
The Mesenchymal Niche in Myelodysplastic Syndromes

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
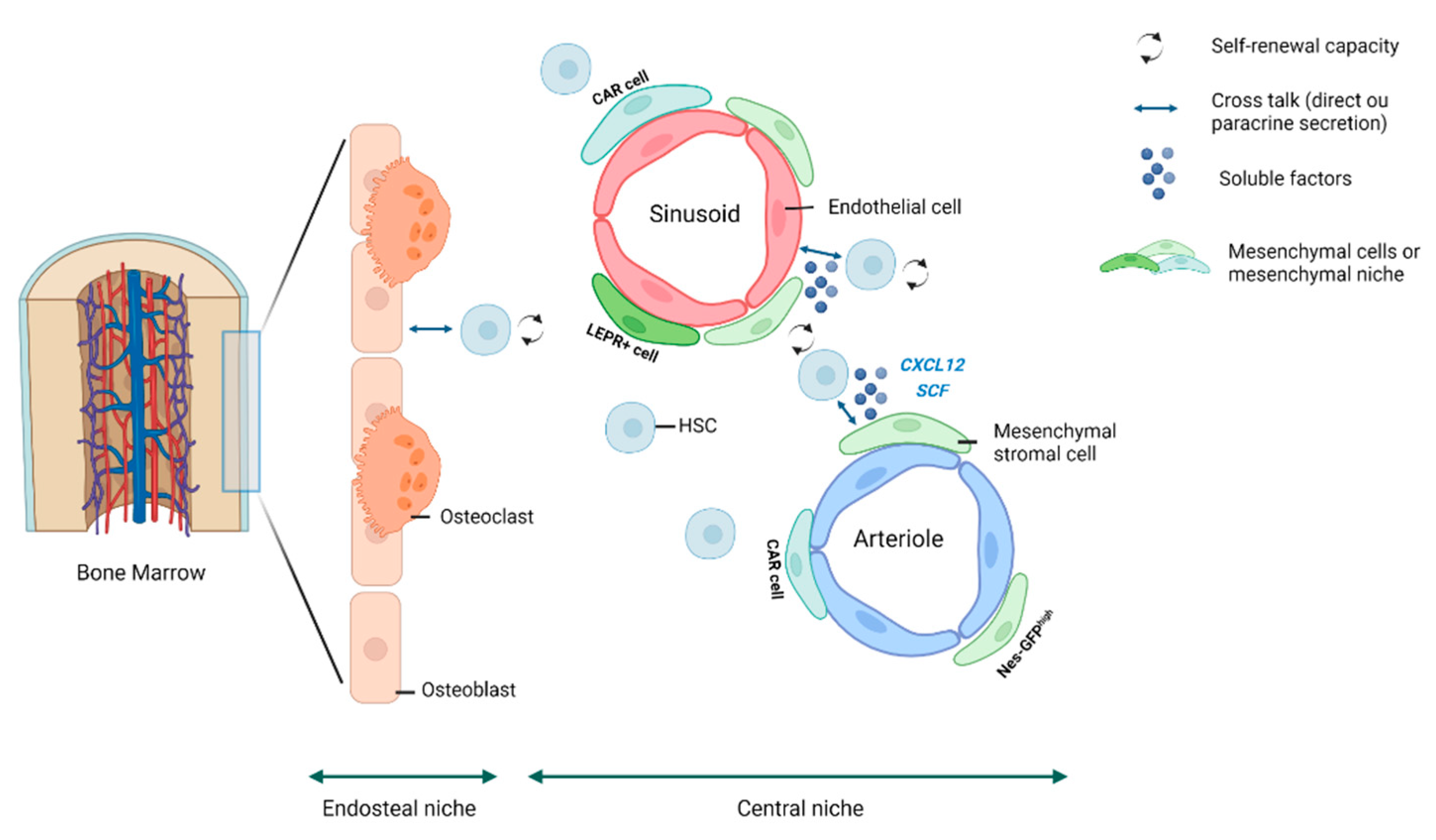
2. The HSC Niche
3. The Mesenchymal Niche
4. The Mesenchymal Niche in MDS
4.1. The Niche-Induced Leukemogenesis Model
4.2. The Niche-Facilitated Leukemogenesis Model: The Fundamental Role of Inflammation
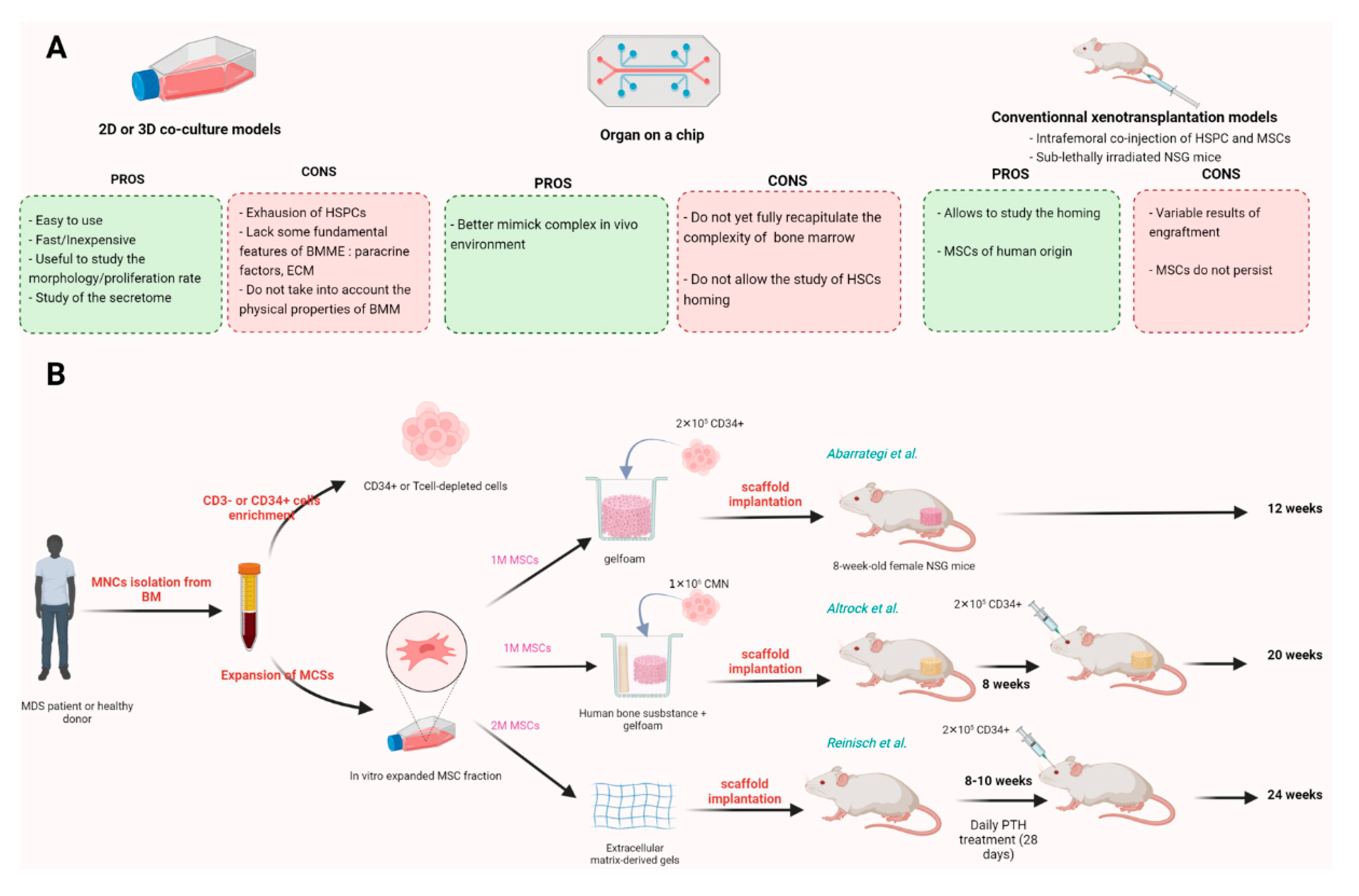
5. In Vitro and In Vivo MSCs Based Models
5.1. In Vitro Models
5.2. In Vivo Models
5.2.1. Xenotransplantation Models
5.2.2. Humanized Bone Marrow-like Structures
6. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Doulatov, S.; Notta, F.; Laurenti, E.; Dick, J.E. Hematopoiesis: A Human Perspective. Cell Stem Cell 2012, 10, 120–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orford, K.W.; Scadden, D.T. Deconstructing stem cell self-renewal: Genetic insights into cell-cycle regulation. Nat. Rev. Genet. 2008, 9, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Schofield, R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 1978, 4, 4–25. [Google Scholar]
- Kiel, M.J.; Yilmaz, O.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. SLAM Family Receptors Distinguish Hematopoietic Stem and Progenitor Cells and Reveal Endothelial Niches for Stem Cells. Cell 2005, 121, 1109–1121. [Google Scholar] [CrossRef] [Green Version]
- Laurenti, E.; Gottgens, B. From haematopoietic stem cells to complex differentiation landscapes. Nature 2018, 553, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; MacArthur, B.; Lira, S.A.; Scadden, D.T.; Ma’Ayan, A.; Enikolopov, G.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef]
- Kunisaki, Y.; Bruns, I.; Scheiermann, C.; Ahmed, J.; Pinho, S.; Zhang, D.; Mizoguchi, T.; Wei, Q.; Lucas, D.; Ito, K.; et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 2013, 502, 637–643. [Google Scholar] [CrossRef] [Green Version]
- Taichman, R.S.; Reilly, M.J.; Emerson, S.G. Human osteoblasts support human hematopoietic progenitor cells in vitro bone marrow cultures. Blood 1996, 87, 518–524. [Google Scholar] [CrossRef]
- Visnjic, D.; Kalajzic, Z.; Rowe, D.W.; Katavic, V.; Lorenzo, J.; Aguila, H.L. Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood 2004, 103, 3258–3264. [Google Scholar] [CrossRef]
