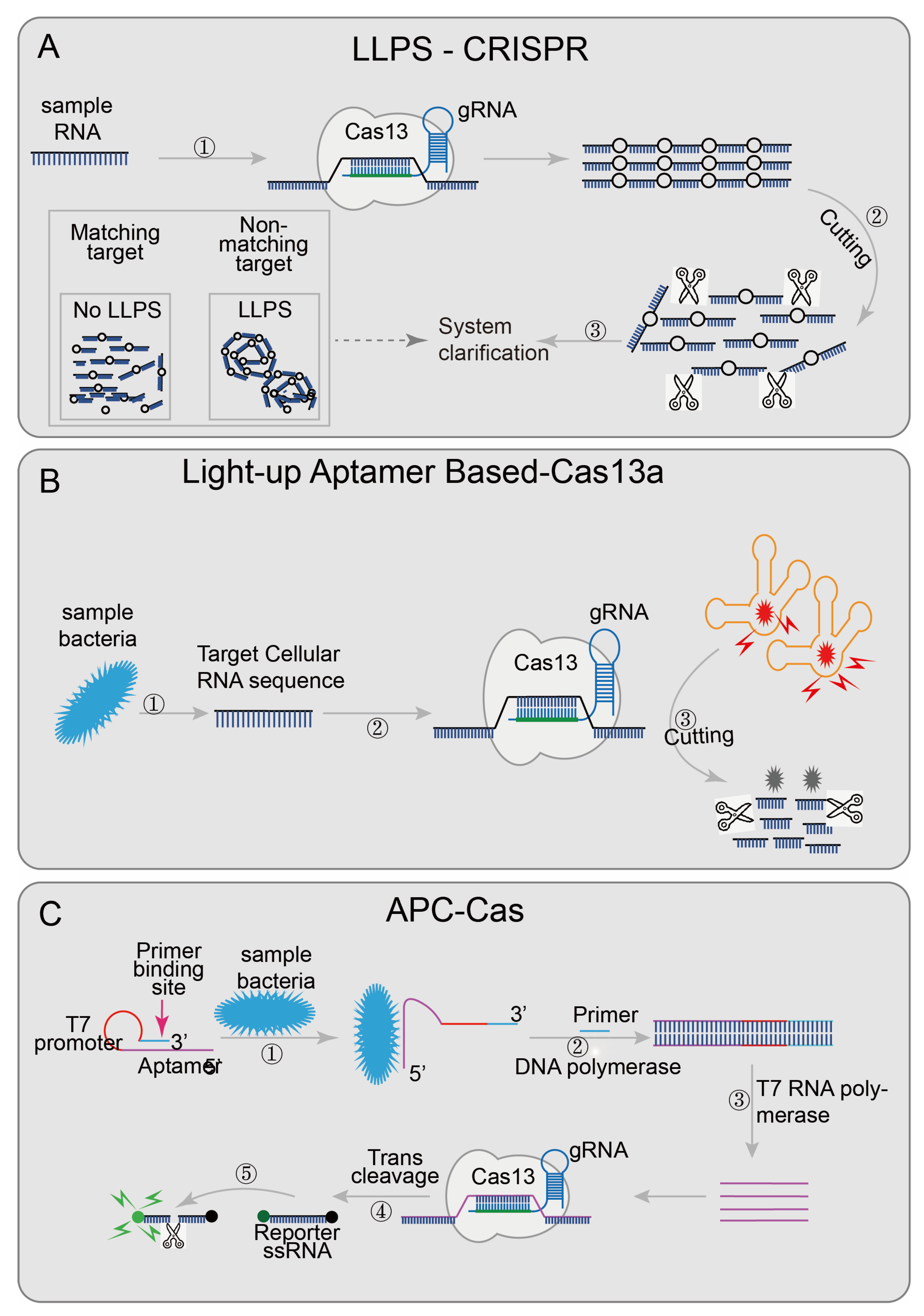
5.1. CRISPR/Cas13-Based Biosensing Systems
5.2. Evaluation of CRISPR/Cas13-Based Biosensing Systems
6. CRISPR/Cas 14 Systems
6.1. CRISPR/Cas14-Based Biosensing Systems
6.2. Evaluation of CRISPR/Cas14-Based Biosensing Systems
7. CRISPR/Cas 10 Systems
7.1. CRISPR/Cas10-Based Biosensing Systems
7.2. Evaluation of CRISPR/Cas10-Based Biosensing Systems
8. Application of the CRISPR/Cas System in SARS-CoV-2 Nucleic Acid Detection
8.1. CRISPR/Cas9 Systems in SARS-CoV-2 Detection
8.2. CRISPR/Cas12 Systems in SARS-CoV-2 Detection
8.3. CRISPR/Cas13 Systems in SARS-CoV-2 Detection
8.4. CRISPR/Cas10 Systems in SARS-CoV-2 Detection
9. Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hwang, H.; Hwang, B.Y.; Bueno, J. Biomarkers in Infectious Diseases. Dis. Markers 2018, 2018, 8509127. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, A.A.Z. Zika, the Newest TORCH Infectious Disease in the Americas. Clin. Infect. Dis. 2020, 70, 2673–2674. [Google Scholar] [CrossRef] [PubMed]
- Jacob, S.T.; Crozier, I.; Fischer, W.A., 2nd; Hewlett, A.; Kraft, C.S.; Vega, M.A.; Soka, M.J.; Wahl, V.; Griffiths, A.; Bollinger, L.; et al. Ebola virus disease. Nat. Rev. Dis. Primers 2020, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- De, W.E.; Van, D.N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar]
- Velavan, T.P.; Meyer, C.G. The COVID-19 epidemic. Trop. Med. Int. Health 2020, 25, 278–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Binda, G.; Misra, C.S.; Rath, D. The era of Cas12 and Cas13 CRISPR-based disease diagnosis. Crit. Rev. Microbiol. 2022, 1–16. [Google Scholar] [CrossRef]
- Marraffini, L.A.; Sontheimer, E.J. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat. Rev. Genet. 2010, 11, 181–190. [Google Scholar] [CrossRef]
- Shivram, H.; Cress, B.F.; Knott, G.J.; Doudna, J.A. Controlling and enhancing CRISPR systems. Nat. Chem. Biol. 2021, 17, 10–19. [Google Scholar] [CrossRef]
- Barrangou, R. The roles of CRISPR-Cas systems in adaptive immunity and beyond. Curr. Opin. Immunol. 2015, 32, 36–41. [Google Scholar] [CrossRef]
- Peters, J.E.; Makarova, K.S.; Shmakov, S.; Koonin, E.V. Recruitment of CRISPR-Cas systems by Tn7-like transposons. Proc. Natl. Acad. Sci. USA 2017, 114, E7358–E7366. [Google Scholar] [CrossRef] [PubMed]
- McGinn, J.; Marraffini, L.A. Molecular mechanisms of CRISPR-Cas spacer acquisition. Nat. Rev. Microbiol. 2019, 17, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, F.; Doudna, J.A. CRISPR-Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [Green Version]
- Yadav, N.; Narang, J.; Chhillar, A.K.; Rana, J.S. CRISPR: A new paradigm of theranostics. Nanomedicine 2021, 33, 102350. [Google Scholar] [CrossRef]
- Sharma, G.; Sharma, A.R.; Bhattacharya, M.; Lee, S.S.; Chakraborty, C. CRISPR-Cas9: A Preclinical and Clinical Perspective for the Treatment of Human Diseases. Mol. Ther. 2021, 29, 571–586. [Google Scholar] [CrossRef]
- Zavvar, T.S.; Khoshbin, Z.; Ramezani, M.; Alibolandi, M.; Abnous, K.; Taghdisi, S.M. CRISPR/Cas-engineered technology: Innovative approach for biosensor development. Biosens. Bioelectron. 2022, 214, 114501. [Google Scholar] [CrossRef]
