
Hajdu-Cheney Syndrome: Report of a Case in Spain


 ,
, 
 , and
, and 
Abstract
:1. Introduction
2. Patient Information
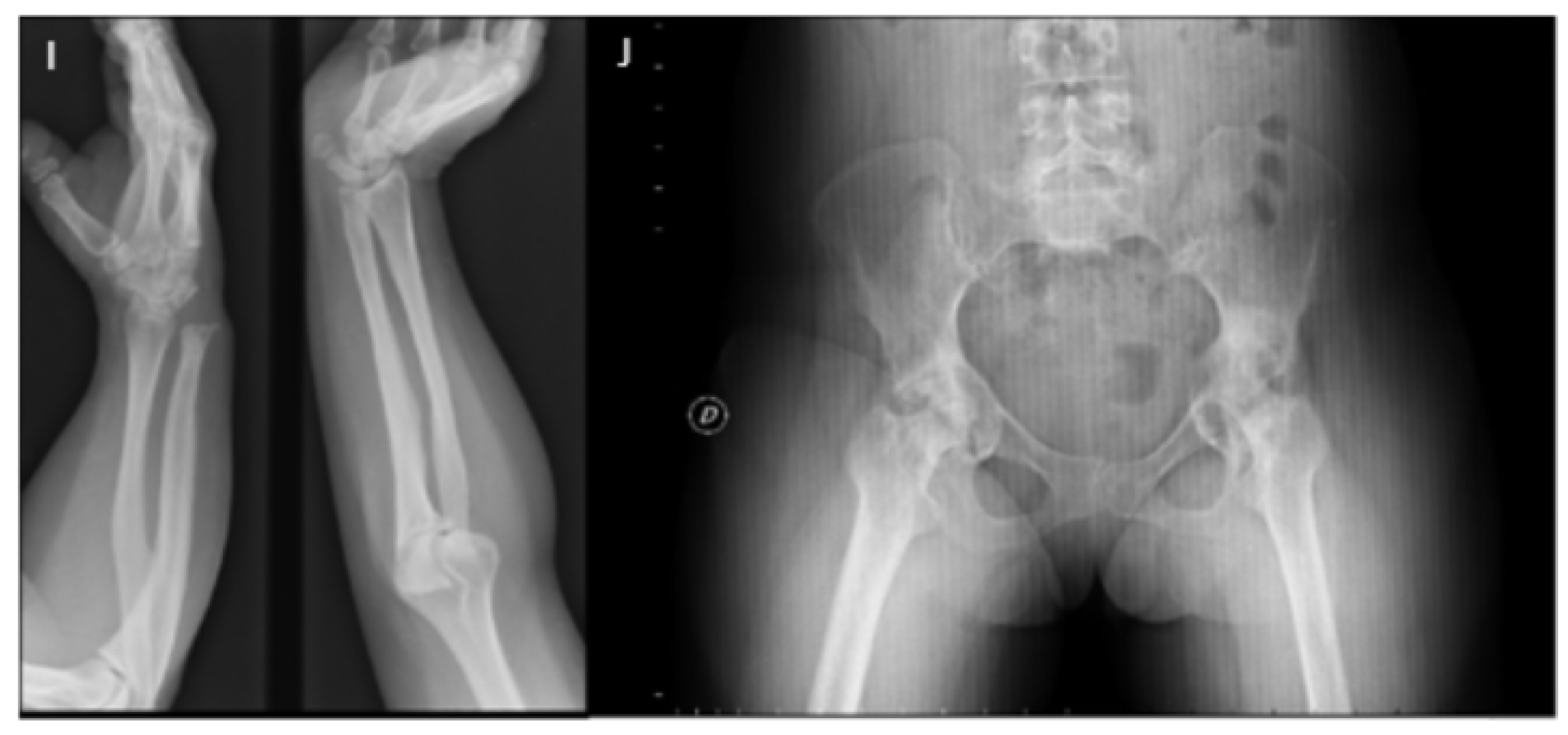
3. Musculoskeletal Features
4. Respiratory Features
5. Ear, Nose and Throat Features
6. Gastroenterology Aspects
7. Gynecology Aspects
8. Maxilofacial Features
9. Hematology Aspects
10. Psychiatric Aspects
11. Psychosocial Aspects and Lifestyle
12. Milestones
13. Diagnosis Related Problems, Differential Diagnosis and Prognosis
14. Therapeutic Interventions
15. Discussion
16. Limitations
17. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HCS | Hajdu-Cheney syndrome |
| CT | Computed tomography |
| VAS | Visual Analog Scale |
| AHI | Apnea–hypopnea index |
| BPM | Beats per minute |
| FEV | Forced expiratory volume |
| FVC | Forced vital capacity |
| IT | Inspiration time |
| PHQ-9 | Patient Health Questionnaire |
| GAD-7 | General Anxiety Disorder scale |
| OSAHS | Oximetric sleep apnea–hypopnea syndrome |
References
- Singh, J.A.; Williams, C.B.; McAlister, W.H. Talo-patello-scaphoid osteolysis, synovitis, and short fourth metacarpals in sisters: A new syndrome? Am. J. Med. Genet. 2003, 121A, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Simpson, M.A.; Irving, M.D.; Asilmaz, E.; Gray, M.J.; Dafou, D.; Elmslie, F.V.; Mansour, S.; Holder, S.E.; Brain, C.E.; Burton, B.K.; et al. Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nat. Genet. 2011, 43, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Majewski, J.; Schwartzentruber, J.A.; Caqueret, A.; Patry, L.; Marcadier, J.; Fryns, J.P.; Boycott, K.M.; Ste-Marie, L.G.; Mckiernan, F.E.; Marik, I.; et al. Mutations in NOTCH2 in families with Hajdu-Cheney syndrome. Hum. Mutat. 2011, 32, 1114–1117. [Google Scholar] [CrossRef] [PubMed]
- Descartes, M.; Rojnueangnit, K.; Cole, L.; Sutton, A.; Morgan, S.L.; Patry, L.; Samuels, M.E. Hajdu-Cheney syndrome: Phenotypical progression with de-novo NOTCH2 mutation. Clin. Dysmorphol. 2014, 23, 88–94. [Google Scholar] [CrossRef]
- Hajdu, N.; Kauntze, R. Cranio-skeletal dysplasia. Br. J. Radiol. 1948, 21, 42–48. [Google Scholar] [CrossRef]
- Cheney, W. Acro-Osteolysis. Am. J. Roentgenol. Radium Ther. Nucl. Med. 1965, 94, 595–607. [Google Scholar]
- Cortés-Martín, J.; Díaz-Rodriguez, L.; Piqueras-Sola, B.; Rodriguez-Blanque, R.; Bermejo-Fernández, A.; Sanchez-García, J.C. Hajdu-Cheney Syndrome: A Systematic Review of the Literature. Int. J. Environ. Res. Public Health 2020, 17, 6174. [Google Scholar] [CrossRef]
- Regev, M.; Pode-Shakked, B.; Jacobson, J.M.; Raas-Rothschild, A.; Goldstein, D.B.; Anikster, Y. Phenotype variability in Hajdu-Cheney syndrome. Eur. J. Med. Genet. 2019, 62, 35–38. [Google Scholar] [CrossRef]