- Zhang, J.; Niu, C.; Ye, L.; Huang, H.; He, X.; Tong, W.-G.; Ross, J.; Haug, J.; Johnson, T.; Feng, J.Q.; et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 2003, 425, 836–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.Y.; Scadden, D.T.; Kronenberg, H.M. Role of the Osteoblast Lineage in the Bone Marrow Hematopoietic Niches. J. Bone Miner. Res. 2009, 24, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Tao, F.; Venkatraman, A.; Li, Z.; Smith, S.E.; Unruh, J.; Chen, S.; Ward, C.; Qian, P.; Perry, J.M.; et al. N-Cadherin-Expressing Bone and Marrow Stromal Progenitor Cells Maintain Reserve Hematopoietic Stem Cells. Cell Rep. 2019, 26, 652–669.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the Hematopoietic Stem Cell Pool by CXCL12-CXCR4 Chemokine Signaling in Bone Marrow Stromal Cell Niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Saunders, T.; Enikolopov, G.; Morrison, S.J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012, 481, 457–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, A.T.; Butler, J.M.; Nolan, D.J.; Kranz, A.; Iida, K.; Kobayashi, M.; Kopp, H.-G.; Shido, K.; Petit, I.; Yanger, K.; et al. Engraftment and Reconstitution of Hematopoiesis Is Dependent on VEGFR2-Mediated Regeneration of Sinusoidal Endothelial Cells. Cell Stem Cell 2009, 4, 263–274. [Google Scholar] [CrossRef] [Green Version]
- Bessy, T.; Itkin, T.; Passaro, D. Bioengineering the Bone Marrow Vascular Niche. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Passaro, D.; Irigoyen, M.; Catherinet, C.; Gachet, S.; Jesus, C.D.C.D.; Lasgi, C.; Quang, C.T.; Ghysdael, J. CXCR4 Is Required for Leukemia-Initiating Cell Activity in T Cell Acute Lymphoblastic Leukemia. Cancer Cell 2015, 27, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Duarte, D.; Hawkins, E.; Akinduro, O.; Ang, H.; De Filippo, K.; Kong, I.Y.; Haltalli, M.; Ruivo, N.; Straszkowski, L.; Vervoort, S.J.; et al. Inhibition of Endosteal Vascular Niche Remodeling Rescues Hematopoietic Stem Cell Loss in AML. Cell Stem Cell 2017, 22, 64–77.e6. [Google Scholar] [CrossRef] [Green Version]
- Passaro, D.; Di Tullio, A.; Abarrategi, A.; Rouault-Pierre, K.; Foster, K.; Ariza-McNaughton, L.; Montaner, B.; Chakravarty, P.; Bhaw, L.; Diana, G.; et al. Increased Vascular Permeability in the Bone Marrow Microenvironment Contributes to Disease Progression and Drug Response in Acute Myeloid Leukemia. Cancer Cell 2017, 32, 324–341.e6. [Google Scholar] [CrossRef] [Green Version]
- Frieedenstein, A.J.; Petrakova, K.V.; Kurolesova, A.L.; Frolova, G.P. Heterotopic of bone marrow. Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation 1968, 6, 230–247. [Google Scholar]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Loss of Cxcl12/Sdf-1 in Adult Mice Decreases the Quiescent State of Hematopoietic Stem/Progenitor Cells and Alters the Pattern of Hematopoietic Regeneration after Myelosuppression. Available online: https://pubmed-ncbi-nlm-nih-gov.proxy.insermbiblio.inist.fr/20833981/ (accessed on 2 June 2022).