- Kirby, E.N.; Shue, B.; Thomas, P.Q.; Beard, M.R. CRISPR Tackles Emerging Viral Pathogens. Viruses 2021, 13, 2157. [Google Scholar] [CrossRef]
- Aman, R.; Mahas, A.; Mahfouz, M. Nucleic Acid Detection Using CRISPR/Cas Biosensing Technologies. ACS Synth. Biol. 2020, 9, 1226–1233. [Google Scholar] [CrossRef]
- Freije, C.A.; Sabeti, P.C. Detect and destroy: CRISPR-based technologies for the response against viruses. Cell Host Microbe 2021, 29, 689–703. [Google Scholar] [CrossRef]
- Chaudhuri, A.; Halder, K.; Datta, A. Classification of CRISPR/Cas system and its application in tomato breeding. Theor. Appl. Genet. 2022, 135, 367–387. [Google Scholar] [CrossRef]
- Liu, G.; Lin, Q.; Jin, S.; Gao, C. The CRISPR-Cas toolbox and gene editing technologies. Mol Cell. 2022, 82, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Frangos, A.; Hall, L.N.; Nemudraia, A.; Nemudryi, A.; Krishna, P.; Wiegand, T.; Wilkinson, R.A.; Snyder, D.T.; Hedges, J.F.; Cicha, C.; et al. Intrinsic signal amplification by type III CRISPR-Cas systems provides a sequence-specific SARS-CoV-2 diagnostic. Cell Rep. Med. 2021, 2, 100319. [Google Scholar] [CrossRef] [PubMed]
- Pardee, K.; Green, A.A.; Takahashi, M.K.; Braff, D.; Lambert, G.; Lee, J.W.; Ferrante, T.; Ma, D.; Donghia, N.; Fan, M.; et al. Rapid, Low-Cost Detection of Zika Virus Using Programmable Biomolecular Components. Cell 2016, 165, 1255–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.; Zhou, X.; Wang, H.; Xing, D. Clustered Regularly Interspaced Short Palindromic Repeats/Cas9 Triggered Isothermal Amplification for Site-Specific Nucleic Acid Detection. Anal. Chem. 2018, 90, 2193–2200. [Google Scholar] [CrossRef]
- Quan, J.; Langelier, C.; Kuchta, A.; Batson, J.; Teyssier, N.; Lyden, A.; Caldera, S.; McGeever, A.; Dimitrov, B.; King, R.; et al. FLASH: A next-generation CRISPR diagnostic for multiplexed detection of antimicrobial resistance sequences. Nucleic Acids Res. 2019, 47, e83. [Google Scholar] [CrossRef] [Green Version]
- Marsic, T.; Ali, Z.; Tehseen, M.; Mahas, A.; Hamdan, S.; Mahfouz, M. Vigilant: An Engineered VirD2-Cas9 Complex for Lateral Flow Assay-Based Detection of SARS-CoV-2. Nano Lett. 2021, 21, 3596–3603. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Y.; Zhang, L.; Liu, S.; Zhang, M.; Wang, J.; Ning, N.; Peng, Y.; He, J.; Hu, Y.; et al. CRISPR-Cas9 Triggered Two-Step Isothermal Amplification Method for E. coli O157:H7 Detection Based on a Metal-Organic Framework Platform. Anal. Chem. 2020, 92, 3032–3041. [Google Scholar] [CrossRef]
- Zhang, Y.; Qian, L.; Wei, W.; Wang, Y.; Wang, B.; Lin, P.; Liu, W.; Xu, L.; Li, X.; Liu, D.; et al. Paired Design of dCas9 as a Systematic Platform for the Detection of Featured Nucleic Acid Sequences in Pathogenic Strains. ACS Synth. Biol. 2017, 6, 211–216. [Google Scholar] [CrossRef]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da, C.M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef] [Green Version]