- Brennan, A.M.; Pauli, R.M. Hajdu-Cheney syndrome: Evolution of phenotype and clinical problems. Am. J. Med. Genet. 2001, 100, 292–310. [Google Scholar] [CrossRef]
- Brown, D.M.; Bradford, D.S.; Gorlin, R.J.; Desnick, R.J.; Langer, L.O.; Jowsey, J.; Sauk, J.J. The acro-osteolysis syndrome: Morphologic and biochemical studies. J. Pediatr. 1976, 88, 573–580. [Google Scholar] [CrossRef]
- Sawin, P.D.; Menezes, A.H. Basilar invagination in osteogenesis imperfecta and related osteochondrodysplasias: Medical and surgical management. J. Neurosurg. 1997, 86, 950–960. [Google Scholar] [CrossRef] [PubMed]
- Ades, L.C.; Morris, L.L.; Haan, E.A. Hydrocephalus in Hajdu-Cheney syndrome. J. Med. Genet. 1993, 30, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, G.; Aoaki, K.; Haga, N.; Hasegawa, T.; Aoki, K.; Haga, N.; Hasegawa, T. Syringohydromyelia in Hajdu-Cheney syndrome. Pediatr. Radiol. 1996, 26, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Bazopoulou-Kyrkanidou, E.; Vrahopoulos, T.P.; Eliades, G.; Vastardis, H.; Tosios, K.; Vrotsos, I.A. Periodontitis Associated with Hajdu-Cheney Syndrome. J. Periodontol. 2007, 78, 1831–1838. [Google Scholar] [CrossRef]
- Letchumanan, P.; Thumboo, J.; Leong, R.T.K. A patient with progressive shortening of the fingers. J. Rheumatol. 2009, 36, 198–199. [Google Scholar] [CrossRef]
- Stathopoulos, I.P.; Trovas, G.; Lampropoulou-Adamidou, K.; Koromila, T.; Kollia, P.; Papaioannou, N.A.; Lyritis, G. Severe osteoporosis and mutation in NOTCH2 gene in a woman with Hajdu-Cheney syndrome. Bone 2013, 52, 366–371. [Google Scholar] [CrossRef]
- Sargin, G.; Cildag, S.; Senturk, T. Hajdu-Cheney syndrome with ventricular septal defect. Kaohsiung J. Med. Sci. 2013, 29, 343–344. [Google Scholar] [CrossRef]
- Currarino, G. Hajdu-Cheney syndrome associated with serpentine fibulae and polycystic kidney disease. Pediatr. Radiol. 2009, 39, 47–52. [Google Scholar] [CrossRef]
- Isidor, B.; Lindenbaum, P.; Pichon, O.; Bézieau, S.; Dina, C.; Jacquemont, S.; Martin-Coignard, D.; Thauvin-Robinet, C.; Le Merrer, M.; Mandel, J.L.; et al. Truncating mutations in the last exon of NOTCH2 cause a rare skeletal disorder with osteoporosis. Nat. Genet. 2011, 43, 306–308. [Google Scholar] [CrossRef]
- Gibofsky, A. Genetics of the hajdu-cheney syndrome. Arthritis Rheum. 1987, 30, 718. [Google Scholar] [CrossRef]
