
Differentiating Polycystic Ovary Syndrome from Adrenal Disorders

Abstract
:1. Introduction
2. Pathophysiology of Hyperandrogenism
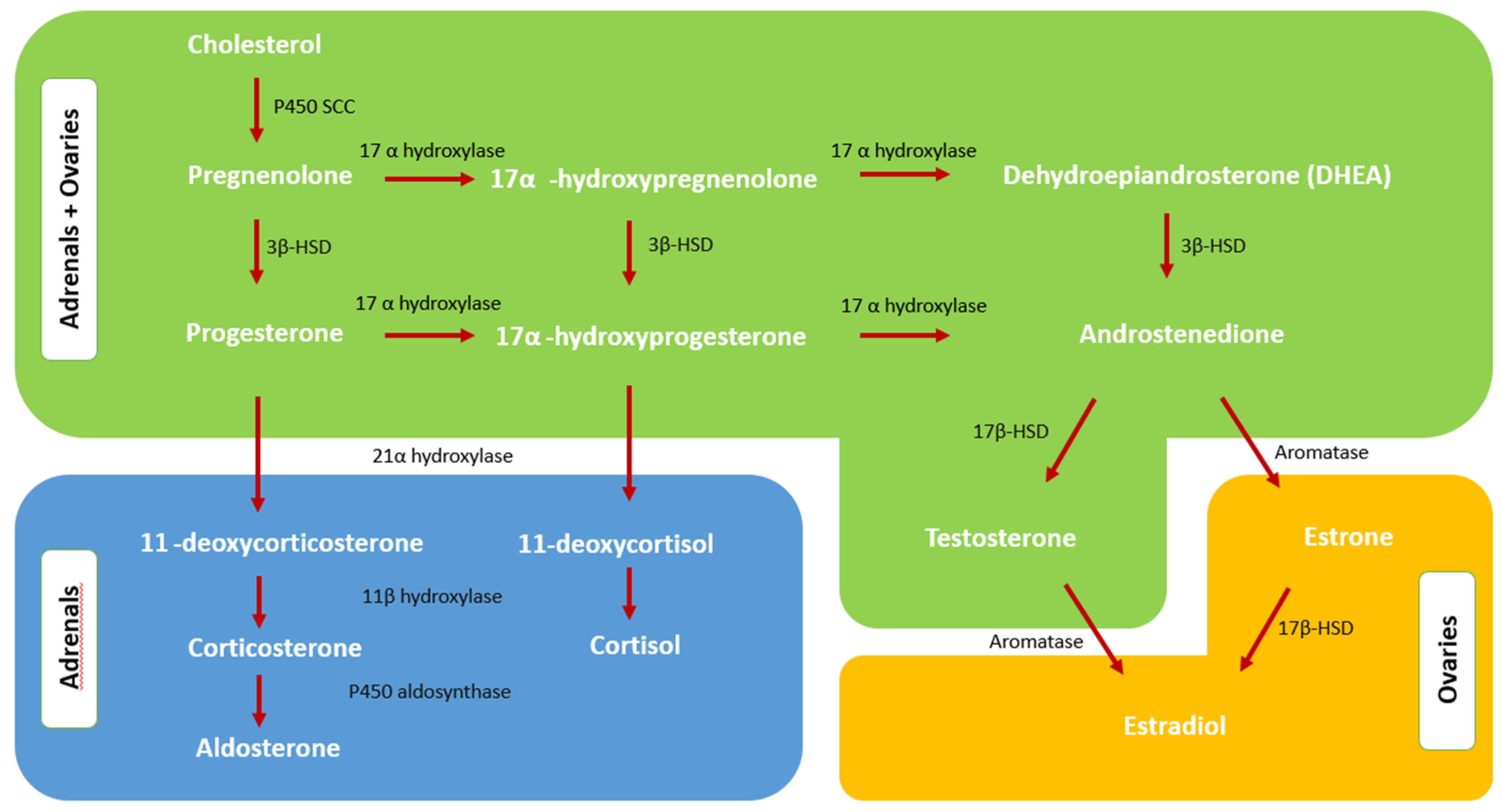
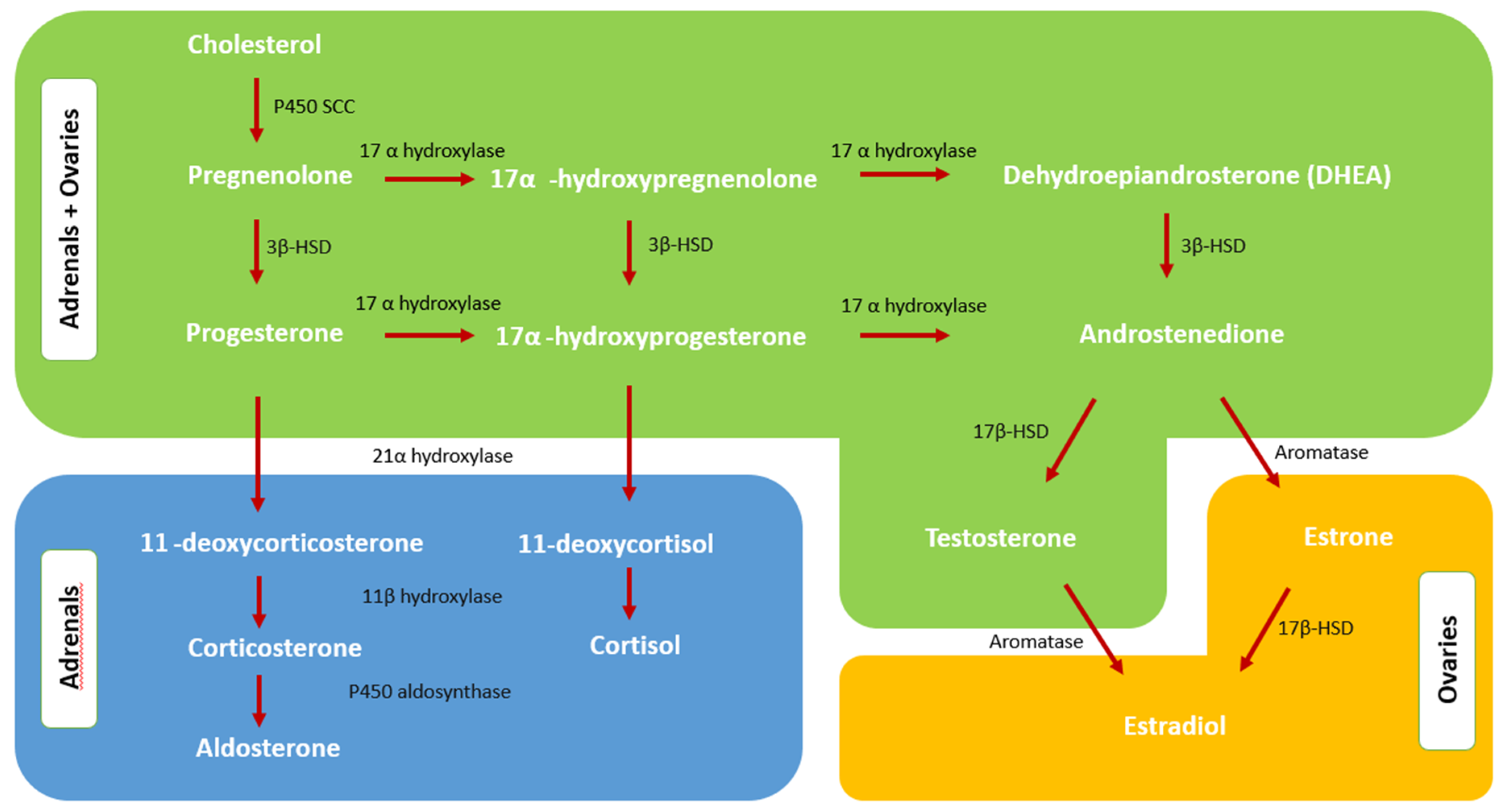
2.1. Normal Androgen Metabolism
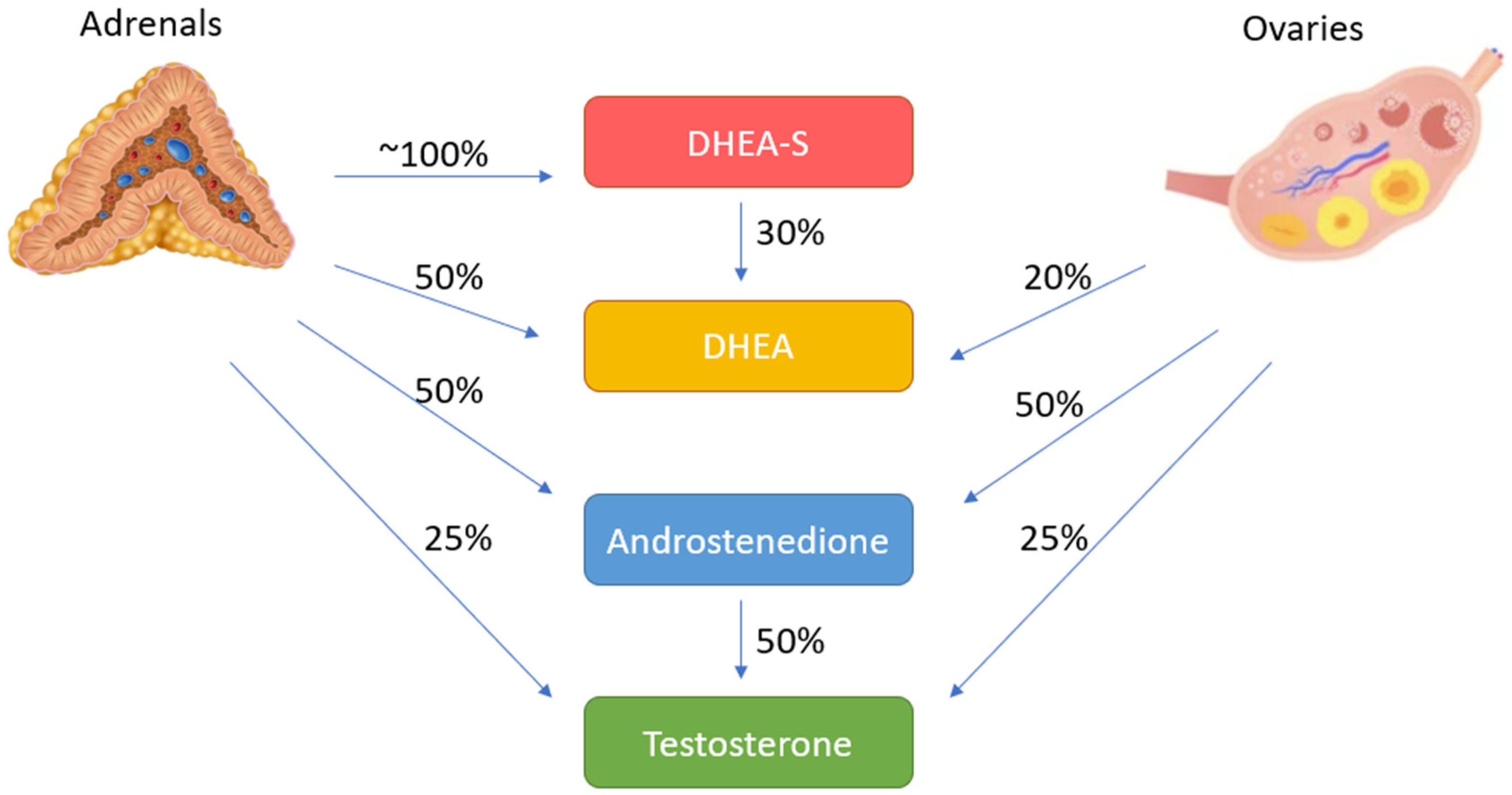
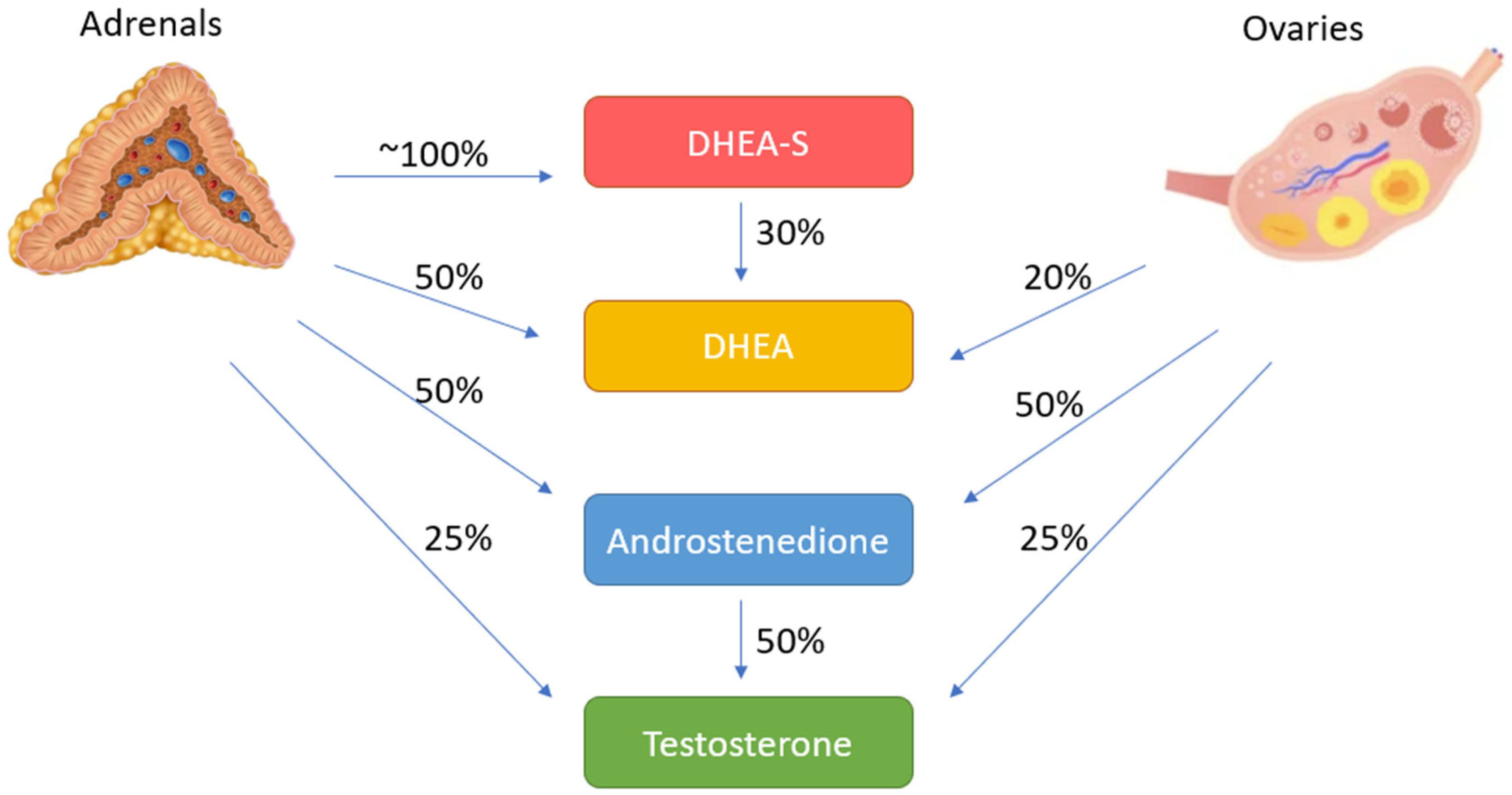
2.2. Adrenal Steroidogenesis and Androgen Excess of Adrenal Origin
2.3. Androgen Excess Mechanism in PCOS
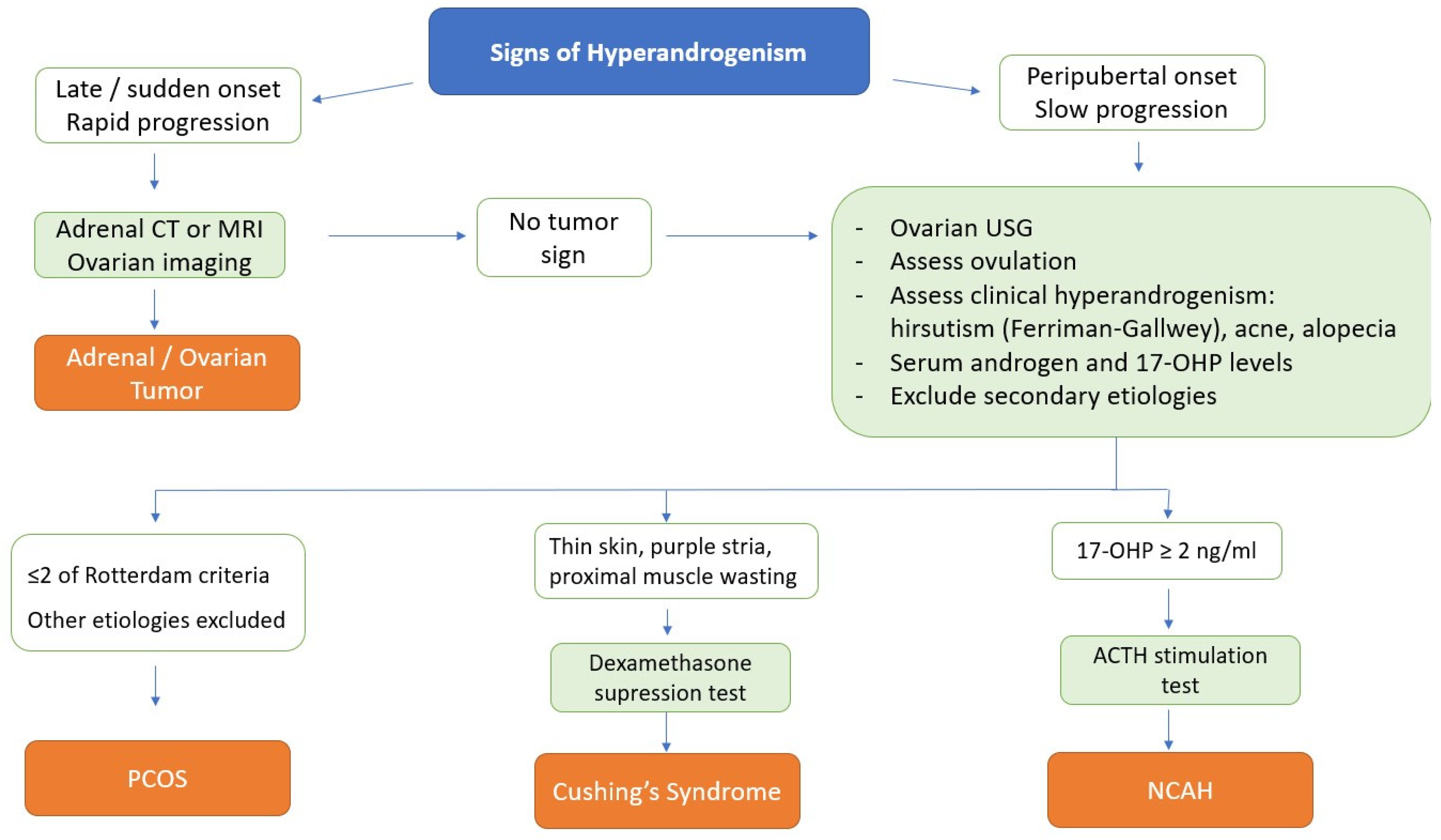
3. Differential Diagnosis
3.1. Non-Classical Congenital Adrenal Hyperplasia (NCAH)
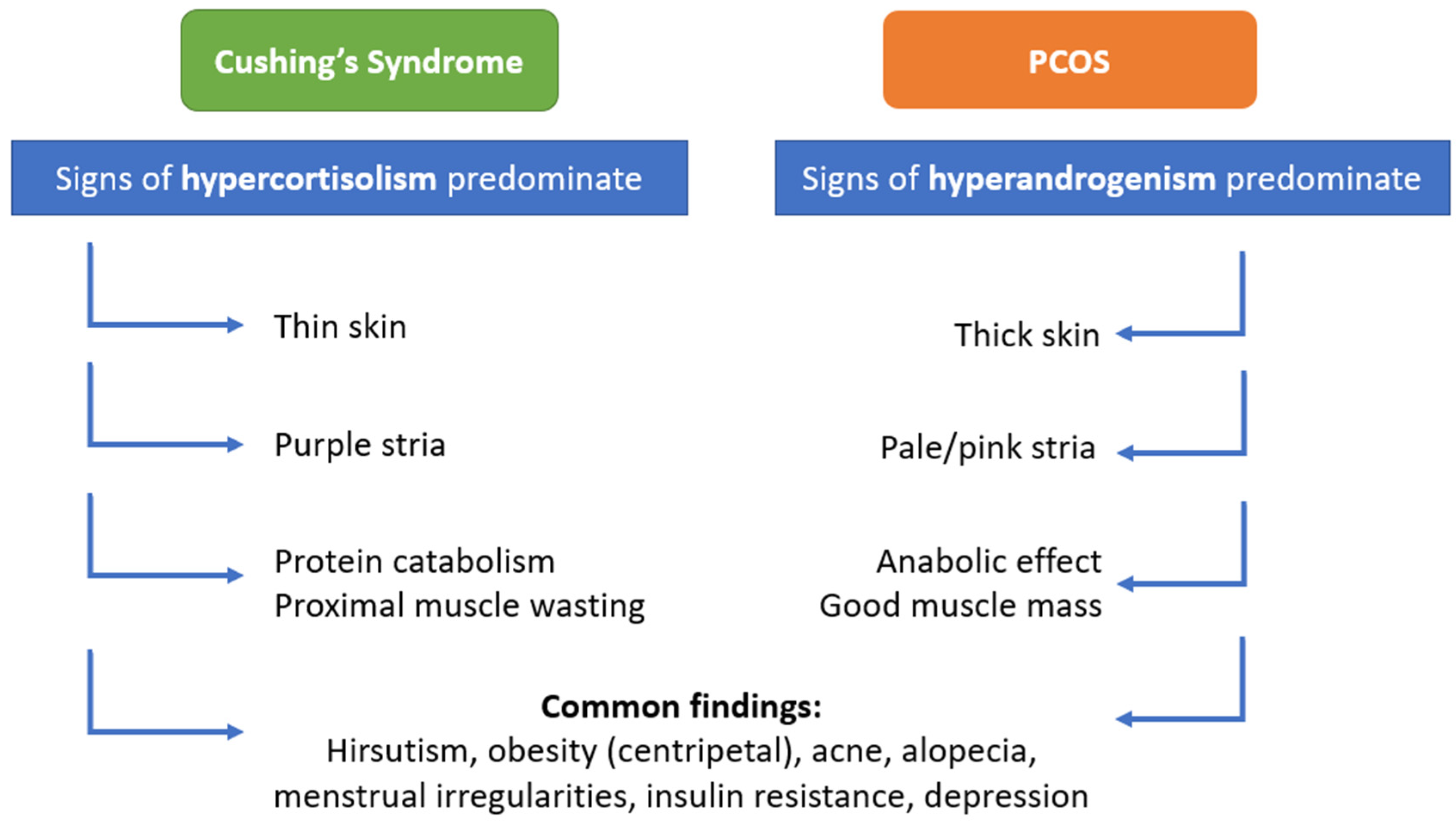
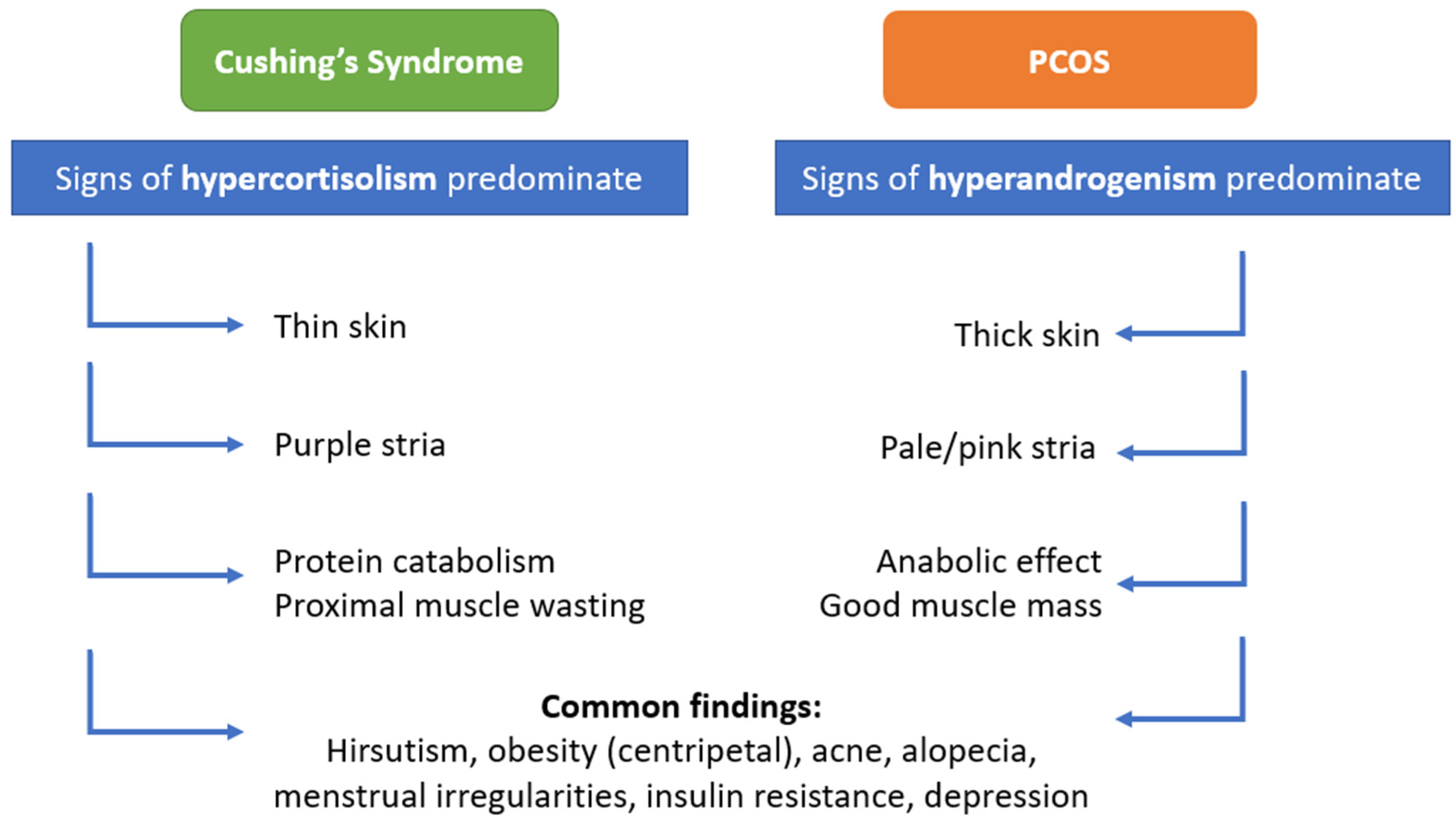
3.2. Cushing’s Syndrome
3.3. Androgen Secreting Adrenal Tumors
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Riestenberg, C.; Jagasia, A.; Markovic, D.; Buyalos, R.P.; Azziz, R. Health care-related economic burden of polycystic ovary syndrome in the United States: Pregnancy-related and long-term health consequences. J. Clin. Endocrinol. Metab. 2022, 107, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Conway, G.; Dewailly, D.; Diamanti-Kandarakis, E.; Escobar-Morreale, H.; Franks, S.; Gambineri, A.; Kelestimur, F.; Macut, D.; Micic, D.; Pasquali, R.; et al. The polycystic ovary syndrome: A position statement from the European Society of Endocrinology. Eur. J. Endocrinol. 2014, 171, P1–P29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yildiz, B.O.; Bozdag, G.; Yapici, Z.; Esinler, I.; Yarali, H. Prevalence, phenotype and cardiometabolic risk of polycystic ovary syndrome under different diagnostic criteria. Hum. Reprod. 2012, 27, 3067–3073. [Google Scholar] [CrossRef] [PubMed]
- Della Corte, L.; Foreste, V.; Barra, F.; Gustavino, C.; Alessandri, F.; Centurioni, M.G.; Ferrero, S.; Bifulco, G.; Giampaolino, P. Current and experimental drug therapy for the treatment of polycystic ovarian syndrome. Expert Opin. Investig. Drugs 2020, 29, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Celik, O.; Yildiz, B.O. Adrenal and Polycystic Ovary Syndrome; Cambridge University Press: Cambridge, UK, 2022; p. 67. [Google Scholar]
- Sahin, Y.; Kelestimur, F. The frequency of late-onset 21-hydroxylase and 11 beta-hydroxylase deficiency in women with polycystic ovary syndrome. Eur. J. Endocrinol. 1997, 137, 670–674. [Google Scholar] [CrossRef] [Green Version]
- Karaca, Z.; Acmaz, B.; Acmaz, G.; Tanriverdi, F.; Unluhizarci, K.; Aribas, S.; Sahin, Y.; Kelestimur, F.; Ünlühızarcı, K. Routine screening for Cushing’s syndrome is not required in patients presenting with hirsutism. Eur. J. Endocrinol. 2013, 168, 379–384. [Google Scholar] [CrossRef] [Green Version]