- Li, S.Y.; Cheng, Q.X.; Wang, J.M.; Li, X.Y.; Zhang, Z.L.; Gao, S.; Cao, R.B.; Zhao, G.P.; Wang, J. CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 2018, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Huyke, D.A.; Sharma, E.; Sahoo, M.K.; Huang, C.; Banaei, N.; Pinsky, B.A.; Santiago, J.G. Electric field-driven microfluidics for rapid CRISPR-based diagnostics and its application to detection of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 29518–29525. [Google Scholar] [CrossRef]
- Li, F.; Ye, Q.; Chen, M.; Xiang, X.; Zhang, J.; Pang, R.; Xue, L.; Wang, J.; Gu, Q.; Lei, T.; et al. Cas12aFDet: A CRISPR/Cas12a-based fluorescence platform for sensitive and specific detection of Listeria monocytogenes serotype 4c. Anal. Chim. Acta 2021, 1151, 338248. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ke, Y.; Liu, W.; Sun, Y.; Ding, X. A One-Pot Toolbox Based on Cas12a/crRNA Enables Rapid Foodborne Pathogen Detection at Attomolar Level. ACS Sens. 2020, 5, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Zhang, P.; Wu, G.; Tan, Y.; Zhao, Y.; Cao, S.; Song, Y.; Yang, R.; Du, Z. Highly Specific and Sensitive Detection of Yersinia pestis by Portable Cas12a-UPTLFA Platform. Front. Microbiol. 2021, 12, 700016. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ye, Q.; Chen, M.; Zhou, B.; Zhang, J.; Pang, R.; Xue, L.; Wang, J.; Zeng, H.; Wu, S.; et al. An ultrasensitive CRISPR/Cas12a based electrochemical biosensor for Listeria monocytogenes detection. Biosens. Bioelectron. 2021, 179, 11307. [Google Scholar] [CrossRef]
- Bonini, A.; Poma, N.; Vivaldi, F.; Biagini, D.; Bottai, D.; Tavanti, A.; Francesco, F.D. A label-free impedance biosensing assay based on CRISPR/Cas12a collateral activity for bacterial DNA detection. J. Pharm. Biomed. Anal. 2021, 204, 114268. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, S.; Wu, N.; Wu, J.; Wang, G.; Zhao, G.; Wang, J. HOLMESv2: A CRISPR-Cas12b-Assisted Platform for Nucleic Acid Detection and DNA Methylation Quantitation. ACS Synth. Biol. 2019, 8, 2228–2237. [Google Scholar] [CrossRef]
- Teng, F.; Guo, L.; Cui, T.; Wang, X.G.; Xu, K.; Gao, Q.; Zhou, Q.; Li, W. CDetection: CRISPR-Cas12b-based DNA detection with sub-attomolar sensitivity and single-base specificity. Genome Biol. 2019, 20, 132. [Google Scholar] [CrossRef] [PubMed]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Zhou, X.; Shan, Y.; Yue, H.; Huang, R.; Hu, J.; Xing, D. Sensitive detection of a bacterial pathogen using allosteric probe-initiated catalysis and CRISPR-Cas13a amplification reaction. Nat. Commun. 2020, 11, 267. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, C.M.; Myhrvold, C.; Thakku, S.G.; Freije, C.A.; Metsky, H.C.; Yang, D.K.; Ye, S.H.; Boehm, S.K.; Kosoko-Thoroddsen, T.F.; Kehe, J.; et al. Massively multiplexed nucleic acid detection with Cas13. Nature 2020, 582, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Dong, X.; Wang, Y.; Yang, L.; Cai, K.; Zhang, X.; Kou, Z.; He, L.; Sun, S.; Li, T.; et al. Sensitive and Easy-Read CRISPR Strip for COVID-19 Rapid Point-of-Care Testing. CRISPR J. 2021, 4, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Spoelstra, W.K.; Jacques, J.M.; Gonzalez-Linares, R.; Nobrega, F.L.; Haagsma, A.C.; Dogterom, M.; Meijer, D.H.; Idema, T.; Brouns, S.J.J.; Reese, L. CRISPR-based DNA and RNA detection with liquid-liquid phase separation. Biophys. J. 2021, 120, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhou, W.; Lin, X.; Khan, M.R.; Deng, S.; Zhou, M.; He, G.; Wu, C.; Deng, R.; He, Q. Light-up RNA aptamer signaling-CRISPR-Cas13a-based mix-and-read assays for profiling viable pathogenic bacteria. Biosens. Bioelectron. 2021, 176, 112906. [Google Scholar] [CrossRef]
