
Novel CSF Biomarkers Tracking Autoimmune Inflammatory and Neurodegenerative Aspects of CNS Diseases



Abstract
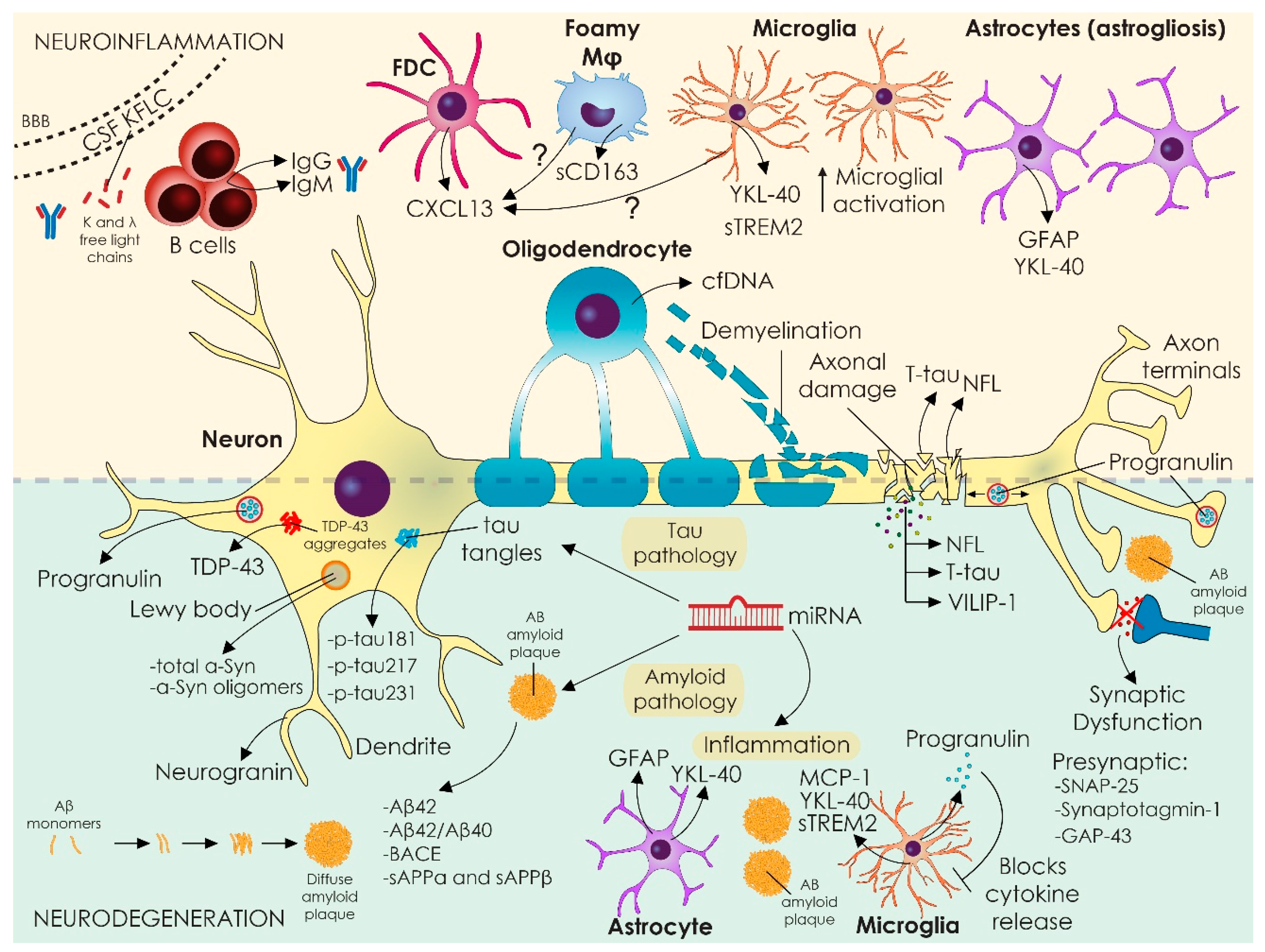
:1. Introduction
2. Biomarkers Tracking Inflammatory Aspects
2.1. Intrathecal Free Light Chain Synthesis
2.2. YKL-40
2.3. sTREM2
2.4. Soluble CD136 and CXCL13
2.5. IL-6
2.6. Monocyte Chemoattractant Protein-1 (MCP-1/CCL2)
2.7. Glial Fibrillary Acidic Protein (Astrogliosis Marker)
3. Biomarkers Tracking Neurodegenerative Aspects
3.1. Neurofilament Light Chain
3.2. VILIP-1
3.3. Ubiquitin C-Terminal Hydrolase L1
4. Biomarkers Tracking Synaptic Pathology
4.1. Neurogranin
4.2. SNAP-25
4.3. GAP-43
5. Biomarkers Tracking Disease-Specific Proteins
5.1. Alpha-Synuclein
5.2. TAR DNA-Binding Protein of 43kDa (TDP-43)
5.3. Progranulin
6. Molecular Biomarkers
6.1. MicroRNAs
6.2. Cell-Free DNA (Genomic and Mitochondrial Origin)
7. Future Directions
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Abs | autoantibodies |
| AD | Alzheimer’s disease |
| ALS | amyotrophic lateral sclerosis |
| APP | Amyloid precurcor protein |
| AQP4 | aquaporin-4 |
| α-Syn | alpha-synuclein |
| BACE1 | Beta-secretase 1, also known as beta-site amyloid precursor protein cleaving enzyme 1 |
| BBB | blood–brain barrier |
| bvFTD | behavioral variant of FTD |
| CBD | corticobasal degeneration |
| cfDNA | cell-free DNA |
| cf-mtDNA | mitochondrial cell-free DNA |
| CHMP2B | charged multivesicular body protein 2B |
| CIS | clinically isolated syndrome |
| CJD | Creutzfeldt–Jacob disease |
| CSF | cerebrospinal fluid |
| CXCL13 | chemokine (C-X-C motif) ligand 13 |
| ddPCR | droplet digital polymerase chain reaction |
| DLB | Lewy bodies |
| DMT | disease-modifying therapies |
| EDSS | Expanded Disability Status Scale |
| ELISA | enzyme-linked immunosorbent assay |
| FDG-PET | fluorodeoxyglucose PET |
| FLC | free light chains |
| FTD | frontotemporal dementia |
| FTLDs | frontotemporal lobar degenerations |
| FTLD-U | ubiquitin-positive frontotemporal lobar degeneration |
| FUS | fused in sarcoma protein |
| GAP-43 | growth-associated protein 43 |
| GFAP | glial fibrillary acidic protein |
| GRNL | granulin |
| IL-13 | interleukin-13 |
| IL-17 | interleukin-17 |
| IL-5 | interleukin-5 |
| IL-6 | interleukin-6 |
| IL-8 | interleukin-8 |
| KFLC | kappa free light chain |
| KCSF | kappa free light chain of cerebrospinal fluid |
| LFLC | lambda free light chain |
| MCI | mild cognitive impairment |
| MCP-1 | monocyte chemoattractant protein-1 |
| miRNAs | microRNAs |
| MOGAD | myelin oligodendrocyte glycoprotein antibody disease |
| MRI | magnetic resonance imaging |
| MS | multiple sclerosis |
| MSA | multiple system atrophy |
| NAWM | normal-appearing white matter |
| NDDs | neurodegenerative diseases |
| NF | neurofilaments |
| NFH | heavy neurofilament chains |
| NFL | neurofilament light protein |
| Ng | neurogranin |
| NGS | next-generation sequencing |
| NIDs | neuroinflammatory diseases |
| NMOSD | NMO spectrum disorders |
| NMO | neuromyelitis optica |
| OBs | oligoclonal bands |
| PD | Parkinson’s disease |
| PDD | PD dementia |
| PET | emission tomography |
| PGRN | progranulin |
| PMCA | protein-misfolding cyclic amplification |
| PPA | primary progressive aphasia |
| PSP | progressive supranuclear palsy |
| p-tau | phospho-tau protein |
| qMSP | quantitative methylation-specific PCR |
| qRT-PCR | real-time quantitative PCR |
| RIS | radiologically isolated syndrome |
| RRMS | relapsing-remitting MS |
| RT-QuIC | real-time quaking-induced conversion |
| SAAs | seed amplification assays |
| sCD163 | soluble cluster of differentiation 163 |
| SiMoA | single molecule array |
| SNAP-25 | synaptosomal associated protein 25 |
| SPMS | secondary progressive MS |
| sTREM2 | soluble TREM2 |
| svFTD | semantic variant of FTD |
| TDP-43 | TAR DNA-binding protein of 43 kDa |
| TGF-β | transforming growth factor beta |
| Th17 | T helper 17 cells |
| Th2 | T helper 2 cells |
| TREM2 | triggering receptor expressed on myeloid cells 2 |
| t-tau | total tau |
| UCH-L | ubiquitin C-terminal hydrolase L1 |
| UPS | ubiquitin proteasome system |
| VaD | vascular dementia |
| VILIP-1 | visinin-like protein 1 |
| YKL-40 | also known as chitinase-3-like protein 1 (CHI3L1), a 40 kD chitin binding protein with a YKL domain |
References