- Schawo, S.; Weber, M.A.; Libicher, M. Junge frau mit rückenschmerzen und akroosteolysen. Radiologe 2006, 46, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Pittaway, J.F.H.; Harrison, C.; Rhee, Y.; Holder-Espinasse, M.; Fryer, A.E.; Cundy, T.; Drake, W.M.; Irving, M.D. Bisphosphonate therapy for spinal osteoporosis in Hajdu-Cheney syndrome—New data and literature review. Orphanet J. Rare Dis. 2018, 13, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baader, T.; Molina, J.; Venezian, S.; Rojas, C.; Farías, R.; Fierro-Freixenet, C.; Backenstrass, M.; Mundt, C. Validación y utilidad de la encuesta PHQ-9 (Patient Health Questionnaire) en el diagnóstico de depresión en pacientes usuarios de atención primaria en Chile. Rev. Chil. Neuropsiquiatr. 2012, 9, 10–22. [Google Scholar] [CrossRef]
- NSpitzer, R.L.; Kroenke, K.; Williams, J.B.W.; Löwe, B. A Brief Measure for Assessing Generalized Anxiety Disorder: The GAD-7. Arch. Intern. Med. 2006, 166, 1092–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilagut, G.; Ferrer, M.; Rajmil, L.; Rebollo, P.; Permanyer-Miralda, G.; Quintana, J.M.; Santed, R.; Valderas, J.M.; Ribera, A.; Domingo-Salvany, A.; et al. The Spanish version of the Short Form 36 Health Survey: A decade of experience and new developments. Gac. Sanit. 2005, 19, 135–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, S.M. Pycnodysostosis: A review. J. Bone Jt. Surg. Am. 1967, 49, 153–163. [Google Scholar] [CrossRef]
- Zeng, C.; Lin, Y.; Lu, Z.; Chen, Z.; Jiang, X.; Mao, X.; Liu, Z.; Lu, X.; Zhang, K.; Yu, Q.; et al. Distinct severity of phenotype in Hajdu-Cheney syndrome: A case report and literature review. BMC Musculoskelet. Disord. 2020, 21, 2–6. [Google Scholar] [CrossRef]
- Graversen, L.; Handrup, M.M.; Irving, M.; Hove, H.; Diness, B.R.; Risom, L.; Svaneby, D.; Aagaard, M.M.; Vogel, I.; Gjørup, H.; et al. Phenotypic presentations of Hajdu-Cheney syndrome according to age—5 distinct clinical presentations. Eur. J. Med. Genet. 2019, 63, 103650. [Google Scholar] [CrossRef]
- Swan, L.; Gole, G.; Sabesan, V.; Cardinal, J.; Coman, D. Congenital Glaucoma: A Novel Ocular Manifestation of Hajdu-Cheney Syndrome. Case Rep. Genet. 2018, 2018, 2508345. [Google Scholar] [CrossRef] [Green Version]
- Takatani, R.; Someya, T.; Kazukawa, I.; Nishimura, G.; Minagawa, M.; Kohno, Y. Hajdu-Cheney syndrome: Infantile onset of hydrocephalus and serpentine fibulae. Pediatr. Int. 2009, 51, 831–833. [Google Scholar] [CrossRef]
- Jirečková, J.; Magner, M.; Lambert, L.; Baxová, A.; Leiská, A.; Kopečková, L.; Fajkusová, L.; Zeman, J. The Age Dependent Progression of Hajdu-Cheney Syndrome in Two Families. Prague Med. Rep. 2018, 119, 156–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harnasch, H. Die Akroosteolysis, ein neues Krankheitsbild. RöFo—Fortschritte auf dem Gebiet der Röntgenstrahlen und der Bildgebenden Verfahren 1949, 72, 352–359. [Google Scholar] [CrossRef]