- Song, F.; Wei, Y.; Wang, P.; Ge, X.; Li, C.; Wang, A.; Yang, Z.; Wan, Y.; Li, J. Combining tag-specific primer extension and magneto-DNA system for Cas14a-based universal bacterial diagnostic platform. Biosens. Bioelectron. 2021, 185, 113262. [Google Scholar] [CrossRef]
- Ge, X.; Meng, T.; Tan, X.; Wei, Y.; Tao, Z.; Yang, Z.; Song, F.; Wang, P.; Wan, Y. Cas14a1-mediated nucleic acid detectifon platform for pathogens. Biosens. Bioelectron. 2021, 189, 113350. [Google Scholar] [CrossRef]
- Sridhara, S.; Goswami, H.N.; Whyms, C.; Dennis, J.H.; Li, H. Virus detection via programmable Type III-A CRISPR-Cas systems. Nat. Commun. 2021, 12, 5653. [Google Scholar] [CrossRef]
- Gruschow, S.; Adamson, C.S.; White, M.F. Specificity and sensitivity of an RNA targeting type III CRISPR complex coupled with a NucC endonuclease effector. Nucleic Acids Res. 2021, 49, 13122–13134. [Google Scholar] [CrossRef]
- Steens, J.A.; Zhu, Y.; Taylor, D.W.; Bravo, J.P.K.; Prinsen, S.H.P.; Schoen, C.D.; Keijser, B.J.K.; Ossendrijver, M.; Hofstra, L.M.; Brouns, S.J.J.; et al. SCOPE enables type III CRISPR-Cas diagnostics using flexible targeting and stringent CARF ribonuclease activation. Nat. Commun. 2021, 12, 5033. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, D.P.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.F.; Li, Y.X.; Wang, J.Z.; Zhao, Y.T.; Wang, Y. Controlling CRISPR-Cas9 by guide RNA engineering. Wiley Interdiscip. Rev. RNA 2022, e1731. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Jiang, F.; Taylor, D.W.; Sternberg, S.H.; Kaya, E.; Ma, E.; Anders, V.; Hauer, M.; Zhou, K.; Lin, S.; et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 2014, 343, 1247997. [Google Scholar] [CrossRef] [Green Version]
- Bolotin, A.; Quinquis, B.; Sorokin, A.; Ehrlich, S.D. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 2005, 151, 2551–2561. [Google Scholar] [CrossRef] [Green Version]
- Makarova, K.S.; Grishin, N.V.; Shabalina, S.A.; Wolf, Y.I.; Koonin, E.V. A putative RNA-interference-based immune system in prokaryotes: Computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol. Direct. 2006, 1, 7. [Google Scholar] [CrossRef] [Green Version]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [Green Version]
- Sternberg, S.H.; Redding, S.; Jinek, M.; Greene, E.C.; Doudna, J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014, 507, 62–67. [Google Scholar] [CrossRef] [Green Version]
- Torres, R.; Martin, M.C.; Garcia, A.; Cigudosa, J.C.; Ramirez, J.C.; Rodriguez-Perales, S. Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR-Cas9 system. Nat. Commun. 2014, 5, 3964. [Google Scholar] [CrossRef] [Green Version]
- Xue, W.; Chen, S.; Yin, H.; Tammela, T.; Papagiannakopoulos, T.; Joshi, N.S.; Cai, W.; Yang, G.; Bronson, R.; Crowley, D.G.; et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 2014, 514, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef]
