
Detection of Unknown and Rare Pathogenic Variants in Antithrombin, Protein C and Protein S Deficiency Using High-Throughput Targeted Sequencing

 ,
,  ,
,
Abstract
1. Introduction
1.1. Factor V Leiden and Prothrombin Mutation
1.2. Anticoagulant Proteins
1.3. Genetic Testing for Genetic Suspicion of Thrombophilia
2. Materials and Methods
2.1. Patient Selection
2.2. DNA Isolation
2.3. Ion Torrent High-Throughput Sequencing
2.3.1. Primer Design
2.3.2. Library Preparation
2.3.3. Templating and Chip Loading
2.3.4. Sequencing
2.3.5. Data Analysis
2.4. MLPA Analysis
2.5. Sanger Sequencing
2.6. Statistical Evaluation
3. Results
3.1. Antithrombin Deficiency
3.2. Protein C Deficiency
3.3. Protein S Deficiency
4. Discussion
4.1. Antithrombin Deficiency
4.2. Protein C Deficiency
4.3. Protein S Deficiency
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Heit, J.A. Epidemiology of venous thromboembolism. Nat. Rev. Cardiol. 2015, 12, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Tagalakis, V.; Patenaude, V.; Kahn, S.R.; Suissa, S. Incidence of and mortality from venous thromboembolism in a real-world population: The Q-VTE Study Cohort. Am. J. Med. 2013, 126, 832.e13–832.e21. [Google Scholar] [CrossRef] [PubMed]
- Prandoni, P.; Lensing, A.W.A.; Cogo, A.; Cuppini, S.; Villalta, S.; Carta, M.; Cattelan, A.M.; Polistena, P.; Bernardi, E.; Prins, M.H. The long-term Clinical course of acute deep venous thrombosis. Ann. Intern. Med. 1996, 125, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Anderson, F.A.; Wheeler, H.B.; Goldberg, R.J.; Hosmer, D.W.; Patwardhan, N.A.; Jovanovic, B.; Forcier, A.; Dalen, J.E. A population-based perspective of the hospital incidence and case-fatality rates of deep vein thrombosis and pulmonary embolism: The Worcester DVT Study. Arch. Intern. Med. 1991, 151, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Souto, J.C.; Almassy, L.; Borell, M.; Blanco-Vaca, F.; Mateo, J.; Soria, J.M.; Coll, I.; Felices, R.; Stone, W.; Fontcuberta, J.; et al. Genetic susceptibility to thrombosis and its relationship to physiological risk factors: The GAIT study. Genetic Analysis of Idiopathic Thrombophilia. Am. J. Hum. Genet. 2000, 67, 1452–1459. [Google Scholar] [CrossRef] [PubMed]
- Stoneham, S.M.; Milne, K.M.; Nuttall, E.; Frew, G.H.; Sturrock, B.R.H.; Sivaloganathan, H.; Ladikou, E.E.; Drage, S.; Phillips, B.; Chevassut, T.J.T.; et al. Thrombotic risk in COVID-19: A case series and case–control study. Clin. Med. 2020, 20, e76–e81. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Cox, M.; Clegg, M.B. World distribution of factor V Leiden. Lancet 1995, 346, 1133–1134. [Google Scholar] [CrossRef]