- Rosenmann, E.; Penchas, S.; Cohen, T.; Aviad, I. Sporadic idiopathic acro-osteolysis with cranio-skeletal dysplasia, polycystic kidneys and glomerulonephritis a case of the hajdu-cheney syndrome. Pediatr. Radiol. 1977, 6, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Elias, A.N.; Pinals, R.S.; Clarke Anderson, H.; Gould, L.V.; Streeten, D.H.P. Hereditary osteodysplasia with acro-osteolysis (the Hajdu-Cheney syndrome). Am. J. Med. 1978, 65, 627–636. [Google Scholar] [CrossRef]
- Leidig-Bruckner, G.; Pfeilschifter, J.; Penning, N.; Limberg, B.; Priemel, M.; Delling, G.; Ziegler, R. Severe osteoporosis in familial Hajdu-Cheney syndrome: Progression of acro-osteolysis and osteoporosis during long-term follow-up. J. Bone Miner. Res. 1999, 14, 2036–2041. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.; Zugibe, F.T.; Gilbert, E.F.; Opitz, J.M. Arthro-Dento-Osteo Dysplasia (Hajdu-Cheney Syndrome). Z. Kinderheilkd. 1973, 114, 93–110. [Google Scholar] [CrossRef]
- Nunziata, V.; di Giovanni, G.; Ballanti, P.; Bonucci, E. High turnover osteoporosis in acro-osteolysis (Hajdu-Cheney syndrome). J. Endocrinol. Investig. 1990, 13, 251–255. [Google Scholar] [CrossRef]
- Siklar, Z.; Tanyer, G.; Dallar, Y.; Aksoy, F.G. Hajdu-Cheney syndrome with growth hormone deficiency and neuropathy. J. Pediatr. Endocrinol. Metab. 2000, 13, 951–954. [Google Scholar] [CrossRef]
- Williams, B. Foramen magnum impaction in a case of acro-osteolysis. Br. J. Surg. 1977, 64, 70–73. [Google Scholar] [CrossRef]
- Weleber, R.G.; Beals, R.K. The Hajdu-Cheney syndrome. Report of two cases and review of the literature. J. Pediatr. 1976, 88, 243–249. [Google Scholar] [CrossRef]
- Jiménez, I.; Medina-Gontier, J.; Caballero, J.; Medina, J. Hand Deformities in Hajdu-Cheney Syndrome: A Case Series of 3 Patients Across 3 Consecutive Generations. J. Hand Surg. Am. 2020, 46, 73.e1–73.e5. [Google Scholar] [CrossRef]
- Shurtleff, D.B.; Sparkes, R.S.; Clawson, D.K.; Guntheroth, W.G.; Mottet, N.K. Hereditary Osteolysis with Hypertension and Nephropathy. JAMA J. Am. Med. Assoc. 1964, 188, 363–368. [Google Scholar] [CrossRef]
- Ventosa, P.; Gorina, N.; Balaguer Santamaria, A.; Riera, L.; Casals, I. Acroosteólisis en un paciente de 4 años: Datos clínicos del síndrome de Hajdu-Cheney. An. Pediatr. 2013, 79, 62–63. [Google Scholar] [CrossRef]
- Greenberg, B.E.; Street, D.M. Idiopathic Non-Familial Aero-Osteolysis. Radiology 1957, 69, 259–262. [Google Scholar] [CrossRef]
- Colmenares Roldán, L.; de la Calle Rodríguez, N. Síndrome de Hajdu Cheney, una enfermedad poco frecuente. Rev. CES Med. 2013, 27, 101–106. [Google Scholar] [CrossRef]