- Gaetani, L.; Paolini Paoletti, F.; Bellomo, G.; Mancini, A.; Simoni, S.; Di Filippo, M.; Parnetti, L. CSF and Blood Biomarkers in Neuroinflammatory and Neurodegenerative Diseases: Implications for Treatment. Trends Pharmacol. Sci. 2020, 41, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Graus, F.; Titulaer, M.J.; Balu, R.; Benseler, S.; Bien, C.G.; Cellucci, T.; Cortese, I.; Dale, R.C.; Gelfand, J.M.; Geschwind, M.; et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016, 15, 391–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- Skillbäck, T.; Farahmand, B.Y.; Rosén, C.; Mattsson, N.; Nägga, K.; Kilander, L.; Religa, D.; Wimo, A.; Winblad, B.; Schott, J.M.; et al. Cerebrospinal fluid tau and amyloid-β1-42 in patients with dementia. Brain 2015, 138, 2716–2731. [Google Scholar] [CrossRef] [Green Version]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Michel, L.; Touil, H.; Pikor, N.B.; Gommerman, J.L.; Prat, A.; Bar-Or, A. B Cells in the Multiple Sclerosis Central Nervous System: Trafficking and Contribution to CNS-Compartmentalized Inflammation. Front. Immunol. 2015, 6, 636. [Google Scholar] [CrossRef] [Green Version]
- Pryce, G.; Baker, D. Oligoclonal bands in multiple sclerosis; Functional significance and therapeutic implications. Does the specificity matter. Mult. Scler. Relat. Disord. 2018, 25, 131–137. [Google Scholar] [CrossRef]
- Matute-Blanch, C.; Villar, L.M.; Álvarez-Cermeño, J.C.; Rejdak, K.; Evdoshenko, E.; Makshakov, G.; Nazarov, V.; Lapin, S.; Midaglia, L.; Vidal-Jordana, A.; et al. Neurofilament light chain and oligoclonal bands are prognostic biomarkers in radiologically isolated syndrome. Brain 2018, 141, 1085–1093. [Google Scholar] [CrossRef] [Green Version]
- Ramsden, D.B. Multiple sclerosis: Assay of free immunoglobulin light chains. Ann. Clin. Biochem. 2017, 54, 5–13. [Google Scholar] [CrossRef]
- Rudick, R.A.; Peter, D.R.; Bidlack, J.M.; Knutson, D.W. Multiple sclerosis: Free light chains in cerebrospinal fluid. Neurology 1985, 35, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Bracco, F.; Gallo, P.; Menna, R.; Battistin, L.; Tavolato, B. Free light chains in the CSF in multiple sclerosis. J. Neurol. 1987, 234, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Presslauer, S.; Milosavljevic, D.; Brücke, T.; Bayer, P.; Hübl, W. Elevated levels of kappa free light chains in CSF support the diagnosis of multiple sclerosis. J. Neurol. 2008, 255, 1508–1514. [Google Scholar] [CrossRef] [PubMed]
- Duranti, F.; Pieri, M.; Centonze, D.; Buttari, F.; Bernardini, S.; Dessi, M. Determination of κFLC and κ Index in cerebrospinal fluid: A valid alternative to assess intrathecal immunoglobulin synthesis. J. Neuroimmunol. 2013, 263, 116–120. [Google Scholar] [CrossRef]
- Hassan-Smith, G.; Durant, L.; Tsentemeidou, A.; Assi, L.K.; Faint, J.M.; Kalra, S.; Douglas, M.R.; Curnow, S.J. High sensitivity and specificity of elevated cerebrospinal fluid kappa free light chains in suspected multiple sclerosis. J. Neuroimmunol. 2014, 276, 175–179. [Google Scholar] [CrossRef]
- Makshakov, G.; Nazarov, V.; Kochetova, O.; Surkova, E.; Lapin, S.; Evdoshenko, E. Diagnostic and Prognostic Value of the Cerebrospinal Fluid Concentration of Immunoglobulin Free Light Chains in Clinically Isolated Syndrome with Conversion to Multiple Sclerosis. PLoS ONE 2015, 10, e0143375. [Google Scholar] [CrossRef] [Green Version]
- Saadeh, R.S.; Bryant, S.C.; McKeon, A.; Weinshenker, B.; Murray, D.L.; Pittock, S.J.; Willrich, M.A.V. CSF Kappa Free Light Chains: Cutoff Validation for Diagnosing Multiple Sclerosis. Mayo Clin. Proc. 2022, 97, 738–751. [Google Scholar] [CrossRef]
- Leurs, C.E.; Twaalfhoven, H.; Lissenberg-Witte, B.I.; van Pesch, V.; Dujmovic, I.; Drulovic, J.; Castellazzi, M.; Bellini, T.; Pugliatti, M.; Kuhle, J.; et al. Kappa free light chains is a valid tool in the diagnostics of MS: A large multicenter study. Mult. Scler. 2020, 26, 912–923. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, M.; Gjelstrup, M.C.; Stilund, M.; Christensen, T.; Petersen, T.; Jon Møller, H. Cerebrospinal fluid free kappa light chains and kappa index perform equal to oligoclonal bands in the diagnosis of multiple sclerosis. Clin. Chem. Lab. Med. 2018, 57, 210–220. [Google Scholar] [CrossRef]
- Zeman, D.; Kušnierová, P.; Bartoš, V.; Hradílek, P.; Kurková, B.; Zapletalová, O. Quantitation of free light chains in the cerebrospinal fluid reliably predicts their intrathecal synthesis. Ann. Clin. Biochem. 2016, 53, 174–176. [Google Scholar] [CrossRef]