- Dahlbäck, B. Advances in understanding pathogenic mechanisms of thrombophilic disorders. Blood 2008, 112, 19–27. [Google Scholar] [CrossRef]
- Emmerich, J.; Rosendaal, F.R.; Cattaneo, M.; Margaglione, M.; De Stefano, V.; Cumming, T.; Arruda, V.; Hillarp, A.; Reny, J.L. Combined effect of factor V Leiden and prothrombin 20210A on the risk of venous thromboembolism. Thromb. Haemost. 2001, 86, 809–816. [Google Scholar]
- Wypasek, E.; Corral, J.; Alhenc-Gelas, M.; Sydor, W.; Iwaniec, T.; Celińska-Lowenhoff, M.; Potaczek, D.P.; Blecharczyk, A.; Zawilska, K.; Musiał, J.; et al. Genetic characterization of antithrombin, protein C, and protein S deficiencies in Polish patients. Pol. Arch. Int. Med. 2017, 127, 512–523. [Google Scholar] [CrossRef][Green Version]
- García de Frutos, P.; Fuentes-Prior, P.; Hurtado, B.; Sala, N. Molecular basis of protein S deficiency. Thromb. Haemost. 2007, 98, 543–556. [Google Scholar] [PubMed]
- De Stefano, V.; Finazzi, G.; Mannucci, P.M. Inherited thrombophilia: Pathogenesis, clinical syndromes, and management. Blood 1996, 87, 3531–3544. [Google Scholar] [CrossRef] [PubMed]
- Di Minno, M.N.D.; Ambrosino, P.; Ageno, W.; Rosendaald, F.; Di Minno, G.; Dentali, F. Natural anticoagulants deficiency and the risk of venous thromboembolism: A meta-analysis of observational studies. Thromb. Res. 2015, 135, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Bucciarelli, P.; Rosendaal, F.R.; Tripodi, A.; Mannucci, P.M.; De Stefano, V.; Palareti, G.; Finazzi, G.; Baudo, F.; Quintavalla, R. Risk of venous thromboembolism and clinical manifestations in carriers of antithrombin, protein C, protein S deficiency, or activated protein C resistance: A multicenter collaborative family study. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1026–1033. [Google Scholar] [CrossRef]
- Colucci, G.; Tsakiris, D.A. Thrombophilia screening revisited: An issue of personalized medicine. J. Thromb. Thrombol. 2020, 49, 618–629. [Google Scholar] [CrossRef]
- Lotta, L.A.; Wang, M.; Yu, J.; Martinelli, I.; Yu, F.; Passamonti, S.M.; Consonni, D.; Pappalardo, E.; Menegatti, M.; Scherer, S.E.; et al. Identification of genetic risk variants for deep vein thrombosis by multiplexed next-generation sequencing of 186 hemostatic/pro-inflammatory genes. BMC Med. Genom. 2012, 5, 7. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Fidalgo, T.; Ribeiro, M.L. Added value of next-generation sequencing for haemostasis diagnosis. Thromb. Haemost. Res. 2017, 1, 1007. [Google Scholar]