- Teede, H.J.; Misso, M.L.; Costello, M.F.; Dokras, A.; Laven, J.; Moran, L.; Piltonen, T.; Norman, R.J. Recommendations from the international evidence-based guideline for the assessment and management of polycystic ovary syndrome. Hum. Reprod. 2018, 33, 1602–1618. [Google Scholar] [CrossRef] [Green Version]
- Abbott, D.H.; Dumesic, D.A.; Levine, J.E. Hyperandrogenic origins of polycystic ovary syndrome–implications for pathophysiology and therapy. Expert Rev. Endocrinol. Metab. 2019, 14, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Vargatu, I. Williams textbook of endocrinology. Acta Endocrinol. 2016, 12, 113. [Google Scholar] [CrossRef]
- Judd, H.L.; Yen, S.S. Serum androstenedione and testosterone levels during the menstrual cycle. J. Clin. Endocrinol. Metab. 1973, 36, 475–481. [Google Scholar] [CrossRef]
- Longcope, C. 1 Adrenal and gonadal androgen secretion in normal females. Clin. Endocrinol. Metab. 1986, 15, 213–228. [Google Scholar] [CrossRef]
- Dunn, J.F.; Nisula, B.C.; Rodbard, D. Transport of Steroid Hormones: Binding of 21 Endogenous Steroids to Both Testosterone-Binding Globulin and Corticosteroid-Binding Globulin in Human Plasma. J. Clin. Endocrinol. Metab. 1981, 53, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Azziz, R.; Carmina, E.; Sawaya, M.E. Idiopathic hirsutism. Endocr. Rev. 2000, 21, 347–362. [Google Scholar] [PubMed] [Green Version]
- Prizant, H.; Gleicher, N.; Sen, A. Androgen actions in the ovary: Balance is key. J. Endocrinol. 2014, 222, R141–R151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef] [Green Version]
- Unluhizarci, K.; Karaca, Z.; Kelestimur, F. Role of insulin and insulin resistance in androgen excess disorders. World J. Diabetes 2021, 12, 616. [Google Scholar] [CrossRef]
- Cadagan, D.; Khan, R.; Amer, S. Thecal cell sensitivity to luteinizing hormone and insulin in polycystic ovarian syndrome. Reprod. Biol. 2016, 16, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Garmey, J.C.; Veldhuis, J.D. Interactive stimulation by luteinizing hormone and insulin of the steroidogenic acute regulatory (StAR) protein and 17α-hydroxylase/17, 20-lyase (CYP17) genes in porcine theca cells. Endocrinology 2000, 141, 2735–2742. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, Y.; Yan, L.-Y.; Qiao, J. Increased expression of P450scc and CYP17 in development of endogenous hyperandrogenism in a rat model of PCOS. Endocrine 2013, 43, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Taylor, H.S.; Pal, L.; Sell, E. Speroff’s Clinical Gynecologic Endocrinology and Infertility; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2019. [Google Scholar]
- Jacobson, L. Hypothalamic–pituitary–adrenocortical axis regulation. Endocrinol. Metab. Clin. 2005, 34, 271–292. [Google Scholar] [CrossRef]