- Gurtner, K.M.; Shosha, E.; Bryant, S.C.; Andreguetto, B.D.; Murray, D.L.; Pittock, S.J.; Willrich, M.A.V. CSF free light chain identification of demyelinating disease: Comparison with oligoclonal banding and other CSF indexes. Clin. Chem. Lab. Med. 2018, 56, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Süße, M.; Hannich, M.; Petersmann, A.; Zylla, S.; Pietzner, M.; Nauck, M.; Dressel, A. Kappa free light chains in cerebrospinal fluid to identify patients with oligoclonal bands. Eur. J. Neurol. 2018, 25, 1134–1139. [Google Scholar] [CrossRef] [PubMed]
- Presslauer, S.; Milosavljevic, D.; Huebl, W.; Parigger, S.; Schneider-Koch, G.; Bruecke, T. Kappa free light chains: Diagnostic and prognostic relevance in MS and CIS. PLoS ONE 2014, 9, e89945. [Google Scholar] [CrossRef] [Green Version]
- Senel, M.; Mojib-Yezdani, F.; Braisch, U.; Bachhuber, F.; Lewerenz, J.; Ludolph, A.C.; Otto, M.; Tumani, H. CSF Free Light Chains as a Marker of Intrathecal Immunoglobulin Synthesis in Multiple Sclerosis: A Blood-CSF Barrier Related Evaluation in a Large Cohort. Front. Immunol. 2019, 10, 641. [Google Scholar] [CrossRef] [Green Version]
- Reiber, H.; Zeman, D.; Kušnierová, P.; Mundwiler, E.; Bernasconi, L. Diagnostic relevance of free light chains in cerebrospinal fluid—The hyperbolic reference range for reliable data interpretation in quotient diagrams. Clin. Chim. Acta 2019, 497, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, R.B.; Emery, J.G.; Connor, J.R.; Dodds, R.; Lysko, P.G.; Rosenberg, M. Induction and expression of human cartilage glycoprotein 39 in rheumatoid inflammatory and peripheral blood monocyte-derived macrophages. Exp. Cell Res. 1997, 237, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Bonneh-Barkay, D.; Wang, G.; Starkey, A.; Hamilton, R.L.; Wiley, C.A. In vivo CHI3L1 (YKL-40) expression in astrocytes in acute and chronic neurological diseases. J. Neuroinflamm. 2010, 7, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cubas-Núñez, L.; Gil-Perotín, S.; Castillo-Villalba, J.; López, V.; Solís Tarazona, L.; Gasqué-Rubio, R.; Carratalá-Boscá, S.; Alcalá-Vicente, C.; Pérez-Miralles, F.; Lassmann, H.; et al. Potential Role of CHI3L1+ Astrocytes in Progression in MS. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e972. [Google Scholar] [CrossRef]
- Hinsinger, G.; Galéotti, N.; Nabholz, N.; Urbach, S.; Rigau, V.; Demattei, C.; Lehmann, S.; Camu, W.; Labauge, P.; Castelnovo, G.; et al. Chitinase 3-like proteins as diagnostic and prognostic biomarkers of multiple sclerosis. Mult. Scler. 2015, 21, 1251–1261. [Google Scholar] [CrossRef]
- Comabella, M.; Fernández, M.; Martin, R.; Rivera-Vallvé, S.; Borrás, E.; Chiva, C.; Julià, E.; Rovira, A.; Cantó, E.; Alvarez-Cermeño, J.C.; et al. Cerebrospinal fluid chitinase 3-like 1 levels are associated with conversion to multiple sclerosis. Brain 2010, 133, 1082–1093. [Google Scholar] [CrossRef]
- Schneider, R.; Bellenberg, B.; Gisevius, B.; Hirschberg, S.; Sankowski, R.; Prinz, M.; Gold, R.; Lukas, C.; Aiden Haghikia, A. Chitinase 3-like 1 and neurofilament light chain in CSF and CNS atrophy in MS. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e906. [Google Scholar] [CrossRef] [PubMed]
- Francisco Pérez-Miralles, F.; Prefasi, D.; García-Merino, A.; Gascón-Giménez, F.; Medrano, N.; Castillo-Villalba, J.; Cubas, L.; Alcalá, C.; Gil-Perotín, S.; Gómez-Ballesteros, R.; et al. CSF Chitinase 3-like-1 association with disability of primary progressive MS. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e815. [Google Scholar] [CrossRef] [PubMed]
- Condello, C.; Yuan, P.; Schain, A.; Grutzendler, J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat. Commun. 2015, 6, 6176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonell, A.; Mansilla, A.; Rami, L.; Lladó, A.; Iranzo, A.; Olives, J.; Balasa, M.; Sánchez-Valle, R.; Molinuevo, J.L. Cerebrospinal fluid level of YKL-40 protein in preclinical and prodromal Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 42, 901–908. [Google Scholar] [CrossRef]
- Wang, L.; Gao, T.; Cai, T.; Li, K.; Zheng, P.; Liu, J.; Alzheimer’s Disease Neuroimaging Initiative. Cerebrospinal fluid levels of YKL-40 in prodromal Alzheimer’s disease. Neurosci. Lett. 2020, 715, 134658. [Google Scholar] [CrossRef]
- Sutphen, C.L.; Jasielec, M.S.; Shah, A.R.; Macy, E.M.; Xiong, C.; Vlassenko, A.G.; Benzinger, T.L.; Stoops, E.E.; Vanderstichele, H.M.; Brix, B.; et al. Longitudinal Cerebrospinal Fluid Biomarker Changes in Preclinical Alzheimer Disease During Middle Age. JAMA Neurol. 2015, 72, 1029–1042. [Google Scholar] [CrossRef] [Green Version]