- Wolock, S.L.; Krishnan, I.; Tenen, D.E.; Matkins, V.; Camacho, V.; Patel, S.; Agarwal, P.; Bhatia, R.; Tenen, D.G.; Klein, A.M.; et al. Mapping Distinct Bone Marrow Niche Populations and Their Differentiation Paths. Cell Rep. 2019, 28, 302–311.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Puerta, G.J.; Marchal, J.A.; López-Ruiz, E.; Gálvez-Martín, P. Role of Mesenchymal Stromal Cells as Therapeutic Agents: Potential Mechanisms of Action and Implications in Their Clinical Use. J. Clin. Med. 2020, 9, 445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Self-Renewing Osteoprogenitors in Bone Marrow Sinusoids Can Organize a Hematopoietic Microenvironment. Available online: https://pubmed-ncbi-nlm-nih-gov.proxy.insermbiblio.inist.fr/17956733/ (accessed on 2 June 2022).
- Reinisch, A.; Thomas, D.; Corces, M.R.; Zhang, X.; Gratzinger, D.; Hong, W.-J.; Schallmoser, K.; Strunk, D.; Majeti, R. A humanized bone marrow ossicle xenotransplantation model enables improved engraftment of healthy and leukemic human hematopoietic cells. Nat. Med. 2016, 22, 812–821. [Google Scholar] [CrossRef]
- Abarrategi, A.; Foster, K.; Hamilton, A.; Mian, S.A.; Passaro, D.; Gribben, J.; Mufti, G.; Bonnet, D. Versatile humanized niche model enables study of normal and malignant human hematopoiesis. J. Clin. Investig. 2017, 127, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Mossner, M.; Jann, J.-C.; Wittig, J.; Nolte, F.; Fey, S.; Nowak, V.; Obländer, J.; Pressler, J.; Palme, I.; Xanthopoulos, C.; et al. Mutational hierarchies in myelodysplastic syndromes dynamically adapt and evolve upon therapy response and failure. Blood 2016, 128, 1246–1259. [Google Scholar] [CrossRef] [Green Version]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Flynn, C.M.; Kaufman, D. Donor cell leukemia: Insight into cancer stem cells and the stem cell niche. Blood 2006, 109, 2688–2692. [Google Scholar] [CrossRef] [Green Version]
- Raaijmakers, M.H.G.P.; Mukherjee, S.; Guo, S.; Zhang, S.; Kobayashi, T.; Schoonmaker, J.A.; Ebert, B.L.; Al-Shahrour, F.; Hasserjian, R.P.; Scadden, E.O.; et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 2010, 464, 852–857. [Google Scholar] [CrossRef] [Green Version]