- Strushkevich, N.; Gilep, A.A.; Shen, L.; Arrowsmith, C.H.; Edwards, A.M.; Usanov, S.A.; Park, H.-W. Structural insights into aldosterone synthase substrate specificity and targeted inhibition. Mol. Endocrinol. 2013, 27, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Cussen, L.; McDonnell, T.; Bennett, G.; Thompson, C.J.; Sherlock, M.; O’Reilly, M.W. Approach to androgen excess in women: Clinical and biochemical insights. Clin. Endocrinol. 2022, 97, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Konings, G.; Brentjens, L.; Delvoux, B.; Linnanen, T.; Cornel, K.; Koskimies, P.; Bongers, M.; Kruitwagen, R.; Xanthoulea, S.; Romano, A. Intracrine regulation of estrogen and other sex steroid levels in endometrium and non-gynecological tissues; pathology, physiology, and drug discovery. Front. Pharmacol. 2018, 9, 940. [Google Scholar] [CrossRef]
- Taylor, A.E.; McCourt, B.; Martin, K.A.; Anderson, E.J.; Adams, J.M.; Schoenfeld, D.; Hall, J.E. Determinants of abnormal gonadotropin secretion in clinically defined women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 1997, 82, 2248–2256. [Google Scholar] [CrossRef]
- Balen, A.H. Hypersecretion of luteinizing hormone and the polycystic ovary syndrome. Hum. Reprod. 1993, 8 (Suppl. S2), 123–128. [Google Scholar] [CrossRef] [PubMed]
- Laven, J.S.; Imani, B.; Eijkemans, M.J.; De Jong, F.H.; Fauser, B.C. Absent biologically relevant associations between serum inhibin B concentrations and characteristics of polycystic ovary syndrome in normogonadotrophic anovulatory infertility. Hum. Reprod. 2001, 16, 1359–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosencrantz, M.A.; Coffler, M.S.; Haggan, A.; Duke, K.B.; Donohue, M.C.; Shayya, R.F.; Su, H.I.; Chang, R.J. Clinical evidence for predominance of delta-5 steroid production in women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2011, 96, 1106–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickenheisser, J.K.; Nelson-DeGrave, V.L.; McAllister, J.M. Dysregulation of cytochrome P450 17α-hydroxylase messenger ribonucleic acid stability in theca cells isolated from women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2005, 90, 1720–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakimiuk, A.J.; Weitsman, S.R.; Navab, A.; Magoffin, D.A. Luteinizing hormone receptor, steroidogenesis acute regulatory protein, and steroidogenic enzyme messenger ribonucleic acids are overexpressed in thecal and granulosa cells from polycystic ovaries. J. Clin. Endocrinol. Metab. 2001, 86, 1318–1323. [Google Scholar]
- Şahin, Y.; Keleştimur, F. 17-Hydroxyprogesterone response to buserelin testing in the polycystic ovary syndrome. Clin. Endocrinol. 1993, 39, 151–155. [Google Scholar] [CrossRef]