- Simeoni, I.; Stephens, J.C.; Hu, F.; Deevi, S.V.V.; Megy, K.; Bariana, T.K.; Lentaigne, C.; Schulman, S.; Sivapalaratnam, S.; Vries, M.J.A.; et al. A high-throughput sequencing test for diagnosing inherited bleeding, thrombotic, and platelet disorders. Blood 2016, 127, 2791–2803. [Google Scholar] [CrossRef]
- Lee, E.J.; Dykas, D.J.; Leavitt, A.D.; Camire, R.M.; Ebberink, E.; García de Frutos, P.; Gnanasambandan, K.; Gu, S.X.; Huntington, J.A.; Lentz, S.R.; et al. Whole-exome sequencing in evaluation of patients with venous thromboembolism. Blood Adv. 2017, 1, 1224–1237. [Google Scholar] [CrossRef]
- Downes, K.; Megy, K.; Duarte, D.; Vries, M.; Gebhart, J.; Hofer, S.; Shamardina, O.; Deevi, S.V.V.; Stephens, J.; Mapeta, R.; et al. Diagnostic high-throughput sequencing of 2396 patients with bleeding, thrombotic, and platelet disorders. Blood 2019, 134, 2082–2091. [Google Scholar] [CrossRef] [PubMed]
- Bereczky, Z.; Gindele, R.; Speker, M.; Kállai, J. Deficiencies of the Natural Anticoagulants—Novel Clinical Laboratory Aspects of Thrombophilia Testing. EJIFCC 2016, 27, 130–146. [Google Scholar] [PubMed]
- Caspers, M.; Pavlova, A.; Driesen, J.; Harbrecht, U.; Klamroth, R.; Kadar, J.; Fischer, R.; Kemkes- Matthes, B.; Oldenburg, J. Deficiencies of antithrombin, protein C and protein S—Practical experience in genetic analysis of a large patient cohort. Thromb. Haemost. 2012, 108, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Castaldo, G.; Cerbone, A.M.; Guida, A.; Tandurella, I.; Ingino, R.; Tufano, A.; Ceglia, C.; Di Minno, M.N.D.; Ruocco, A.L.; Di Minno, G. Molecular analysis and genotype-phenotype correlation in patients with antithrombin deficiency from Southern Italy. Thromb. Haemost. 2012, 107, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Seo, J.Y.; Lee, K.O.; Bang, S.H.; Lee, S.T.; Ki, CH.S.; Kim, J.W.; Jung, CH.W.; Kim, D.K.; Kim, S.H. Distinct frequencies and mutation spectrums of genetic thrombophilia in Korea in comparison with other Asian countries both in patients with thromboembolism and in the general population. Haematologica 2014, 99, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Luxembourg, B.; Delev, D.; Geisen, C.; Spannagl, M.; Krause, M.; Miesbach, W.; Heller, CH.; Bergmann, F.; Schmeink, U.; Grossmann, R.; et al. Molecular basis of antithrombin deficiency. Thromb. Haemost. 2011, 105, 635–646. [Google Scholar]
- Provaznikova, D.; Matyskova, M.; Capova, I.; Grancarova, D.; Drbohlavova, E.; Slechtova, M.; Hrachovinova, I. Seventeen novel SERPINC1 variants causing hereditary antithrombin deficiency in a Czech population. Thromb. Res. 2020, 189, 39–41. [Google Scholar] [CrossRef]