- Vasta, L.M.; Khan, N.E.; Higgs, C.P.; Harney, L.A.; Carr, A.G.; Harris, A.K.; Schultz, K.A.P.; McMaster, M.L.; Stewart, D.R. Hematologic indices in individuals with pathogenic germline DICER1 variants. Blood Adv. 2021, 5, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, C.; Muntion, S.; Roson, B.; Blanco, B.; Lopez-Villar, O.; Carrancio, S.; Sanchez-Guijo, F.M.; Diez-Campelo, M.; Alvarez-Fernandez, S.; Sarasquete, M.E.; et al. Impaired expression of DICER, DROSHA, SBDS and some microRNAs in mesenchymal stromal cells from myelodysplastic syndrome patients. Haematologica 2012, 97, 1218–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zambetti, N.A.; Ping, Z.; Chen, S.; Kenswil, K.J.; Mylona, M.A.; Sanders, M.A.; Hoogenboezem, R.M.; Bindels, E.M.; Adisty, M.N.; Van Strien, P.M.; et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-leukemia. Cell Stem Cell 2016, 19, 613–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geyh, S.; Oz, S.; Cadeddu, R.-P.; Frobel, J.; Bruckner, B.; Kundgen, A.; Fenk, R.; Bruns, I.; Zilkens, C.; Hermsen, D.; et al. Insufficient stromal support in MDS results from molecular and functional deficits of mesenchymal stromal cells. Leukemia 2013, 27, 1841–1851. [Google Scholar] [CrossRef] [Green Version]
- Li, A.J.; Calvi, L.M. The microenvironment in myelodysplastic syndromes: Niche-mediated disease initiation and progression. Exp. Hematol. 2017, 55, 3–18. [Google Scholar] [CrossRef]
- Jann, J.-C. Mesenchymal Stromal Cells (MSCs) from Myelodysplastic Syndromes (MDS) Are Not Clonally Mutated In Vivo. Présenté à 63rd ASH Annual Meeting and Exposition, déc. 2021. Available online: https://ash.confex.com/ash/2021/webprogram/Paper150244.html (accessed on 30 December 2021).
- Blau, O.; Baldus, C.D.; Hofmann, W.-K.; Thiel, G.; Nolte, F.; Burmeister, T.; Türkmen, S.; Benlasfer, O.; Schümann, E.; Sindram, A.; et al. Mesenchymal stromal cells of myelodysplastic syndrome and acute myeloid leukemia patients have distinct genetic abnormalities compared with leukemic blasts. Blood 2011, 118, 5583–5592. [Google Scholar] [CrossRef] [Green Version]
- Blau, O.; Hofmann, W.-K.; Baldus, C.D.; Thiel, G.; Serbent, V.; Schümann, E.; Thiel, E.; Blau, I.W. Chromosomal aberrations in bone marrow mesenchymal stroma cells from patients with myelodysplastic syndrome and acute myeloblastic leukemia. Exp. Hematol. 2007, 35, 221–229. [Google Scholar] [CrossRef]
- Poon, Z.; Dighe, N.; Venkatesan, S.S.; Cheung, A.M.S.; Fan, X.; Bari, S.; Hota, M.; Ghosh, S.; Hwang, W.Y.K. Bone marrow MSCs in MDS: Contribution towards dysfunctional hematopoiesis and potential targets for disease response to hypomethylating therapy. Leukemia 2018, 33, 1487–1500. [Google Scholar] [CrossRef] [Green Version]