- Olsson, B.; Hertze, J.; Lautner, R.; Zetterberg, H.; Nägga, K.; Höglund, K.; Basun, H.; Annas, P.; Lannfelt, L.; Andreasen, N.; et al. Microglial markers are elevated in the prodromal phase of Alzheimer’s disease and vascular dementia. J. Alzheimer’s Dis. 2013, 33, 45–53. [Google Scholar] [CrossRef]
- Oeckl, P.; Weydt, P.; Steinacker, P.; Anderl-Straub, S.; Nordin, F.; Volk, A.E.; Diehl-Schmid, J.; Andersen, P.M.; Kornhuber, J.; Danek, A.; et al. Different neuroinflammatory profile in amyotrophic lateral sclerosis and frontotemporal dementia is linked to the clinical phase. J. Neurol. Neurosurg. Psychiatry 2019, 90, 4–10. [Google Scholar] [CrossRef]
- Llorens, F.; Thüne, K.; Tahir, W.; Kanata, E.; Diaz-Lucena, D.; Xanthopoulos, K.; Kovatsi, E.; Pleschka, C.; Garcia-Esparcia, P.; Schmitz, M.; et al. YKL-40 in the brain and cerebrospinal fluid of neurodegenerative dementias. Mol. Neurodegener 2017, 12, 83. [Google Scholar] [CrossRef] [Green Version]
- Piccio, L.; Buonsanti, C.; Cella, M.; Tassi, I.; Schmidt, R.E.; Fenoglio, C.; Rinker, J., II; Naismith, R.T.; Panina-Bordignon, P.; Passini, N.; et al. Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain 2008, 131, 3081–3091. [Google Scholar] [CrossRef]
- Azzolini, F.; Gilio, L.; Pavone, L.; Iezzi, E.; Dolcetti, E.; Bruno, A.; Buttari, F.; Musella, A.; Mandolesi, G.; Guadalupi, L.; et al. Neuroinflammation is associated with GFAP and sTREM2 levels in multiple sclerosis. Biomolecules 2022, 12, 222. [Google Scholar] [CrossRef] [PubMed]
- Ohrfelt, A.; Axelsson, M.; Malmestrom, C.; Novakova, L.; Heslegrave, A.; Blennow, K.; Lycke, J.; Zetterberg, H. Soluble TREM-2 in cerebrospinal fluid from patients with multiple sclerosis treated with natalizumab or mitoxantrone. Mult. Scler. 2016, 22, 1587–1595. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suárez-Calvet, M.; Kleinberger, G.; Araque Caballero, M.Á.; Brendel, M.; Rominger, A.; Alcolea, D.; Fortea, J.; Lleó, A.; Blesa, R.; Gispert, J.D.; et al. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol. Med. 2016, 8, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Calvet, M.; Araque Caballero, M.Á.; Kleinberger, G.; Bateman, R.J.; Fagan, A.M.; Morris, J.C.; Levin, J.; Danek, A.; Ewers, M.; Haass, C.; et al. Early changes in CSF sTREM2 in dominantly inherited Alzheimer’s disease occur after amyloid deposition and neuronal injury. Sci. Transl. Med. 2016, 8, 369ra178. [Google Scholar] [CrossRef] [Green Version]
- Ewers, M.; Franzmeier, N.; Suárez-Calvet, M.; Morenas-Rodriguez, E.; Caballero, M.A.A.; Kleinberger, G.; Piccio, L.; Cruchaga, C.; Deming, Y.; Dichgans, M.; et al. Alzheimer’s Disease Neuroimaging Initiative. Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer’s disease. Sci. Transl. Med. 2019, 11, eaav6221. [Google Scholar] [CrossRef]
- Heslegrave, A.; Heywood, W.; Paterson, R.; Magdalinou, N.; Svensson, J.; Johansson, P.; Öhrfelt, A.; Blennow, K.; Hardy, J.; Schott, J.; et al. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Henjum, K.; Almdahl, I.S.; Årskog, V.; Minthon, L.; Hansson, O.; Fladby, T.; Nilsson, L.N.G. Cerebrospinal fluid soluble TREM2 in aging and Alzheimer’s disease. Alzheimer’s Res. Ther. 2016, 8, 17. [Google Scholar] [CrossRef] [Green Version]
- Knapskog, A.B.; Henjum, K.; Idland, A.V.; Eldholm, R.S.; Persson, K.; Saltvedt, I.; Watne, L.O.; Engedal, K.; Nilsson, L.N.G. Cerebrospinal fluid sTREM2 in Alzheimer’s disease: Comparisons between clinical presentation and AT classification. Sci. Rep. 2020, 10, 15886. [Google Scholar] [CrossRef]
- Suárez-Calvet, M.; Morenas Rodríguez, E.; Kleinberger, G.; Schlepckow, K.; Caballero, M.Á.A.; Franzmeier, N.; Capell, A.; Fellerer, K.; Nuscher, B.; Eren, E.; et al. Early increase of CSF sTREM2 in Alzheimer’s disease is associated with tau related neurodegeneration but not with amyloid-β pathology. Mol. Neurodegener. 2019, 14, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascoal, T.A.; Benedet, A.L.; Ashton, N.J.; Kang, M.S.; Therriault, J.; Chamoun, M.; Savard, M.; Lussier, F.Z.; Tissot, C.; Karikari, T.K.; et al. Publisher Correction: Microglial activation and tau propagate jointly across Braak stages. Nat. Med. 2021, 27, 2048–2049. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Qiu, J.; Liu, H.; Zhou, M.; Huang, S.; Guo, W.; Lin, Y.; Chen, X.; Li, Z.; Li, G.; et al. Analysis of Cerebrospinal Fluid Soluble TREM2 and Polymorphisms in Sporadic Parkinson’s Disease in a Chinese Population. J. Mol. Neurosci. 2020, 70, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Roos, P.; von Essen, M.R.; Nielsen, T.T.; Johannsen, P.; Stokholm, J.; Bie, A.S.; Waldemar, G.; Simonsen, A.H.; Heslegrave, A.; Zetterberg, H.; et al. Inflammatory markers of CHMP2B-mediated frontotemporal dementia. J. Neuroimmunol. 2018, 324, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Lucena, D.; Kruse, N.; Thüne, K.; Schmitz, M.; Villar-Piqué, A.; da Cunha, J.E.G.; Hermann, P.; López-Pérez, Ó.; Andrés-Benito, P.; Ladogana, A.; et al. TREM2 expression in the brain and biological fluids in prion diseases. Acta Neuropathol. 2021, 141, 841–859. [Google Scholar] [CrossRef] [PubMed]
- Fabriek, B.O.; Moller, H.J.; Vloet, R.P.; van Winsen, L.M.; Hanemaaijer, R.; Teunissen, C.E.; Uitdehaag, B.M.; van den Berg, T.K.; Dijkstra, C.D. Proteolytic shedding of the macrophage scavenger receptor CD163 in multiple sclerosis. J. Neuroimmunol. 2007, 187, 179–186. [Google Scholar] [CrossRef]
- Housley, W.J.; Pitt, D.; Hafler, D.A. Biomarkers in multiple sclerosis. Clin. Immunol. 2015, 161, 51–58. [Google Scholar] [CrossRef]
- Ferraro, D.; Galli, V.; Vitetta, F.; Simone, A.M.; Bedin, R.; Del Giovane, C.; Morselli, F.; Filippini, M.M.; Nichelli, P.F.; Sola, P. Cerebrospinal fluid CXCL13 in clinically isolated syndrome patients: Association with oligoclonal IgM bands and prediction of multiple sclerosis diagnosis. J. Neuroimmunol. 2015, 283, 64–69. [Google Scholar] [CrossRef] [Green Version]
- De Fino, C.; Lucchini, M.; Lucchetti, D.; Nociti, V.; Losavio, F.A.; Bianco, A.; Colella, F.; Ricciardi-Tenore, C.; Sgambato, A.; Mirabella, M. The predictive value of CSF multiple assay in multiple sclerosis: A single center experience. Mult. Scler. Relat. Disord. 2019, 35, 176–181. [Google Scholar] [CrossRef]
- Zhong, X.; Wang, H.; Dai, Y.; Wu, A.; Bao, J.; Xu, W.; Cheng, C.; Lu, Z.; Qiu, W.; Hu, X. Cerebrospinal fluid levels of CXCL13 are elevated in neuromyelitis optica. J. Neuroimmunol. 2011, 240–241, 104–108. [Google Scholar] [CrossRef]
- Kimura, A.; Kishimoto, T. IL-6: Regulator of Treg/Th17 balance. Eur. J. Immunol. 2010, 40, 1830–1835. [Google Scholar] [CrossRef]
- Uzawa, A.; Mori, M.; Sato, Y.; Masuda, S.; Kuwabara, S. CSF interleukin-6 level predicts recovery from neuromyelitis optica relapse. J. Neurol. Neurosurg. Psychiatry 2012, 83, 339–340. [Google Scholar] [CrossRef]
- Uzawa, A.; Mori, M.; Arai, K.; Sato, Y.; Hayakawa, S.; Masuda, S.; Taniguchi, J.; Kuwabara, S. Cytokine and chemokine profiles in neuromyelitis optica: Significance of interleukin-6. Mult. Scler. 2010, 16, 1443–1452. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Tateishi, T.; Isobe, N.; Yonekawa, T.; Yamasaki, R.; Matsuse, D.; Murai, H.; Kira, J. Characteristic cerebrospinal fluid cytokine/chemokine profiles in neuromyelitis optica, relapsing remitting or primary progressive multiple sclerosis. PLoS ONE 2013, 8, e61835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, A.; Takemura, M.; Saito, K.; Serrero, G.; Yoshikura, N.; Hayashi, Y.; Inuzuka, T. Increased cerebrospinal fluid progranulin correlates with interleukin-6 in the acute phase of neuromyelitis optica spectrum disorder. J. Neuroimmunol. 2017, 305, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, Y.; Sun, X.; Lu, T.; Wei, L.; Fang, L.; Chen, C.; Huang, Q.; Hu, X.; Lu, Z.; et al. Cytokine and Chemokine Profiles in Patients with Neuromyelitis Optica Spectrum Disorder. Neuroimmunomodulation 2016, 23, 352–358. [Google Scholar] [CrossRef]
- Zelek, W.M.; Fathalla, D.; Morgan, A.; Touchard, S.; Loveless, S.; Tallantyre, E.; Robertson, N.P.; Morgan, B.P. Cerebrospinal fluid complement system biomarkers in demyelinating disease. Mult. Scler. 2020, 26, 1929–1937. [Google Scholar] [CrossRef]
- Wang, H.H.; Dai, Y.Q.; Qiu, W.; Lu, Z.Q.; Peng, F.H.; Wang, Y.G.; Bao, J.; Li, Y.; Hu, X.Q. Interleukin-17-secreting T cells in neuromyelitis optica and multiple sclerosis during relapse. J. Clin. Neurosci. 2011, 18, 1313–1317. [Google Scholar] [CrossRef]
- Prins, M.; Dutta, R.; Baselmans, B.; Brevé, J.J.; Bol, J.G.; Deckard, S.A.; van der Valk, P.; Amor, S.; Trapp, B.D.; de Vries, H.E.; et al. Discrepancy in CCL2 and CCR2 expression in white versus grey matter hippocampal lesions of Multiple Sclerosis patients. Acta Neuropathol. Commun. 2014, 2, 98. [Google Scholar] [CrossRef]