- Chang, W.; Liu, W.; Shen, H.; Chen, S.; Liao, P.; Liu, Y. Molecular AND logic gate for multiple single-nucleotide mutations detection based on CRISPR/Cas9n system-trigged signal amplification. Anal. Chim. Acta 2020, 1112, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Compton, J. Nucleic acid sequence-based amplification. Nature 1991, 350, 91–92. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zheng, H.; Wei, X.; Lin, Z.; Guo, L.; Qiu, B.; Chen, G. Metal-organic framework (MOF): A novel sensing platform for biomolecules. Chem. Commun. 2013, 49, 1276–1278. [Google Scholar] [CrossRef]
- Cui, L.; Zhu, Z.; Lin, N.; Zhang, H.; Guan, Z.; Yang, C.J. A T7 exonuclease-assisted cyclic enzymatic amplification method coupled with rolling circle amplification: A dual-amplification strategy for sensitive and selective microRNA detection. Chem. Commun. 2014, 50, 1576–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batista, A.C.; Pacheco, L.G.C. Detecting pathogens with Zinc-Finger, TALE and CRISPR-based programmable nucleic acid binding proteins. J. Microbiol. Methods 2018, 152, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Blackstock, D.; Chen, W. Halo-tag mediated self-labeling of fluorescent proteins to molecular beacons for nucleic acid detection. Chem. Commun. 2014, 50, 13735–13738. [Google Scholar] [CrossRef]
- Ali, Z.; Shami, A.; Sedeek, K.; Kamel, R.; Alhabsi, A.; Tehseen, M.; Hassan, N.; Butt, H.; Kababji, A.; Hamdan, S.M.; et al. Fusion of the Cas9 endonuclease and the VirD2 relaxase facilitates homology-directed repair for precise genome engineering in rice. Commun. Biol. 2020, 3, 44. [Google Scholar] [CrossRef] [Green Version]
- Qian, J.; Boswell, S.A.; Chidley, C.; Lu, Z.X.; Pettit, M.E.; Gaudio, B.L.; Fajnzylber, J.M.; Ingram, R.T.; Ward, R.H.; Li, J.Z.; et al. An enhanced isothermal amplification assay for viral detection. Nat. Commun. 2020, 11, 5920. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Crawford, E.D.; O’Donovan, B.D.; Wilson, M.R.; Chow, E.D.; Retallack, H.; DeRisi, J.L. Depletion of Abundant Sequences by Hybridization (DASH): Using Cas9 to remove unwanted high-abundance species in sequencing libraries and molecular counting applications. Genome Biol. 2016, 17, 41. [Google Scholar] [CrossRef] [Green Version]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; Oost, J.V.D.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015, 163, 759–771. [Google Scholar] [CrossRef]
- Li, S.Y.; Cheng, Q.X.; Liu, J.K.; Nie, X.Q.; Zhao, G.P.; Wang, J. CRISPR-Cas12a has both cis- and trans-cleavage activities on single-stranded DNA. Cell Res. 2018, 28, 491–493. [Google Scholar] [CrossRef]
- East-Seletsky, A.; O’Connell, M.R.; Knight, S.C.; Burstein, D.; Cate, J.H.; Tjian, R.; Doudna, J.A. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 2016, 538, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Leung, R.K.; Cheng, Q.X.; Wu, Z.L.; Khan, G.; Liu, Y.; Xia, H.Y.; Wang, J. CRISPR-Cas12-based nucleic acids detection systems. Methods 2022, 203, 276–281. [Google Scholar] [CrossRef]