- Ping, Z.; Chen, S.; Hermans, S.J.F.; Kenswil, K.J.G.; Feyen, J.; Van Dijk, C.; Bindels, E.M.J.; Mylona, A.M.; Adisty, N.M.; Hoogenboezem, R.M.; et al. Activation of NF-κB driven inflammatory programs in mesenchymal elements attenuates hematopoiesis in low-risk myelodysplastic syndromes. Leukemia 2018, 33, 536–541. [Google Scholar] [CrossRef]
- Flores-Figueroa, E.; Montesinos, J.J.; Flores-Guzmán, P.; Gutiérrez-Espíndola, G.; Arana-Trejo, R.M.; Castillo-Medina, S.; Pérez-Cabrera, A.; Hernández-Estévez, E.; Arriaga, L.; Mayani, H. Functional analysis of myelodysplastic syndromes-derived mesenchymal stem cells. Leuk. Res. 2008, 32, 1407–1416. [Google Scholar] [CrossRef]
- Dong, L.; Yu, W.-M.; Zheng, H.; Loh, M.L.; Bunting, S.T.; Pauly, M.; Huang, G.; Zhou, M.; Broxmeyer, H.E.; Scadden, D.T.; et al. Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature 2016, 539, 304–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018, 9, 586. [Google Scholar] [CrossRef] [PubMed]
- Sperling, A.; Gibson, C.J.; Ebert, A.S.S.C.J.G.B.L. The genetics of myelodysplastic syndrome: From clonal haematopoiesis to secondary leukaemia. Nat. Cancer 2017, 17, 5–19. [Google Scholar] [CrossRef] [Green Version]
- Muto, T.; Walker, C.S.; Choi, K.; Hueneman, K.; Smith, M.A.; Gul, Z.; Garcia-Manero, G.; Ma, A.; Zheng, Y.; Starczynowski, D.T. Adaptive response to inflammation contributes to sustained myelopoiesis and confers a competitive advantage in myelodysplastic syndrome HSCs. Nat. Immunol. 2020, 21, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Hormaechea-Agulla, D.; Matatall, K.A.; Le, D.T.; Kain, B.; Long, X.; Kus, P.; Jaksik, R.; Challen, G.A.; Kimmel, M.; King, K.Y. Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFNγ signaling. Cell Stem Cell 2021, 28, 1428–1442.e6. [Google Scholar] [CrossRef] [PubMed]
- Basiorka, A.A.; McGraw, K.L.; Eksioglu, E.A.; Chen, X.; Johnson, J.; Zhang, L.; Zhang, Q.; Irvine, B.A.; Cluzeau, T.; Sallman, D.A.; et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 2016, 128, 2960–2975. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zambetti, N.A.; Bindels, E.M.J.; Kenswill, K.; Mylona, A.M.; Adisty, N.M.; Hoogenboezem, R.M.; Sanders, M.A.; Cremers, E.M.P.; Westers, T.M.; et al. Massive parallel RNA sequencing of highly purified mesenchymal elements in low-risk MDS reveals tissue-context-dependent activation of inflammatory programs. Leukemia 2016, 30, 1938–1942. [Google Scholar] [CrossRef] [Green Version]
- Medyouf, H.; Mossner, M.; Jann, J.-C.; Nolte, F.; Raffel, S.; Herrmann, C.; Lier, A.; Eisen, C.; Nowak, V.; Zens, B.; et al. Myelodysplastic Cells in Patients Reprogram Mesenchymal Stromal Cells to Establish a Transplantable Stem Cell Niche Disease Unit. Cell Stem Cell 2014, 14, 824–837. [Google Scholar] [CrossRef] [Green Version]