- Trait, R.C.; Walker, I.D.; Perry, D.J.; Islam, S.I.A.M.; Daly, M.E.; McCall, F.; Conkie, J.A.; Carrell, R.W. Prevalence of antithrombin deficiency in the healthy population. Br. J. Haematol. 1994, 87, 106–112. [Google Scholar]
- Owen, M.C.; Borg, J.Y.; Soria, C.; Soria, J.; Caen, J.; Carrell, R.W. Heparin binding defect in a new antithrombin III variant: Rouen, 47 Arg to His. Blood 1987, 69, 1275–1279. [Google Scholar] [CrossRef]
- Gandrille, S.; Aiach, M. Identification of Mutations in 90 of 121 Consecutive Symptomatic French Patients With a Type I Protein C Deficiency. Blood 1995, 86, 2598–2605. [Google Scholar] [CrossRef]
- Grundy, C.B.; Chisholm, M.; Kakkar, V.V.; Cooper, D.N. A novel homozygous missense mutation in the protein C (PROC) gene causing recurrent venous thrombosis. Hum. Genet. 1992, 89, 683–684. [Google Scholar] [CrossRef] [PubMed]
- Grundy, C.B.; Melissari, E.; Lindo, V.; Scully, M.F.; Kakkar, V.V.; Cooper, D.N. Late-onset homozygous protein C deficiency. Lancet 1991, 338, 575–576. [Google Scholar] [CrossRef]
- Fidalgo, T.; Martinho, P.; Salvado, R.; Manco, L.; Oliveira, A.C.; Pinto, C.S.; Gonçalves, E.; Marques, D.; Sevivas, T.; Martins, N.; et al. Familial thrombotic risk based on the genetic background of Protein C Deficiency in a Portuguese Study. Eur. J. Hum. Genet. 2015, 95, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Long, G.L. Identification of two novel point mutations in the human protein S gene associated with familial protein S deficiency and thrombosis. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1407–1415. [Google Scholar] [CrossRef]
- Choi, J.; Kim, H.J.; Chang, M.H.; Choi, J.R.; Yoo, J.H. A Rare Splicing Mutation in the PROS1 Gene of a Korean Patient with Type I Hereditary Protein S Deficiency. Ann. Clin. Lab. Sci. 2011, 41, 397–400. [Google Scholar]



{kind=link}
{kind=link}
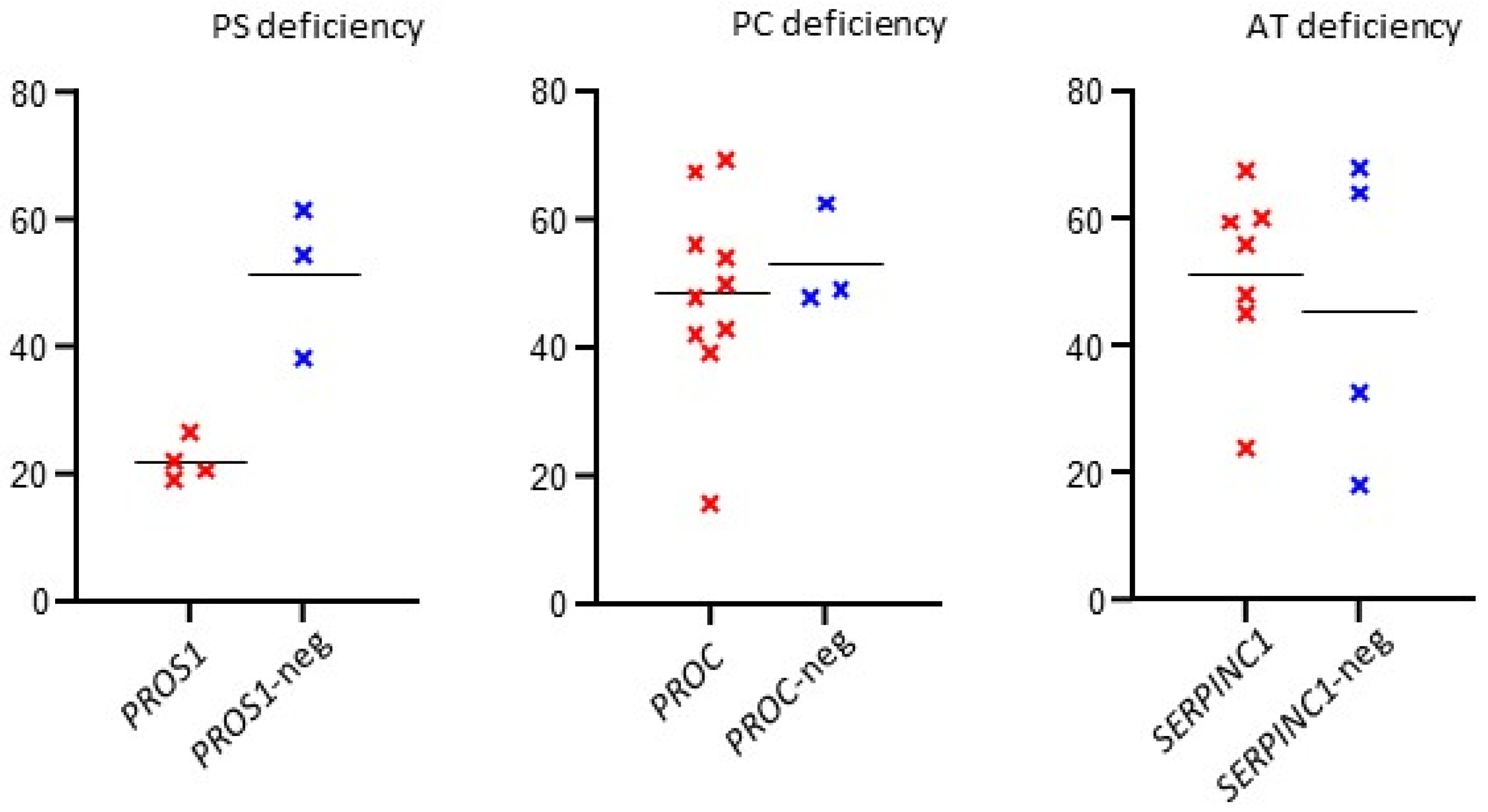
| Indication | Positive vs. Negative (t-Test) | Tested/(Variant Found) | Missense | Nonsense | Frameshift | Splicing | Stop Loss | Novel Variant |
|---|---|---|---|---|---|---|---|---|
| ATD | 0.6241 | 11 (7) 63.6% | 6 | 1 | – | – | – | 1 |
| PCD | 0.6286 | 13 (10) 76.9% | 8 | 1 | – | – | 1 | 3 |
| PSD | 0.0047 | 7 (4) 57.1% | 2 | – | 1 | 1 | – | 2 |
| Total | - | 31 (21) 67.7% | 16 (76.2%) | 2 (9.5%) | 1 (4.8%) | 1 (4.8%) | 1 (4.8%) | 6 (28.6%) |