- Vissarionov, S.V.; Filippova, A.N.; Zhurbitskaia, M.V.; Khusainov, N.O.; Belyanchikov, S.M. Surgical treatment of a patient with multiple fractures of the thoracic and lumbar vertebrae associated with Hajdu-Cheney syndrome. Hir. Pozvonochnika 2019, 16, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Chawla, B.S. Cranio-skeletal dysplasia with acro-osteolysis. Br. J. Radiol. 1964, 37, 702–705. [Google Scholar] [CrossRef]
- Fryns, J.P. Serpentine fibula syndrome: A variant clinical presentation of Hajdu-Cheney syndrome? Clin. Dysmorphol. 1997, 6, 287–288. [Google Scholar] [CrossRef]
- Ramos, F.J.; Kaplan, B.S.; Bellah, R.D.; Zackai, E.H.; Kaplan, P. Further evidence that the Hajdu-Cheney syndrome and the “serpentine fibula-polycystic kidney syndrome” are a single entity. Am. J. Med. Genet. 1998, 78, 474–481. [Google Scholar] [CrossRef]
- Gray, M.J.; Kim, C.A.; Bertola, D.R.; Arantes, P.R.; Stewart, H.; Simpson, M.A.; Irving, M.D.; Robertson, S.P. Serpentine fibula polycystic kidney syndrome is part of the phenotypic spectrum of Hajdu-Cheney syndrome. Eur. J. Hum. Genet. 2012, 20, 122–124. [Google Scholar] [CrossRef] [Green Version]
- Iwaya, T.; Taniguchi, K.; Watanabe, J.; Iinuma, K.; Hamazaki, Y.; Yoshikawa, S. Hajdu-Cheney syndrome. Arch. Orthop. Trauma Surg. 1979, 95, 293–302. [Google Scholar] [CrossRef]
- Sasaki, K.; Ito, Y.; Kawame, H.; Kikuchi, A.; Tanaka, H. Fatal case of Hajdu-Cheney syndrome with idiopathic pulmonary hemosiderosis. Pediatr. Int. 2019, 61, 190–192. [Google Scholar] [CrossRef]
- Nozaki, T.; Ihara, K.; Makimura, M.; Kinjo, T.; Hara, T. A girl with Hajdu-Cheney syndrome and premature ovarian failure. J. Pediatr. Endocrinol. Metab. 2012, 25, 171–173. [Google Scholar] [CrossRef]
- Shaw, D.G. Acro-osteolysis and bone fragility. Br. J. Radiol. 1969, 42, 934–936. [Google Scholar] [CrossRef]
- Lee, J.W.; Kim, Y.J.; Kang, J.; Shin, T.J.; Hyun, H.-K.; Kim, Y.-J.; Lee, S.-H.; Lee, Z.H.; Kim, J.-W. Dental implications in Hajdu-Cheney syndrome: A novel case report and review of the literature. Oral Dis. 2018, 24, 1037–1041. [Google Scholar] [CrossRef]
- Antoniades, K.; Kaklamanos, E.; Kavadia, S.; Hatzistilianou, M.; Antoniades, V. Hajdu-Cheney syndrome (acro-osteolysis): A case report of dental interest. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2003, 95, 725–731. [Google Scholar] [CrossRef]
- Vingerhoedt, E.; Bailleul-Forestier, I.; Fellus, P.; Schoenaers, J.; Frijns, J.P.; Carels, C. Syndrome of Hajdu-Cheney: Three case reports of orofacial interest. Cleft Palate Craniofac. J. 2010, 47, 645–653. [Google Scholar] [CrossRef]
- European Organisation for Rare Diseases (EURORDIS). Enfermedades Raras: El conocimiento de esta prioridad de la Salud Pública; EURORDIS: Paris, France, 2005. [Google Scholar]