- Geyh, S.; Rodríguez-Paredes, M.; Jager, P.; Koch, A.; Bormann, F.; Gutekunst, J.; Zilkens, C.; Germing, U.; Kobbe, G.; Lyko, F.; et al. Transforming growth factor β1-mediated functional inhibition of mesenchymal stromal cells in myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2018, 103, 1462–1471. [Google Scholar] [CrossRef] [Green Version]
- Ham, J.; Lever, L.; Fox, M.; Reagan, M.R. In Vitro 3D Cultures to Reproduce the Bone Marrow Niche. JBMR Plus 2019, 3, e10228. [Google Scholar] [CrossRef] [Green Version]
- Vernon, A.R.; Pemberton, R.M.; Morse, H.R. A novel in vitro 3D model of the human bone marrow to bridge the gap between in vitro and in vivo genotoxicity testing. Mutagenesis 2022, 37, 112–129. [Google Scholar] [CrossRef] [PubMed]
- Rosalem, G.S.; Torres, L.A.G.; Casas, E.B.D.L.; Mathias, F.A.S.; Ruiz, J.C.; Carvalho, M.G.R. Microfluidics and organ-on-a-chip technologies: A systematic review of the methods used to mimic bone marrow. PLoS ONE 2020, 15, e0243840. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E. Is it Time for Reviewer 3 to Request Human Organ Chip Experiments Instead of Animal Validation Studies? Adv. Sci. 2020, 7, 2002030. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Clappier, E.; Gerby, B.; Sigaux, F.; Delord, M.; Touzri, F.; Hernandez, L.; Ballerini, P.; Baruchel, A.; Pflumio, F.; Soulier, J. Clonal selection in xenografted human T cell acute lymphoblastic leukemia recapitulates gain of malignancy at relapse. J. Exp. Med. 2011, 208, 653–661. [Google Scholar] [CrossRef] [Green Version]
- Mian, S.A.; Rouault-Pierre, K.; Smith, A.E.; Seidl, T.; Pizzitola, I.; Kizilors, A.; Kulasekararaj, A.G.; Bonnet, D.; Mufti, G.J. SF3B1 mutant MDS-initiating cells may arise from the haematopoietic stem cell compartment. Nat. Commun. 2015, 6, 10004. [Google Scholar] [CrossRef]
- Nilsson, L.; Åstrand-Grundström, I.; Anderson, K.; Arvidsson, I.; Hokland, P.; Bryder, D.; Kjeldsen, L.; Johansson, B.; Hellström-Lindberg, E.; Hast, R.; et al. Involvement and functional impairment of the CD34+CD38−Thy-1+ hematopoietic stem cell pool in myelodysplastic syndromes with trisomy 8. Blood 2002, 100, 259–267. [Google Scholar] [CrossRef]
- Kerbauy, D.M.B.; Lesnikov, V.; Torok-Storb, B.; Bryant, E.; Deeg, H.J. Engraftment of distinct clonal MDS-derived hematopoietic precursors in NOD/SCID-β2-microglobulin-deficient mice after intramedullary transplantation of hematopoietic and stromal cells. Blood 2004, 104, 2202–2203. [Google Scholar] [CrossRef]