- Eid, C.; Santiago, J.G. Isotachophoresis applied to biomolecular reactions. Lab. Chip. 2017, 18, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Bercovici, M.; Han, C.M.; Liao, J.C.; Santiago, J.G. Rapid hybridization of nucleic acids using isotachophoresis. Proc. Natl. Acad. Sci. USA 2012, 109, 11127–11132. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Somoza, R.A.; Wang, L.; Welter, J.F.; Li, Y.; Caplan, A.I.; Liu, C.C. Exploring the Trans-Cleavage Activity of CRISPR-Cas12a (cpf1) for the Development of a Universal Electrochemical Biosensor. Angew. Chem. Int. Ed. Engl. 2019, 58, 17399–17405. [Google Scholar] [CrossRef]
- Strong, M.E.; Richards, J.R.; Torres, M.; Beck, C.M.; Belle, J.T.L. Faradaic electrochemical impedance spectroscopy for enhanced analyte detection in diagnostics. Biosens. Bioelectron. 2021, 177, 112949. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Gao, P.; Rajashankar, K.R.; Patel, D.J. PAM-Dependent Target DNA Recognition and Cleavage by C2c1 CRISPR-Cas Endonuclease. Cell 2016, 167, 1814–1828. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Jiang, H.; Zhu, Z.; Liu, J.; Li, B. Integrating CRISPR/Cas within isothermal amplification for point-of-Care Assay of nucleic acid. Talanta 2022, 243, 123388. [Google Scholar] [CrossRef] [PubMed]
- Shmakov, S.; Abudayyeh, O.O.; Makarova, K.S.; Wolf, Y.I.; Gootenberg, J.S.; Semenova, E.; Minakhin, M.; Joung, J.; Konermann, S.; Severinov, K.; et al. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol. Cell 2015, 60, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Chen, Z.; Zhang, W.; Zhang, J. Engineering CRISPR/Cas13 System against RNA Viruses: From Diagnostics to Therapeutics. Bioengineering 2022, 9, 291. [Google Scholar] [CrossRef] [PubMed]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef] [Green Version]
- Spoelstra, W.K.; EO, V.D.S.; Dogterom, M.; Reese, L. Nonspherical Coacervate Shapes in an Enzyme-Driven Active System. Langmuir 2020, 36, 1956–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neubacher, S.; Hennig, S. RNA Structure and Cellular Applications of Fluorescent Light-Up Aptamers. Angew. Chem. Int. Ed. Engl. 2019, 58, 1266–1279. [Google Scholar] [CrossRef] [PubMed]
- Vinkenborg, J.L.; Karnowski, N.; Famulok, M. Aptamers for allosteric regulation. Nat. Chem. Biol. 2011, 7, 519–527. [Google Scholar] [CrossRef]
- Zhang, D.Y.; Winfree, E. Dynamic allosteric control of noncovalent DNA catalysis reactions. J. Am. Chem. Soc. 2008, 130, 13921–13926. [Google Scholar] [CrossRef]
- Harrington, L.B.; Burstein, D.; Chen, J.S.; Paez-Espino, D.; Ma, E.; Witte, I.P.; Cofsky, J.C.; Kyrpides, N.C.; Banfield, J.F.; Doudna, J.A. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science 2018, 362, 839–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; An, X. Adaptation by Type III CRISPR-Cas Systems: Breakthrough Findings and Open Questions. Front. Microbiol. 2022, 13, 876174. [Google Scholar] [CrossRef] [PubMed]