- Sahin, Y.; Keleştimur, F. 17-Hydroxyprogesterone responses to gonadotrophin-releasing hormone agonist buserelin and adrenocorticotrophin in polycystic ovary syndrome: Investigation of adrenal and ovarian cytochrome P450c17alpha dysregulation. Hum. Reprod. 1997, 12, 910–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapanainen, J.S.; Koivunen, R.; Fauser, B.C.; Taylor, A.E.; Clayton, R.N.; Rajkowa, M.; White, D.; Franks, S.; Anttila, L.; Pettersson, K.S.; et al. A new contributing factor to polycystic ovary syndrome: The genetic variant of luteinizing hormone. J. Clin. Endocrinol. Metab. 1999, 84, 1711–1715. [Google Scholar] [CrossRef] [PubMed]
- McAllister, J.M.; Legro, R.S.; Modi, B.; Strauss, J.F. Functional genomics of PCOS: From GWAS to molecular mechanisms. Trends Endocrinol. Metab. 2015, 26, 118–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azziz, R.; Black, V.; Hines, G.A.; Fox, L.M.; Boots, L.R. Adrenal androgen excess in the polycystic ovary syndrome: Sensitivity and responsivity of the hypothalamic-pituitary-adrenal axis. J. Clin. Endocrinol. Metab. 1998, 83, 2317–2323. [Google Scholar] [CrossRef]
- Puurunen, J.; Piltonen, T.; Jaakkola, P.; Ruokonen, A.; Morin-Papunen, L.; Tapanainen, J.S. Adrenal androgen production capacity remains high up to menopause in women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2009, 94, 1973–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Techatraisak, K.; Conway, G.; Rumsby, G. Frequency of a polymorphism in the regulatory region of the 17α-hydroxylase-17, 20-lyase (CYP17) gene in hyperandrogenic states. Clin. Endocrinol. 1997, 46, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Keleştimur, F.; Şahin, Y. Alternate pathway 17, 20-lyase enzyme activity in the adrenals is enhanced in patients with polycystic ovary syndrome. Fertil. Steril. 1999, 71, 1075–1078. [Google Scholar] [CrossRef]
- Dunaif, A.; Book, C.B. Insulin resistance in the polycystic ovary syndrome. Clin. Res. Diabetes Obes. 1997, 15, 249–274. [Google Scholar]
- Nestler, J.E.; Jakubowicz, D.J.; Falcon de Vargas, A.; Brik, C.; Quintero, N.; Medina, F. Insulin stimulates testosterone biosynthesis by human thecal cells from women with polycystic ovary syndrome by activating its own receptor and using inositolglycan mediators as the signal transduction system. J. Clin. Endocrinol. Metab. 1998, 83, 2001–2005. [Google Scholar] [PubMed] [Green Version]
- Nelson-Degrave, V.L.; Wickenheisser, J.K.; Hendricks, K.L.; Asano, T.; Fujishiro, M.; Legro, R.S.; Kimball, S.R.; Strauss, J.F.; McAllister, J.M. Alterations in mitogen-activated protein kinase kinase and extracellular regulated kinase signaling in theca cells contribute to excessive androgen production in polycystic ovary syndrome. Mol. Endocrinol. 2005, 19, 379–390. [Google Scholar] [CrossRef] [Green Version]