- Thanopoulou, E.; Cashman, J.; Kakagianne, T.; Eaves, A.; Zoumbos, N.; Eaves, C. Engraftment of NOD/SCID-β2 microglobulin null mice with multilineage neoplastic cells from patients with myelodysplastic syndrome. Blood 2004, 103, 4285–4293. [Google Scholar] [CrossRef] [Green Version]
- Benito, A.I.; Bryant, E.; Loken, M.R.; Sale, G.E.; Nash, R.A.; Gass, M.J.; Deeg, H. NOD/SCID mice transplanted with marrow from patients with myelodysplastic syndrome (MDS) show long-term propagation of normal but not clonal human precursors. Leuk. Res. 2002, 27, 425–436. [Google Scholar] [CrossRef]
- Côme, C.; Balhuizen, A.; Bonnet, D.; Porse, B.T. Myelodysplastic syndrome patient-derived xenografts: From no options to many. Haematologica 2020, 105, 864–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Rongvaux, A.; Taylor, A.; Jiang, T.; Tebaldi, T.; Balasubramanian, K.; Bagale, A.; Terzi, Y.K.; Gbyli, R.; Wang, X.; et al. A highly efficient and faithful MDS patient-derived xenotransplantation model for pre-clinical studies. Nat. Commun. 2019, 10, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouault-Pierre, K.; Mian, S.A.; Goulard, M.; Abarrategi, A.; Di Tulio, A.; Smith, A.E.; Mohamedali, A.; Best, S.; Nloga, A.-M.; Kulasekararaj, A.G.; et al. Preclinical modeling of myelodysplastic syndromes. Leukemia 2017, 31, 2702–2708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altrock, E.; Sens-Albert, C.; Jann, J.-C.; Flach, J.; Riabov, V.; Schmitt, N.; Xu, Q.; Mehralivand, A.; Hecht, A.; Steiner, L.; et al. Humanized three-dimensional scaffold xenotransplantation models for myelodysplastic syndromes. Exp. Hematol. 2021, 107, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Mian, S.A.; Abarrategi, A.; Kong, K.L.; Rouault-Pierre, K.; Wood, H.; Oedekoven, C.A.; Smith, A.E.; Batsivari, A.; Ariza-McNaughton, L.; Johnson, P.; et al. Ectopic Humanized Mesenchymal Niche in Mice Enables Robust Engraftment of Myelodysplastic Stem Cells. Blood Cancer Discov. 2020, 2, 135–145. [Google Scholar] [CrossRef]
- Antonelli, A.; Noort, W.A.; Jaques, J.; De Boer, B.; De Jong-Korlaar, R.; Brouwers-Vos, A.Z.; Lubbers-Aalders, L.; Van Velzen, J.F.; Bloem, A.C.; Yuan, H.; et al. Establishing human leukemia xenograft mouse models by implanting human bone marrow–like scaffold-based niches. Blood 2016, 128, 2949–2959. [Google Scholar] [CrossRef] [Green Version]
- Freeman, F.; McNamara, L.M. Endochondral Priming: A Developmental Engineering Strategy for Bone Tissue Regeneration. Tissue Eng. Part B Rev. 2017, 23, 128–141. [Google Scholar] [CrossRef]