| Proband Age (Sex) | Functional Test (%) | Functional Test Median (%) | Gene | Variant | Exon/Intron | Coordinate hg19 | Variant Effect | Zygosity/Novel * | In Silico Predictions | Family History | Classification |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1981 (F) | 60; 61; 63 | 60 | SERPINC1 | c.79T > C p.Trp27Arg | 2 | 1:173884020 rs1165816584 | missense | het | damaging | na | likely pathogenic |
| 1983 (F) | 67; 68 | 67.5 | SERPINC1 | c.133C > T p.Arg45Trp | 2 | 1:173883966 rs768704768 | missense | het | damaging | no | likely pathogenic |
| 1993 (F) | 56 | 56 | SERPINC1 | c.391C > T p.Leu131Phe | 2 | 1:173883708 rs121909567 | missense | het | damaging | yes | pathogenic |
| 1973 (M) | 54; 65 | 59.5 | SERPINC1 | c.236G > A p.Arg79His | 2 | 1:173883863 rs121909552 | missense | het | damaging | yes | likely pathogenic |
| 1971 (F) | 48 | 48 | SERPINC1 | c.236G > A p.Arg79His | 2 | 1:173883863 rs121909552 | missense | het | damaging | yes | likely pathogenic |
| 2004 (M) | 42; 45; 47 | 45 | SERPINC1 | c.398A > T p.Gln133Leu | 2 | 1:173883701 | missense | het * | damaging | no | pathogenic |
| 1932 (M) | 24 | 24 | SERPINC1 | c.685C > T p.Arg229Ter | 4 | 1:173879969 | nonsense | het | _ | yes | likely pathogenic |
| 1980 (F) | 50 | 50 | PROC | c.450C > A p.Tyr150Ter | 6 | 2:128180899 | nonsense | het * | _ | yes | likely pathogenic |
| 1971 (M) | 39; 45 | 42 | PROC | c.677A > T p.Gln226Leu | 7 | 2:128183802 | missense | het | damaging | yes | pathogenic |
| 1976 (F) | 41; 45 | 43 | PROC | c.677A > T p.Gln226Leu | 7 | 2:128183802 | missense | het | damaging | yes | pathogenic |
| 1973 (F) | 49; 56; 57 | 56 | PROC | c.715G > C p.Gly239Arg | 8 | 2:128184717 | missense | het * | damaging | yes | pathogenic |
| 1988 (F) | 54 | 54 | PROC | c.759C > A p.His253Gln | 8 | 2:128184761 rs1458669732 | missense | het | damaging | yes | likely pathogenic |
| 1971 (F) | 67; 68; 71; 71 | 69.5 | PROC | c.1301T > C p.Val434Ala | 9 | 2:128186437 | missense | het | damaging | yes | pathogenic |
| 1988 (F) | 65; 70 | 67.5 | PROC | c.1384T > C p.Ter462Gln | 9 | 2:128186520 rs370298954 | stop loss | het | _ | yes | likely pathogenic |
| 1978 (F) | 47; 48; 59 | 48 | PROC | c.1106C > T p.Pro369Leu | 9 | 2:128186242 rs1211098698 | missense | het | damaging/ tolerated | no | pathogenic |
| 1953 (M) | 4.4; 27.1 | 15.75 | PROC | c.866C > G p.Pro289Arg | 9 | 2:128186002 | missense | hom * | damaging | yes | likely pathogenic |
| 1994 (F) | 39 | 39 | PROC | c.1019C > T p.Thr340Met | 9 | 2:128186155 rs766261022 | missense | het | damaging | yes | pathogenic |
| 1963 (M) | 22 | 22 | PROS1 | c.1155 + 5G > A | 10 | 3:93611772 | splicing | het | _ | yes | pathogenic |