- Franks, S.; Gilling-Smith, C.; Watson, H.; Willis, D. Insulin action in the normal and polycystic ovary. Endocrinol. Metab. Clin. N. Am. 1999, 28, 361–378. [Google Scholar] [CrossRef]
- Nestler, J.E.; Jakubowicz, D.J. Decreases in ovarian cytochrome P450c17 alpha activity and serum free testosterone after reduction of insulin secretion in polycystic ovary syndrome. N. Engl. J. Med. 1996, 335, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Nestler, J.E.; Barlascini, C.; Matt, D.W.; Steingold, K.A.; Plymate, S.R.; Clore, J.N.; Blackard, W.G. Suppression of serum insulin by diazoxide reduces serum testosterone levels in obese women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 1989, 68, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.C.; Hickman, R. Insulin resistance and diminished glucose tolerance in powerlifters ingesting anabolic steroids. J. Clin. Endocrinol. Metab. 1987, 64, 960–963. [Google Scholar] [CrossRef]
- Weenen, C.; Laven, J.S.; Von Bergh, A.R.; Cranfield, M.; Groome, N.P.; Visser, J.A.; Kramer, P.; Fauser, B.C.; Themmen, A.P. Anti-Müllerian hormone expression pattern in the human ovary: Potential implications for initial and cyclic follicle recruitment. MHR Basic Sci. Reprod. Med. 2004, 10, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Durlinger, A.L.; Gruijters, M.J.; Kramer, P.; Karels, B.; Kumar, T.R.; Matzuk, M.M.; Rose, U.M.; de Jong, F.H.; Uilenbroek, J.T.J.; Grootegoed, J.A.; et al. Anti-Mullerian hormone attenuates the effects of FSH on follicle development in the mouse ovary. Endocrinology 2001, 142, 4891–4899. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.M.; Klausen, C.; Leung, P.C. Antimüllerian hormone inhibits follicle-stimulating hormone-induced adenylyl cyclase activation, aromatase expression, and estradiol production in human granulosa-lutein cells. Fertil. Steril. 2013, 100, 585–592.e1. [Google Scholar] [CrossRef]
- Cedars, M.I.; Steingold, K.A.; de Ziegler, D.; Lapolt, P.S.; Chang, R.J.; Judd, H.L. Long-term administration of gonadotropin-releasing hormone agonist and dexamethasone: Assessment of the adrenal role in ovarian dysfunction. Fertil. Steril. 1992, 57, 495–500. [Google Scholar] [CrossRef]
- Nestler, J.E.; Powers, L.P.; Matt, D.W.; Steingold, K.A.; Plymate, S.R.; Rittmaster, R.S.; Clore, J.N.; Blackard, W.G. A direct effect of hyperinsulinemia on serum sex hormone-binding globulin levels in obese women with the polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 1991, 72, 83–89. [Google Scholar] [CrossRef]
- Derksen, J.; Nagesser, S.K.; Meinders, A.E.; Haak, H.R.; van de Velde, C. Identification of virilizing adrenal tumors in hirsute women. N. Engl. J. Med. 1994, 331, 968–973. [Google Scholar] [CrossRef]