- Sakka, S.; Gafni, R.I.; Davies, J.H.; Clarke, B.; Tebben, P.; Samuels, M.; Saraff, V.; Klaushofer, K.; Fratzl-Zelman, N.; Roschger, P.; et al. Bone Structural Characteristics and Response to Bisphosphonate Treatment in Children with Hajdu-Cheney Syndrome. J. Clin. Endocrinol. Metab. 2017, 102, 4163–4172. [Google Scholar] [CrossRef]
- Adami, G.; Rossini, M.; Gatti, D.; Orsolini, G.; Idolazzi, L.; Viapiana, O.; Scarpa, A.; Canalis, E. Hajdu Cheney Syndrome; report of a novel NOTCH2 mutation and treatment with denosumab. Bone 2016, 92, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Galli-Tsinopoulou, A.; Kyrgios, I.; Giza, S.; Giannopoulou, E.Z.; Maggana, I.; Laliotis, N. Two-year cyclic infusion of pamidronate improves bone mass density and eliminates risk of fractures in a girl with osteoporosis due to Hajdu-Cheney syndrome. Minerva Endocrinol. 2012, 37, 283–289. [Google Scholar]
- Al-Mayouf, S.M.; Madi, S.M.; Bin-Abbas, B.S. Cyclic intravenous pamidronate treatment in children with nodulosis, arthropathy and osteolysis syndrome. Ann. Rheum. Dis. 2006, 65, 1672–1673. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.; Shin, D.Y.; Moon, S.H.; Lee, E.J.; Lim, S.K.; Kim, O.H.; Rhee, Y. Effect of Zoledronic Acid on Acro-Osteolysis and Osteoporosis in a Patient with Hajdu-Cheney Syndrome. Yonsei Med. J. 2011, 52, 543–546. [Google Scholar] [CrossRef] [Green Version]
- McKiernan, F.E. Integrated anti-remodeling and anabolic therapy for the osteoporosis of Hajdu-Cheney syndrome. Osteoporos. Int. 2007, 18, 245–249. [Google Scholar] [CrossRef]
- McKiernan, F.E. Integrated anti-remodeling and anabolic therapy for the osteoporosis of Hajdu-Cheney syndrome: 2-Year follow-up. Osteoporos. Int. 2008, 19, 379–380. [Google Scholar] [CrossRef]
- Kaczoruk-Wieremczuk, M.; Adamska, P.; Adamski, Ł.J.; Wychowański, P.; Jereczek-Fossa, B.A.; Starzyńska, A. Oral surgery procedures in a patient with hajdu-cheney syndrome treated with denosumab—A rare case report. Int. J. Environ. Res. Public Health 2021, 18, 9099. [Google Scholar] [CrossRef]
- Mattei, T.A.; Rehman, A.A.; Issawi, A.; Fassett, D.R. Surgical challenges in the management of cervical kyphotic deformity in patients with severe osteoporosis: An illustrative case of a patient with Hajdu-Cheney syndrome. Eur. Spine J. 2015, 24, 2746–2753. [Google Scholar] [CrossRef]
- Murtagh-Schaffer, C.; Moquin, R.R. Spinal reconstruction in Hajdu-Cheney syndrome. JAAPA 2008, 21, 29–33. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Nakamura, K.; Takahashi, Y. A case report of anesthesia for a child with Hajdu-Cheney syndrome. J. Anesth. 2013, 27, 949–950. [Google Scholar] [CrossRef]