- Moreira, M.A.; Souza, A.L.; Lana-Peixoto, M.A.; Teixeira, M.M.; Teixeira, A.L. Chemokines in the cerebrospinal fluid of patients with active and stable relapsing-remitting multiple sclerosis. Braz. J. Med. Biol. Res. 2006, 39, 441–445. [Google Scholar] [CrossRef]
- Malmeström, C.; Andersson, B.A.; Haghighi, S.; Lycke, J. IL-6 and CCL2 levels in CSF are associated with the clinical course of MS: Implications for their possible immunopathogenic roles. J. Neuroimmunol. 2006, 175, 176–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westin, K.; Buchhave, P.; Nielsen, H.; Minthon, L.; Janciauskiene, S.; Hansson, O. CCL2 is associated with a faster rate of cognitive decline during early stages of Alzheimer’s disease. PLoS ONE 2012, 7, e30525. [Google Scholar] [CrossRef] [PubMed]
- Lycke, J.; Zetterberg, H. The role of blood and CSF biomarkers in the evaluation of new treatments against multiple sclerosis. Expert Rev. Clin. Immunol. 2017, 13, 1143–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axelsson, M.; Malmeström, C.; Nilsson, S.; Haghighi, S.; Rosengren, L.; Lycke, J. Glial fibrillary acidic protein: A potential biomarker for progression in multiple sclerosis. J. Neurol. 2011, 258, 882–888. [Google Scholar] [CrossRef]
- Lucchinetti, C.F.; Brück, W.; Rodriguez, M.; Lassmann, H. Distinct patterns of multiple sclerosis pathology indicates heterogeneity on pathogenesis. Brain Pathol. 1996, 6, 259–274. [Google Scholar] [CrossRef]
- Ozawa, K.; Suchanek, G.; Breitschopf, H.; Brück, W.; Budka, H.; Jellinger, K.; Lassmann, H. Patterns of oligodendroglia pathology in multiple sclerosis. Brain 1994, 117, 1311–1322. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Liu, N.; Xie, Q.; Li, X.; Sun, J.; Wang, H.; Wang, M. A Candidate biomarker of glial fibrillary acidic protein in csf and blood in differentiating multiple sclerosis and its subtypes: A systematic review and meta-analysis. Mult. Scler. Relat. Disord. 2021, 51, 102870. [Google Scholar] [CrossRef]
- Petzold, A.; Eikelenboom, M.J.; Gveric, D.; Keir, G.; Chapman, M.; Lazeron, R.H.; Cuzner, M.L.; Polman, C.H.; Uitdehaag, B.M.; Thompson, E.J.; et al. Markers for different glial cell responses in multiple sclerosis: Clinical and pathological correlations. Brain 2002, 125, 1462–1673. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Chang, H.; Li, X.; Wang, H.; Du, L.; Zhou, H.; Xu, W.; Ma, Y.; Yin, L.; Zhang, X. Cytokines and Tissue Damage Biomarkers in First-Onset Neuromyelitis Optica Spectrum Disorders: Significance of Interleukin-6. Neuroimmunomodulation 2018, 25, 215–224. [Google Scholar] [CrossRef]
- Wei, Y.; Chang, H.; Li, X.; Du, L.; Xu, W.; Cong, H.; Yao, Y.; Zhang, X.; Yin, L. CSF-S100B Is a Potential Candidate Biomarker for Neuromyelitis Optica Spectrum Disorders. Biomed. Res. Int. 2018, 2018, 5381239. [Google Scholar] [CrossRef]
- Kaneko, K.; Sato, D.K.; Nakashima, I.; Ogawa, R.; Akaishi, T.; Takai, Y.; Nishiyama, S.; Takahashi, T.; Misu, T.; Kuroda, H.; et al. CSF cytokine profile in MOG-IgG+ neurological disease is similar to AQP4-IgG+ NMOSD but distinct from MS: A cross-sectional study and potential therapeutic implications. J. Neurol. Neurosurg. Psychiatry 2018, 89, 927–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, K.; Sato, D.K.; Nakashima, I.; Nishiyama, S.; Tanaka, S.; Marignier, R.; Hyun, J.W.; Oliveira, L.M.; Reindl, M.; Seifert-Held, T.; et al. Myelin injury without astrocytopathy in neuroinflammatory disorders with MOG antibodies. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1257–1259. [Google Scholar] [CrossRef] [PubMed]
- Fujii, C.; Tokuda, T.; Ishigami, N.; Mizuno, T.; Nakagawa, M. Usefulness of serum S100B as a marker for the acute phase of aquaporin-4 autoimmune syndrome. Neurosci. Lett. 2011, 494, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Misu, T.; Takano, R.; Fujihara, K.; Takahashi, T.; Sato, S.; Itoyama, Y. Marked increase in cerebrospinal fluid glial fibrillar acidic protein in neuromyelitis optica: An astrocytic damage marker. J. Neurol. Neurosurg. Psychiatry 2009, 80, 575–577. [Google Scholar] [CrossRef] [PubMed]