- Azziz, R.; Sanchez, L.A.; Knochenhauer, E.S.; Moran, C.; Lazenby, J.; Stephens, K.C.; Taylor, K.; Boots, L.R. Androgen excess in women: Experience with over 1000 consecutive patients. J. Clin. Endocrinol. Metab. 2004, 89, 453–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azziz, R.; Dewailly, D.; Owerbach, D. Clinical review 56: Nonclassic adrenal hyperplasia: Current concepts. J. Clin. Endocrinol. Metab. 1994, 78, 810–815. [Google Scholar] [PubMed]
- Carmina, E.; Dewailly, D.; Escobar-Morreale, H.F.; Kelestimur, F.; Moran, C.; Oberfield, S.; Witchel, S.F.; Azziz, R. Non-classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency revisited: An update with a special focus on adolescent and adult women. Hum. Reprod. Update 2017, 23, 580–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmina, E. Pathogenesis and treatment of hirsutism in late-onset congenital adrenal hyperplasia. Reprod. Med. Rev. 1995, 4, 179–187. [Google Scholar] [CrossRef]
- Oberfield, S.E.; Sopher, A.B.; Gerken, A.T. Approach to the girl with early onset of pubic hair. J. Clin. Endocrinol. Metab. 2011, 96, 1610–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dacou-Voutetakis, C.; Dracopoulou, M. High incidence of molecular defects of the CYP21 gene in patients with premature adrenarche. J. Clin. Endocrinol. Metab. 1999, 84, 1570–1574. [Google Scholar] [CrossRef]
- Kohn, B.; Levine, L.S.; Pollack, M.S.; Pang, S.; Lorenzen, F.; Levy, D.; Lerner, A.J.; Rondanini, G.F.; Dupont, B.; New, M.I. Late-onset steroid 21-hydroxylase deficiency: A variant of classical congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 1982, 55, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Moran, C.; Azziz, R.; Carmina, E.; Dewailly, D.; Fruzzetti, F.; Ibanez, L.; Knochenhauer, E.S.; Marcondes, J.A.; Mendonca, B.; Pignatelli, D.; et al. 21-Hydroxylase–deficient nonclassic adrenal hyperplasia is a progressive disorder: A multicenter study. Am. J. Obstet. Gynecol. 2000, 183, 1468–1474. [Google Scholar] [CrossRef]
- New, M.I. Nonclassical 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 2006, 91, 4205–4214. [Google Scholar] [CrossRef]
- Loh, V.; Krishnan, B.; Prentice, M.; Panahloo, A.; Seal, L. Late onset congenital adrenal hyperplasia masquerading as subclinical cushing. In Endocrine Abstracts; Bioscientifica: Bristol, UK, 2009. [Google Scholar]
- Sanchez, L.; Knochenhauer, E.; Gatlin, R.; Moran, C.; Azziz, R. Differential diagnosis of clinically evident hyperandrogenism: Experience with over 1000 consecutive patients. Fertil. Steril. 2001, 76, S111. [Google Scholar] [CrossRef]