- August, D.A.; Ramos, D.C. Anesthesia for a child with Hajdu-Cheney syndrome. Paediatr. Anaesth. 2009, 19, 649–650. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Date | Age | Dmo (g/cm2) | Difference (%) | Difference/DE |
|---|---|---|---|---|
| 10 October 2005 | 38 | 0.819 | ||
| 7 May 2008 | 40.5 | 0.872 | 6.5 | 5.3 |
| 2 December 2011 | 44.1 | 0.914 | 11.6 | 9.5 |
| 29 May 2014 | 46.6 | 0.856 | 4.5 | 3.7 |
| 10 May 2016 | 48.5 | 0.814 | −0.6 | −0.5 |
| Date | Age | Dmo (g/cm2) | Difference (%) | Difference/DE |
|---|---|---|---|---|
| 10 October 2005 | 38 | 0.846 | ||
| 2 December 2011 | 44.1 | 1.933 | 128.5 | 77.6 |
| 29 May 2014 | 46.6 | 1.508 | 78.3 | 47.3 |
| 10 May 2016 | 48.5 | 1.300 | 53.7 | 32.4 |
| 10 December 2018 | 51.1 | 1.035 | 22.3 | 13.5 |
| Important Milestones | Year |
|---|---|
| Birth | 1967 |
| First medical registry | 1973 |
| Diagnostic suspicion | 1973 |
| Definitive diagnosis | 1977 |
| Pes valgus surgery | 1978 |
| Walks using crutches | 1996 |
| Diagnosis of breast fibroadenomas | 2001 |
| Begins treatment with bisphosphonates | 2006 |
| Wheelchair | 2012 |
| Ends treatment with bisphosphonates | 2012 |
| Evaluation for possible hip replacement | 2016 |
| Traffic accident | 2017 |
| Dependency for activities of daily living | 2017 |
| Diagnosis of prolonged adjustment disorder with clinical manifestations of depression and anxiety secondary to the pain and loss of autonomy after accident | 2017 |
| Diagnosis of neutropenia | 2018 |
| Diagnosis of OSAHS | 2019 |
| Scleroderma related to HCS | 2019 |
| PHARMACOLOGICAL | |
|---|---|
| PAINKILLERS | |
| DICLOFENAC 50 mg every 12 h | Treatment for pain management is established following the recommendations of the World Health Organization (WHO) in their analgesic ladder. The strategy consists of a series of drugs administered regularly and rescue medications for the occasions when routine treatment is insufficient for pain control. |
| PARACETAMOL 650 mg every 4 h | |
| TRAMADOL HYDROCHLORIDE 50 mg every 6 h (rescue medication) | |
| DAFALGAN (paracetamol) 1 gr every 6 h (rescue medication) | |
| METAMIZOLE 575 mg every 6 h | |
| FENTANYL patch 12 mcg every 72 h | |
| TRAMADOL HYDROCHLORIDE 200 mg at breakfast | |
| CELECOXIB 200 mg every 24 h | |
| SUPPLEMENTS | |
| OSSOPAN® 400 mg every 24 h | Supplements of calcium and vitamin D are administered together to boost their effect against osteoporosis. |
| SUPRADYN® PROTOVIT 9 drops every 24 h | |
| ORAL CALCIUM 1 gr every 24 h | |
| VITAMIN D3 8000 U every 24 h | |
| IDEOS® 500 mg every 12 h | |
| HIDROFEROL® 0.266 mg every 15 days | |
| ANTI-ACIDS | |
| OMEPRAZOLE 20 mg every 24 h | Due to the diagnosis of gastritis and the polypharmacy the patient receives, an antiacid is required to protect her stomach lining. |