- Takano, R.; Misu, T.; Takahashi, T.; Sato, S.; Fujihara, K.; Itoyama, Y. Astrocytic damage is far more severe than demyelination in NMO: A clinical CSF biomarker study. Neurology 2010, 75, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Petzold, A.; Marignier, R.; Verbeek, M.M.; Confavreux, C. Glial but not axonal protein biomarkers as a new supportive diagnostic criteria for Devic neuromyelitis optica? Preliminary results on 188 patients with different neurological diseases. J. Neurol. Neurosurg. Psychiatry 2011, 82, 467–469. [Google Scholar] [CrossRef]
- Petzold, A. Neurofilament phosphoforms: Surrogate markers for axonal injury, degeneration and loss. J. Neurol. Sci. 2005, 233, 183–198. [Google Scholar] [CrossRef] [Green Version]
- Mattsson, N.; Cullen, N.C.; Andreasson, U.; Zetterberg, H.; Blennow, K. Association between longitudinal plasma neurofilament light and neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 2019, 76, 791–799. [Google Scholar] [CrossRef]
- Khalil, M.; Teunissen, C.E.; Otto, M.; Piehl, F.; Sormani, M.P.; Gattringer, T.; Barro, C.; Kappos, L.; Comabella, M.; Fazekas, F.; et al. Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol. 2018, 14, 577–589. [Google Scholar] [CrossRef]
- Novakova, L.; Zetterberg, H.; Sundström, P.; Axelsson, M.; Khademi, M.; Gunnarsson, M.; Malmeström, C.; Svenningsson, A.; Olsson, T.; Piehl, F.; et al. Monitoring disease activity in multiple sclerosis using serum neurofilament light protein. Neurology 2017, 89, 2230–2237. [Google Scholar] [CrossRef]
- Kuhle, J.; Kropshofer, H.; Hearing, D.A.; Kundu, U.; Meinert, R.; Barro, C.; Dahlke, F.; Tomic, D.; Leppert, D.; Kappos, L. Blood neurofilament light chain as a biomarker of MS disease activity and treatment response. Neurology 2019, 92, e1007–e1015. [Google Scholar] [CrossRef] [PubMed]
- Mariotto, S.; Gastaldi, M.; Grazian, L.; Mancinelli, C.; Capra, R.; Marignier, R.; Alberti, D.; Zanzoni, S.; Schanda, K.; Franciotta, D.; et al. NfL levels predominantly increase at disease onset in MOG-Abs-associated disorders. Mult. Scler. Relat. Disord. 2021, 50, 102833. [Google Scholar] [CrossRef] [PubMed]
- Vakrakou, A.G.; Tzartos, J.S.; Strataki, E.; Boufidou, F.; Dimou, E.; Pyrgelis, E.S.; Constantinides, V.C.; Paraskevas, G.P.; Kapaki, E. Neuronal and neuroaxonal damage cerebrospinal fluid biomarkers in autoimmune encephalitis associated or not with the presence of tumor. Biomedicines 2022, 10, 1262. [Google Scholar] [CrossRef]
- Miyazawa, I.; Nakashima, I.; Petzold, A.; Fujihara, K.; Sato, S.; Itoyama, Y. High CSF neurofilament heavy chain levels in neuromyelitis optica. Neurology 2007, 68, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, C.; Qiu, W.; Lu, Z.; Hu, X.; Wang, K. Cerebrospinal fluid light and heavy neurofilaments in neuromyelitis optica. Neurochem. Int. 2013, 63, 805–808. [Google Scholar] [CrossRef]
- Lijun Peng, Chongfeng Bi, Deyu Xia, Linling Mao, Hairong Qian Increased cerebrospinal fluid neurofilament light chain in central nervous system inflammatory demyelinating disease. Mult. Scler. Relat. Disord. 2019, 30, 123–128. [CrossRef] [Green Version]
- Pereira, J.B.; Westman, E.; Hansson, O.; Alzheimer’s Disease Neuroimaging Initiative. Association between cerebrospinal fluid and plasma neurodegeneration biomarkers with brain atrophy in Alzheimer’s disease. Neurobiol. Aging 2017, 58, 14–29. [Google Scholar] [CrossRef]
- Bos, I.; Vos, S.; Verhey, F.; Scheltens, P.; Teunissen, C.; Engelborghs, S.; Sleegers, K.; Frisoni, G.; Blin, O.; Richardson, J.C.; et al. Cerebrospinal fluid biomarkers of neurodegeneration, synaptic integrity, and astroglial activation across the clinical Alzheimer’s disease spectrum. Alzheimer’s Dement. 2019, 15, 644–654. [Google Scholar] [CrossRef]
- Lehnert, S.; Costa, J.; de Carvalho, M.; Kirby, J.; Kuzma-Kozakiewicz, M.; Morelli, C.; Robberecht, W.; Shaw, P.; Silani, V.; Steinacker, P.; et al. Multicentre quality control evaluation of different biomarker candidates for amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal. Degener. 2014, 15, 344–350. [Google Scholar] [CrossRef]
- Oeckl, P.; Jardel, C.; Salachas, F.; Lamari, F.; Andersen, P.M.; Bowser, R.; de Carvalho, M.; Costa, J.; van Damme, P.; Gray, E.; et al. Multicenter validation of CSF neurofilaments as diagnostic biomarkers for ALS. Amyotroph. Lateral Scler. Frontotemporal. Degener. 2016, 17, 404–413. [Google Scholar] [CrossRef]