| 1981 (F) | 9; 9; 11; 12; 19; 20; 23; 27; 29 | 19 | PROS1 | c.1468delA p.Ile490fs | 12 | 3:93603596 | Frameshift Deletion (INDEL) | het * | _ | yes | likely pathogenic |
| 1953 (F) | 18; 23 | 20.5 | PROS1 | c.1916G > A p.Cys639Tyr | 15 | 3:93593204 | missense | het | damaging | yes | likely pathogenic |
| 2002 (M) | 22.1; 26.5; 27.9 | 26.5 | PROS1 | c.1931T > A p.Ile644Asn | 15 | 3:93593189 | missense | het * | possibly damaging/ damaging | yes | pathogenic |
| Proband Age (Sex) | Functional Test (%) | Functional Test Median (%) | Gene | MLPA/CNV |
|---|---|---|---|---|
| 1982 (F) | 24; 30; 30; 31; 31; 32; 33; 35; 35; 36; 37; 38 | 32.5 | SERPINC1 | neg |
| 1984 (F) | 58; 60; 64; 66; 68; 68; 69; 70; 70 | 68 | SERPINC1 | neg |
| 2019 (M) | 18 | 18 | SERPINC1 | neg |
| 1974 (F) | 64 | 64 | SERPINC1 | neg |
| 1999 (F) | 44; 54 | 49 | PROC | neg |
| 2003 (M) | 62; 63 | 62.5 | PROC | neg |
| 1993 (F) | 41; 45; 48; 48; 49; 49 | 48 | PROC | neg |
| 1983 (F) | 31; 45.6 | 38.3 | PROS1 | neg |
| 1952 (M) | 61.2; 61.9 | 61.55 | PROS1 | neg |
| 1990 (F) | 45; 64 | 54.5 | PROS1 | neg |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vrtel, P.; Slavik, L.; Vodicka, R.; Stellmachova, J.; Prochazka, M.; Prochazkova, J.; Ulehlova, J.; Rohon, P.; Simurda, T.; Stasko, J.; et al. Detection of Unknown and Rare Pathogenic Variants in Antithrombin, Protein C and Protein S Deficiency Using High-Throughput Targeted Sequencing. Diagnostics 2022, 12, 1060. https://doi.org/10.3390/diagnostics12051060
Vrtel P, Slavik L, Vodicka R, Stellmachova J, Prochazka M, Prochazkova J, Ulehlova J, Rohon P, Simurda T, Stasko J, et al. Detection of Unknown and Rare Pathogenic Variants in Antithrombin, Protein C and Protein S Deficiency Using High-Throughput Targeted Sequencing. Diagnostics. 2022; 12(5):1060. https://doi.org/10.3390/diagnostics12051060
Chicago/Turabian StyleVrtel, Petr, Ludek Slavik, Radek Vodicka, Julia Stellmachova, Martin Prochazka, Jana Prochazkova, Jana Ulehlova, Peter Rohon, Tomas Simurda, Jan Stasko, and et al. 2022. "Detection of Unknown and Rare Pathogenic Variants in Antithrombin, Protein C and Protein S Deficiency Using High-Throughput Targeted Sequencing" Diagnostics 12, no. 5: 1060. https://doi.org/10.3390/diagnostics12051060
APA StyleVrtel, P., Slavik, L., Vodicka, R., Stellmachova, J., Prochazka, M., Prochazkova, J., Ulehlova, J., Rohon, P., Simurda, T., Stasko, J., Martinkova, I., & Vrtel, R. (2022). Detection of Unknown and Rare Pathogenic Variants in Antithrombin, Protein C and Protein S Deficiency Using High-Throughput Targeted Sequencing. Diagnostics, 12(5), 1060. https://doi.org/10.3390/diagnostics12051060