- Samai, P.; Pyenson, N.; Jiang, W.; Goldberg, G.W.; Hatoum-Aslan, A.; Marraffini, L.A. Co-transcriptional DNA and RNA Cleavage during Type III CRISPR-Cas Immunity. Cell 2015, 161, 1164–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athukoralage, J.S.; Graham, S.; Rouillon, C.; Gruschow, S.; Czekster, C.M.; White, M.F. The dynamic interplay of host and viral enzymes in type III CRISPR-mediated cyclic nucleotide signalling. eLife 2020, 9, e55852. [Google Scholar] [CrossRef]
- Del, R.C.; Malani, P.N. COVID-19-New Insights on a Rapidly Changing Epidemic. JAMA 2020, 323, 1339–1340. [Google Scholar] [CrossRef] [Green Version]
- Fauci, A.S.; Lane, H.C.; Redfield, R.R. COVID-19—Navigating the Uncharted. N. Engl. J. Med. 2020, 382, 1268–1269. [Google Scholar] [CrossRef]
- Taleghani, N.; Taghipour, F. Diagnosis of COVID-19 for controlling the pandemic: A review of the state-of-the-art. Biosens. Bioelectron. 2021, 174, 112830. [Google Scholar] [CrossRef]
- Safari, F.; Afarid, M.; Rastegari, B.; Borhani-Haghighi, A.; Barekati-Mowahed, M.; Behzad-Behbahani, A. CRISPR systems: Novel approaches for detection and combating COVID-19. Virus Res. 2021, 294, 198282. [Google Scholar] [CrossRef]
- Javalkote, V.S.; Kancharla, N.; Bhadra, B.; Shukla, M.; Soni, B.; Sapre, A.; Goodin, M.; Bandyopadhyay, A.; Dasgupta, S. CRISPR-based assays for rapid detection of SARS-CoV-2. Methods 2022, 203, 594–603. [Google Scholar] [CrossRef]
- Fapohunda, F.O.; Qiao, S.; Pan, Y.; Wang, H.; Liu, Y.; Chen, Q.; Lu, P. CRISPR Cas system: A strategic approach in detection of nucleic acids. Microbiol. Res. 2022, 259, 127000. [Google Scholar] [CrossRef]
- Wu, X.; Scott, D.A.; Kriz, A.J.; Chiu, A.C.; Hsu, P.D.; Dadon, D.B.; Cheng, A.W.; Trevino, A.E.; Konermann, S.; Chen, S.; et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 2014, 32, 670–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.S.; Dagdas, Y.S.; Kleinstiver, B.P.; Welch, M.M.; Sousa, A.A.; Harrington, L.B.; Sternberg, S.H.; Joung, J.K.; Yildiz, A.; Doudna, J.A. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 2017, 550, 407–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification | System Name | Effector | Signal Amplification | Sensitivity b | Specificity | Quantitative | Multiplex | Detection Indicator | Detection Time | Target Type | Infectious Disease Target |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cas9-based class | NASBACC [24] | SpCas9 | NASBA | fM | 1 nt | Y | Y | paper sensors | <4 h | RNA | Zika virus |
| CAS-EXPAR [25] | SpCas9 | EXPAR | aM | 1 nt | N | N | fluorescence | <1 h | DNA | L. monocytogenes | |
| FLASH-NGS [26] | SpCas9 | FLASH | NA | NA | N | Y | sequencing | NA | DNA | S. aureus | |
| Vigilant [27] | SpCas9 | RT-RPA | 2.5 copies | NA | N | N | lateral flow analysis | 35 min | DNA | SARS-CoV-2 | |
| UiO66-platform-based Cas9 [28] | SpCas9 | RCA | fM | NA | N | N | fluorescence | 2 h | DNA | E. coli O157:H7 | |
| dCas9-based class | PC Reporter System [29] | SpdCas9 | PCR | One copy | NA | N | N | fluorescence | PCR + 10 min | DNA | Mycobacterium tuberculosis |
| Cas12a-based class | DETECTR [30] | LbCas12a | RPA | aM | 6 nt | N | N | fluorescence | 2 h | DNA | HPV |
| HOLMES [31] | LbCas12a | PCR | aM | 1nt | N | N | fluorescence | NA | DNA | pseudorabies virus and Japanese encephalitis virus | |