- Moran, C.; Azziz, R. 21-hydroxylase-deficient nonclassic adrenal hyperplasia: The great pretender. Semin. Reprod. Med. 2003, 21, 295–300. [Google Scholar] [PubMed]
- Unluhizarci, K.; Kaltsas, G.; Kelestimur, F. Non polycystic ovary syndrome–related endocrine disorders associated with hirsutism. Eur. J. Clin. Investig. 2012, 42, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Maffazioli, G.D.N.; Bachega, T.A.S.S.; Hayashida, S.A.Y.; Gomes, L.G.; Valassi, H.P.L.; Marcondes, J.A.M.; Mendonca, B.B.; Baracat, E.C.; Maciel, G.A.R. Steroid Screening Tools Differentiating Nonclassical Congenital Adrenal Hyperplasia and Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2020, 105, e2895–e2902. [Google Scholar] [CrossRef] [PubMed]
- Makrantonaki, E.; Zouboulis, C.C. Hyperandrogenismus, adrenale Dysfunktion und Hirsutismus. Hautarzt 2020, 71, 752–761. [Google Scholar] [CrossRef]
- Tatsi, C.; Flippo, C.; Stratakis, C.A. Cushing syndrome: Old and new genes. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101418. [Google Scholar] [CrossRef]
- Trementino, L.; Appolloni, G.; Concettoni, C.; Cardinaletti, M.; Boscaro, M.; Arnaldi, G. Association of glucocorticoid receptor polymorphism A3669G with decreased risk of developing diabetes in patients with Cushing’s syndrome. Eur. J. Endocrinol. 2012, 166, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Arnaldi, G.; Martino, M. Androgens in Cushing’s Syndrome. Front. Horm. Res. 2019, 53, 77–91. [Google Scholar] [PubMed]
- Valassi, E.; Santos, A.; Yaneva, M.; Tóth, M.; Strasburger, C.J.; Chanson, P.; Wass, J.A.; Chabre, O.; Pfeifer, M.; Feelders, R.A.; et al. The European Registry on Cushing’s syndrome: 2-year experience. Baseline demographic and clinical characteristics. Eur. J. Endocrinol. 2011, 165, 383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnaldi, G.; Angeli, A.; Atkinson, A.B.; Bertagna, X.; Cavagnini, F.; Chrousos, G.P.; Fava, G.A.; Findling, J.W.; Gaillard, R.C.; Grossman, A.B.; et al. Diagnosis and complications of Cushing’s syndrome: A consensus statement. J. Clin. Endocrinol. Metab. 2003, 88, 5593–5602. [Google Scholar] [CrossRef] [Green Version]
- Lado-Abeal, J.; Rodriguez-Arnao, J.; Newell-Price, J.D.C.; Perry, L.A.; Grossman, A.B.; Besser, G.M.; Trainer, P.J. Menstrual abnormalities in women with Cushing’s disease are correlated with hypercortisolemia rather than raised circulating androgen levels. J. Clin. Endocrinol. Metab. 1998, 83, 3083–3088. [Google Scholar]
- Brzana, J.; Yedinak, C.G.; Hameed, N.; Plesiu, A.; McCartney, S.; Fleseriu, M. Polycystic ovarian syndrome and Cushing’s syndrome: A persistent diagnostic quandary. Eur. J. Obstet. Gynecol. Reprod. Biol. 2014, 175, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, A.; Feelders, R.A.; Stratakis, C.A.; Nieman, L.K. Cushing’s syndrome. Lancet 2015, 386, 913–927. [Google Scholar] [CrossRef]