| BISPHOSPHONATES | |
| ACREL® 35 mg weekly (risedronic acid) | The treatment with bisphosphonates began in 2006 and was cut off in 2017 after an improvement in densitometric parameters. |
| ADROVANCE 70 mg weekly (alendronic acid/colecalciferol) | |
| ANTIDEPRESSANTS | |
| XERISTAR® 60 mg every 12 h (duloxetine) | Antidepressant treatment begins in 2017 after the diagnosis of a prolonged adjustment disorder with clinical manifestations of depression and anxiety secondary to the pain and loss of autonomy after her traffic accident. |
| DEPRAX® 100 mg every 24 h in the evening (sertraline) | |
| DIAZEPAM 5 mg every 12 h | |
| STILNOX® 10 mg every 24 h in the evening (zolpidem tartrate) | |
| VENLAFAXINE 37.5 mg every 24 h | |
| ZARELIS® 75 every 24 h | |
| LAXATIVE | |
| MOVENTING® 25 mg at breakfast (Naloxegol) | The treatment of chronic constipation requires the regular use of laxatives. |
| SURGICAL | |
| Pes planus valgus | Surgical intervention in 1978 of the patient’s pes planus valgus determines the use of orthopedic insoles and footwear. |
| Surgical lumpectomy of a benign breast lesion | In the context of a fibrocystic mastopathy with fibroadenomas, surgical intervention is required for the correct evaluation of a breast lump. |
| SELF-CARE | |
| Daily basic walk Avoid overweight Maintain physical activity Intellectual activity | Self-care recommendations are designed to maintain as much physical and intellectual activity as possible and to avoid overweight as a preventative method against disease progression. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortés-Martín, J.; Sánchez-García, J.C.; Piqueras-Sola, B.; Rodríguez-Blanque, R.; Tovar-Gálvez, M.I.; Díaz-Rodríguez, L. Hajdu-Cheney Syndrome: Report of a Case in Spain. Diagnostics 2022, 12, 566. https://doi.org/10.3390/diagnostics12030566
Cortés-Martín J, Sánchez-García JC, Piqueras-Sola B, Rodríguez-Blanque R, Tovar-Gálvez MI, Díaz-Rodríguez L. Hajdu-Cheney Syndrome: Report of a Case in Spain. Diagnostics. 2022; 12(3):566. https://doi.org/10.3390/diagnostics12030566
Chicago/Turabian StyleCortés-Martín, Jonathan, Juan Carlos Sánchez-García, Beatriz Piqueras-Sola, Raquel Rodríguez-Blanque, María Isabel Tovar-Gálvez, and Lourdes Díaz-Rodríguez. 2022. "Hajdu-Cheney Syndrome: Report of a Case in Spain" Diagnostics 12, no. 3: 566. https://doi.org/10.3390/diagnostics12030566
APA StyleCortés-Martín, J., Sánchez-García, J. C., Piqueras-Sola, B., Rodríguez-Blanque, R., Tovar-Gálvez, M. I., & Díaz-Rodríguez, L. (2022). Hajdu-Cheney Syndrome: Report of a Case in Spain. Diagnostics, 12(3), 566. https://doi.org/10.3390/diagnostics12030566