| ITP–CRISPR [32] | LbCas12a | RT-LAMP | 10 copies | NA | N | N | fluorescence | 35 min | DNA | SARS-CoV-2 | |
| Cas12aFDet [33] | LbCas12a | PCR/RAA | aM | NA | N | N | fluorescence | 1 h | DNA | L. monocytogenes | |
| OCTOPUS [34] | LbCas12a | RPA | aM | NA | N | N | fluorescence | 50 min | DNA | E. coli O157:H7 and Streptococcus aureus | |
| Cas12a-UPTLFA [35] | LbCas12a | RPA | pM | 1 nt | N | N | fluorescence | 80 min | DNA | Yersinia pestis | |
| RAA-based E-CRISPR [36] | AsCas12a | NA | aM | NA | N | N | Current signal | NA | DNA | L. monocytogenes | |
| EIS- CRISPR [37] | LbCas12a | PCR | nM | NA | N | N | Current signal | 1.5 h | DNA | E. coli and Staphylococcus aureus | |
| Cas12b-based class | HOLMESv2 [38] | AacCas12b | LAMP/PCR/RT-LAMP | aM | 1 nt | Y | N | fluorescence | 1 h | DNA | NA |
| CDetection [39] | AaCas12b | RPA | aM | 1 nt | N | N | fluorescence | NA | DNA | HPV | |
| Cas13-based class | SHERLOCK [40] | LwCas13a | RPA | aM | 1 nt | N | N | fluorescence | 2 h | DNA/ RNA | Zika and dengue virus |
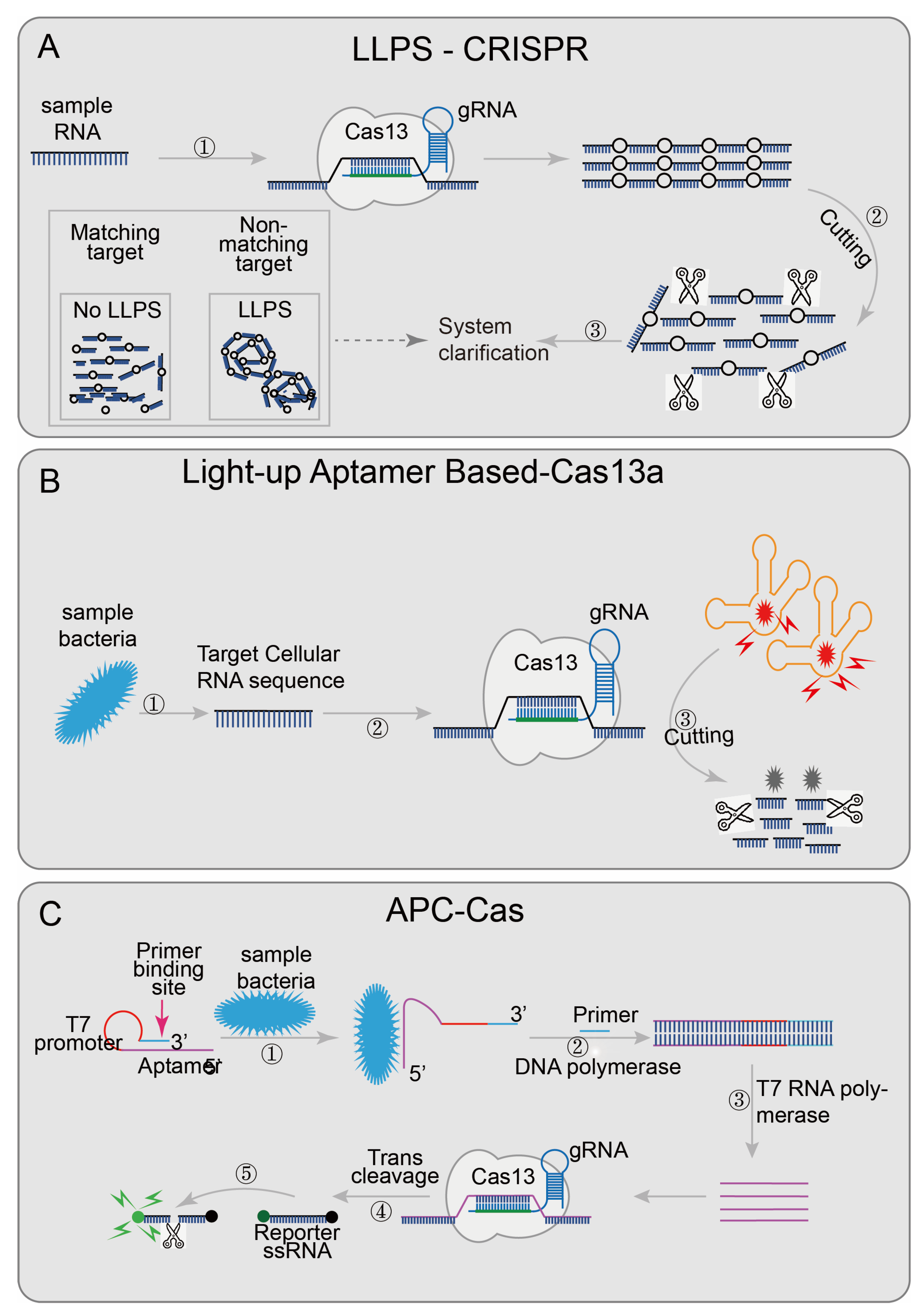
| APC-Cas [41] | LwCas13a | PCR | 1CFU | NA | Y | N | fluorescence | 140 min | bacterial pathogen | Salmonella Enteritidis | |
| CARMEN [42] | LwCas13a | PCR/RPA | NA | NA | N | Y | fluorescence | NA | DNA/ RNA | SARS-CoV-2 | |
| ERASE [43] | LwCas13a | RT-RAA | 1copy | NA | N | N | Lateral flow strip | 60 min | RNA | SARS-CoV-2 | |
| LLPS-CRISPR [44] | LwCas13a | PCR/RPA | aM | NA | N | N | System turbidity | 60 min | DNA/ RNA | NA | |
| Light-up Aptamer based-Cas13a [45] | LwCas13a | NA | 10CFU | NA | N | N | fluorescence | NA | bacterial pathogen | B. cereus | |
| Cas 14-based class | TSPE-Cas14a [46] | LbCas14a1 | PCR | aM | 1 nt | N | N | fluorescence | NA | RNA | six pathogenic species |
| CMP [47] | LbCas14a1 | PCR | aM | NA | N | Y | fluorescence | 80 min | RNA | Streptococcus pyogenes and Eberthella typhi | |
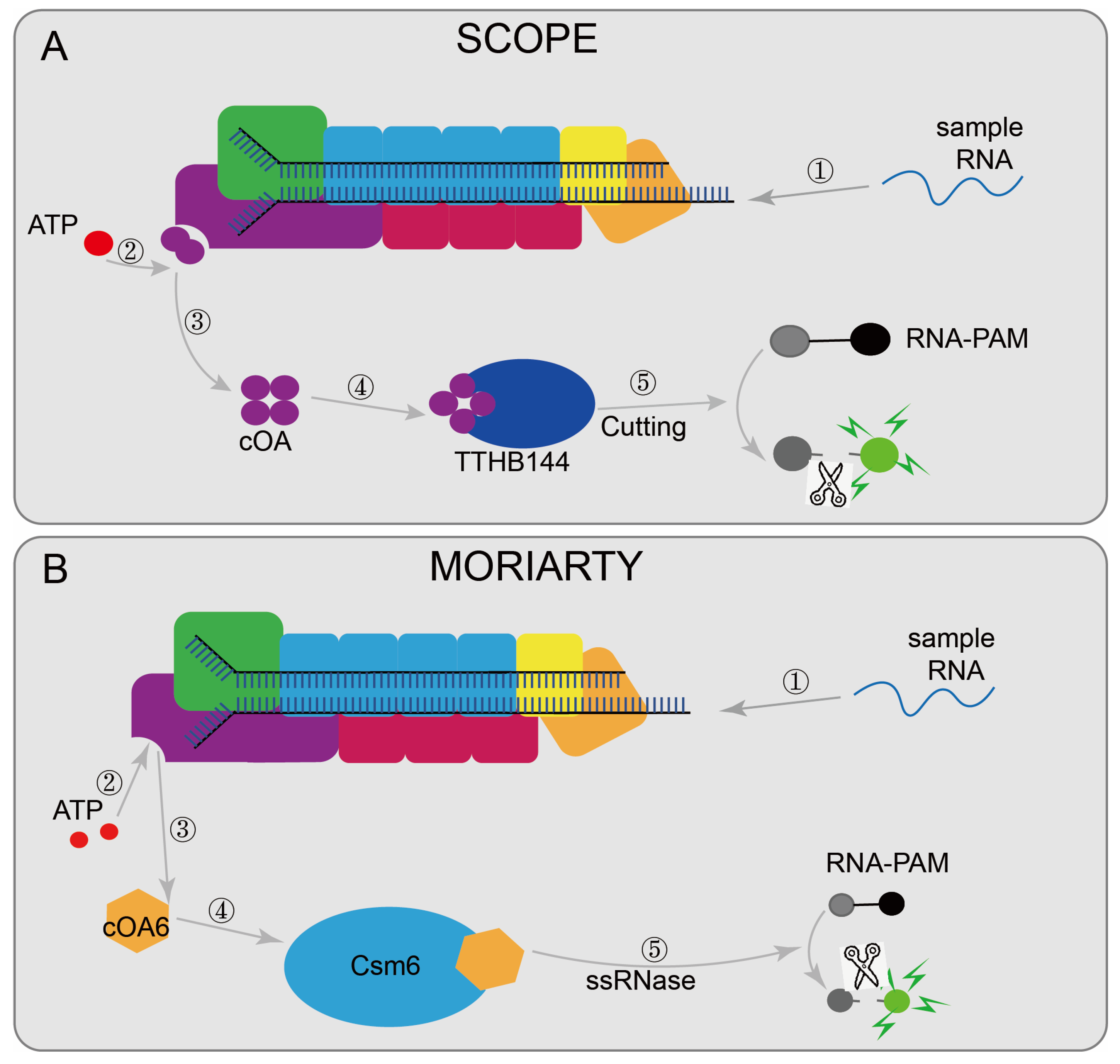
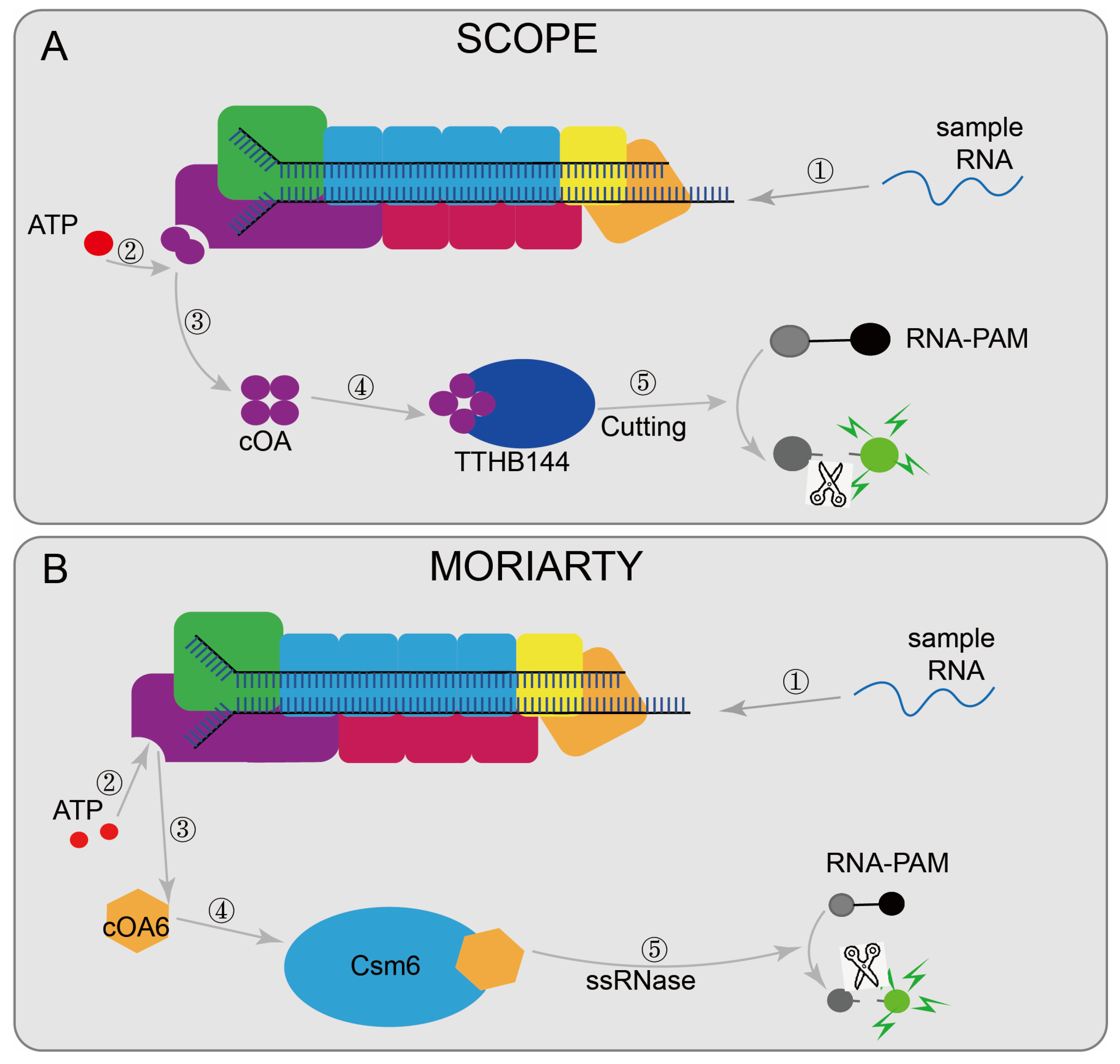
| Cas10 -based class | MORIARTY [48] | LlCsm | RT-RPA | aM | 1 nt | N | N | fluorescence | 50 min | RNA | SARS-CoV-2 |
| VmeCmr–NucC coupled assay [49] | VmeCmr | RT-PCR | fM | 1 nt | N | N | fluorescence | 30 min | RNA | SARS-CoV-2 | |
| SCOPE [50] | TtCmr | RT-LAMP | aM | 1 nt | N | N | fluorescence | 35 min | RNA | SARS-CoV-2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Wang, Y.; Wang, B.; Lou, J.; Ni, P.; Jin, Y.; Chen, S.; Duan, G.; Zhang, R. Application of CRISPR/Cas Systems in the Nucleic Acid Detection of Infectious Diseases. Diagnostics 2022, 12, 2455. https://doi.org/10.3390/diagnostics12102455
Li J, Wang Y, Wang B, Lou J, Ni P, Jin Y, Chen S, Duan G, Zhang R. Application of CRISPR/Cas Systems in the Nucleic Acid Detection of Infectious Diseases. Diagnostics. 2022; 12(10):2455. https://doi.org/10.3390/diagnostics12102455
Chicago/Turabian StyleLi, Junwei, Yuexia Wang, Bin Wang, Juan Lou, Peng Ni, Yuefei Jin, Shuaiyin Chen, Guangcai Duan, and Rongguang Zhang. 2022. "Application of CRISPR/Cas Systems in the Nucleic Acid Detection of Infectious Diseases" Diagnostics 12, no. 10: 2455. https://doi.org/10.3390/diagnostics12102455