- Nieman, L.K.; Biller, B.M.; Findling, J.W.; Newell-Price, J.; Savage, M.O.; Stewart, P.M.; Montori, V.M. The diagnosis of Cushing’s syndrome: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2008, 93, 1526–1540. [Google Scholar] [CrossRef] [PubMed]
- Gardner, D.G.; Shoback, D.M. Greenspan’s Basic and Clinical Endocrinology; McGraw-Hill Education: New York, NY, USA, 2017. [Google Scholar]
- Di Dalmazi, G. Hyperandrogenism and Adrenocortical Tumors. Hyperandrogenism Women 2019, 53, 92–99. [Google Scholar]
- Carmina, E.; Rosato, F.; Jannì, A.; Rizzo, M.; Longo, R.A. Relative prevalence of different androgen excess disorders in 950 women referred because of clinical hyperandrogenism. J. Clin. Endocrinol. Metab. 2006, 91, 2–6. [Google Scholar] [CrossRef] [Green Version]
- Giampaolino, P.; Della Corte, L.; De Rosa, N.; Mercorio, A.; Bruzzese, D.; Bifulco, G. Ovarian volume and PCOS: A controversial issue. Gynecol. Endocrinol. 2018, 34, 229–232. [Google Scholar] [CrossRef]
- Papadakis, G.; Kandaraki, E.A.; Tseniklidi, E.; Papalou, O.; Diamanti-Kandarakis, E. Polycystic ovary syndrome and NC-CAH: Distinct characteristics and common findings. A systematic review. Front. Endocrinol. 2019, 10, 388. [Google Scholar] [CrossRef]
- Rachoń, D. Differential diagnosis of hyperandrogenism in women with polycystic ovary syndrome. Exp. Clin. Endocrinol. Diabetes 2012, 120, 205–209. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Adrenal Diseases | ACTH Hypersecretion | Pregnancy | Other Causes |
|---|---|---|---|
|
|
|
|
| Androgen | Ovary | Adrenal | Peripheral Conversion |
|---|---|---|---|
| Testosterone | 60% | 5% | 35% (from androstenedione) |
| Androstenedione | 60% | 35% | 5% (from DHEAS) |
| DHEAS | <5% | >95% | 0% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yesiladali, M.; Yazici, M.G.K.; Attar, E.; Kelestimur, F. Differentiating Polycystic Ovary Syndrome from Adrenal Disorders. Diagnostics 2022, 12, 2045. https://doi.org/10.3390/diagnostics12092045
Yesiladali M, Yazici MGK, Attar E, Kelestimur F. Differentiating Polycystic Ovary Syndrome from Adrenal Disorders. Diagnostics. 2022; 12(9):2045. https://doi.org/10.3390/diagnostics12092045
Chicago/Turabian StyleYesiladali, Mert, Melis G. K. Yazici, Erkut Attar, and Fahrettin Kelestimur. 2022. "Differentiating Polycystic Ovary Syndrome from Adrenal Disorders" Diagnostics 12, no. 9: 2045. https://doi.org/10.3390/diagnostics12092045
APA StyleYesiladali, M., Yazici, M. G. K., Attar, E., & Kelestimur, F. (2022). Differentiating Polycystic Ovary Syndrome from Adrenal Disorders. Diagnostics, 12(9), 2045. https://doi.org/10.3390/diagnostics12092045