- Burastero, G.; Scarfì, S.; Ferraris, C.; Fresia, C.; Sessarego, N.; Fruscione, F.; Monetti, F.; Scarfò, F.; Schupbach, P.; Podestà, M.; et al. The association of human mesenchymal stem cells with BMP-7 improves bone regeneration of critical-size segmental bone defects in athymic rats. Bone 2010, 47, 117–126. [Google Scholar] [CrossRef]
- Xu, C.; Gao, X.; Wei, Q.; Nakahara, F.; Zimmerman, S.E.; Mar, J.; Frenette, P.S. Stem cell factor is selectively secreted by arterial endothelial cells in bone marrow. Nat. Commun. 2018, 9, 2449. [Google Scholar] [CrossRef]
- Cooper, T.; Sefton, M. Fibronectin coating of collagen modules increases in vivo HUVEC survival and vessel formation in SCID mice. Acta Biomater. 2011, 7, 1072–1083. [Google Scholar] [CrossRef] [Green Version]
- Koike, N.; Fukumura, D.; Gralla, O.; Au, P.; Schechner, J.S.; Jain, R.K. Creation of long-lasting blood vessels. Nature 2004, 428, 138–139. [Google Scholar] [CrossRef] [PubMed]
- Passaro, D.; Abarrategi, A.; Foster, K.; Ariza-McNaughton, L.; Bonnet, D. Bioengineering of Humanized Bone Marrow Microenvironments in Mouse and Their Visualization by Live Imaging. J. Vis. Exp. 2017, e55914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Manero, G.; Daver, N.G.; Xu, J.; Chao, M.; Chung, T.; Tan, A.; Wang, V.; Wei, A.; Vyas, P.; Sallman, D.A. Magrolimab + azacitidine versus azacitidine + placebo in untreated higher risk (HR) myelodysplastic syndrome (MDS): The phase 3, randomized, ENHANCE study. J. Clin. Oncol. 2021, 39, TPS7055. [Google Scholar] [CrossRef]
- Ball, B.; Famulare, C.A.; Stein, E.M.; Tallman, M.S.; Derkach, A.; Roshal, M.; Gill, S.I.; Manning, B.M.; Koprivnikar, J.; McCloskey, J.; et al. Venetoclax and hypomethylating agents (HMAs) induce high response rates in MDS, including patients after HMA therapy failure. Blood Adv. 2020, 4, 2866–2870. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef] [PubMed]
- Kubasch, A.S.; Fenaux, P.; Platzbecker, U. Development of luspatercept to treat ineffective erythropoiesis. Blood Adv. 2021, 5, 1565–1575. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friedrich, C.; Kosmider, O. The Mesenchymal Niche in Myelodysplastic Syndromes. Diagnostics 2022, 12, 1639. https://doi.org/10.3390/diagnostics12071639
Friedrich C, Kosmider O. The Mesenchymal Niche in Myelodysplastic Syndromes. Diagnostics. 2022; 12(7):1639. https://doi.org/10.3390/diagnostics12071639
Chicago/Turabian StyleFriedrich, Chloé, and Olivier Kosmider. 2022. "The Mesenchymal Niche in Myelodysplastic Syndromes" Diagnostics 12, no. 7: 1639. https://doi.org/10.3390/diagnostics12071639