- Steinacker, P.; Feneberg, E.; Weishaupt, J.; Brettschneider, J.; Tumani, H.; Andersen, P.M.; von Arnim, C.A.; Böhm, S.; Kassubek, J.; Kubisch, C.; et al. Neurofilaments in the diagnosis of motoneuron diseases: A prospective study on 455 patients. J. Neurol. Neurosurg. Psychiatry 2016, 87, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Weydt, P.; Oeckl, P.; Huss, A.; Müller, K.; Volk, A.E.; Kuhle, J.; Knehr, A.; Andersen, P.M.; Prudlo, J.; Steinacker, P.; et al. Neurofilament levels as biomarkers in asymptomatic and symptomatic familial amyotrophic lateral sclerosis. Ann. Neurol. 2016, 79, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Abu-Rumeileh, S.; Parchi, P. Cerebrospinal fluid and blood neurofilament light chain protein in prion disease and other rapidly progressive dementias: Current state of the art. Front. Neurosci. 2021, 15, 648743. [Google Scholar] [CrossRef]
- Bernstein, H.G.; Baumann, B.; Danos, P.; Diekmann, S.; Bogerts, B.; Gundelfinger, E.D.; Braunewell, K.H. Regional and cellular distribution of neural visinin-like protein immunoreactivities (VILIP-1 and VILIP-3) in human brain. J. Neurocytol. 1999, 28, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Schnurra, I.; Bernstein, H.G.; Riederer, P.; Braunewell, K.H. The neuronal calcium sensor protein VILIP-1 is associated with amyloid plaques and extracellular tangles in Alzheimer’s disease and promotes cell death and tau phosphorylation in vitro: A link between calcium sensors and Alzheimer’s disease? Neurobiol. Dis. 2001, 8, 900–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunewell, K.H. The visinin-like proteins VILIP-1 and VILIP-3 in Alzheimer’s disease-old wine in new bottles. Front. Mol. Neurosci. 2012, 5, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Ng, K.P.; Therriault, J.; Kang, M.S.; Pascoal, T.A.; Rosa-Neto, P.; Gauthier, S.; Alzheimer’s Disease Neuroimaging Initiative. Cerebrospinal fluid phosphorylated tau, visinin-like protein-1, and chitinase-3-like protein 1 in mild cognitive impairment and Alzheimer’s disease. Transl. Neurodegener. 2018, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Mavroudis, I.A.; Petridis, F.; Chatzikonstantinou, S.; Karantali, E.; Kazis, D. A meta-analysis on the levels of VILIP-1 in the CSF of Alzheimer’s disease compared to normal controls and other neurodegenerative conditions. Aging Clin. Exp. Res. 2021, 33, 265–272. [Google Scholar] [CrossRef]
- Tarawneh, R.; Lee, J.M.; Ladenson, J.H.; Morris, J.C.; Holtzman, D.M. CSF VILIP-1 predicts rates of cognitive decline in early Alzheimer disease. Neurology 2012, 78, 709–719. [Google Scholar] [CrossRef] [Green Version]
- Tarawneh, R.; Head, D.; Allison, S.; Buckles, V.; Fagan, A.M.; Ladenson, J.H.; Morris, J.C.; Holtzman, D.M. Cerebrospinal fluid markers of neurodegeneration and rates of brain atrophy in early Alzheimer disease. JAMA Neurol. 2015, 72, 656–665. [Google Scholar] [CrossRef]
- Bishop, P.; Rocca, D.; Henley, J.M. Ubiquitin C-terminal hydrolase L1 (UCH-L1): Structure, distribution and roles in brain function and dysfunction. Biochem. J. 2016, 473, 2453–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setsuie, R.; Wada, K. The functions of UCH-L1 and its relation to neurodegenerative diseases. Neurochem. Int. 2007, 51, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Jung, Y.K. Alzheimer’s disease meets the ubiquitin-proteasome system. Trends Mol. Med. 2004, 10, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Han, Y.; Yu, Q.; Wang, X.; Wang, S.; Liao, X. UCH-L1 inhibition decreases the microtubule-binding function of tau protein. J. Alzheimer’s Dis. 2016, 49, 353–363. [Google Scholar] [CrossRef]
- Öhrfelt, A.; Johansson, P.; Wallin, A.; Andreasson, U.; Zetterberg, H.; Blennow, K.; Svensson, J. Increased Cerebrospinal Fluid Levels of Ubiquitin Carboxyl-Terminal Hydrolase L1 in Patients with Alzheimer’s Disease. Dement. Geriatr. Cogn. Dis. Extra 2016, 6, 283–294. [Google Scholar] [CrossRef]
- Barschke, P.; Oeckl, P.; Steinacker, P.; Al Shweiki, M.R.; Weishaupt, J.H.; Landwehrmeyer, G.B.; Anderl-Straub, S.; Weydt, P.; Diehl-Schmid, J.; Danek, A.; et al. Different CSF protein profiles in amyotrophic lateral sclerosis and frontotemporal dementia with C9orf72 hexanucleotide repeat expansion. J. Neurol. Neurosurg. Psychiatry 2020, 91, 503–511. [Google Scholar] [CrossRef]
- Dobson, R.; Topping, J.; Davis, A.; Thompson, E.; Giovannoni, G. Cerebrospinal fluid and urinary biomarkers in multiple sclerosis. Acta Neurol. Scand. 2013, 128, 321–327. [Google Scholar] [CrossRef]
- Kester, M.I.; Teunissen, C.E.; Crimmins, D.L.; Herries, E.M.; Ladenson, J.H.; Scheltens, P.; van der Flier, W.M.; Morris, J.C.; Holtzman, D.M.; Fagan, A.M. Neurogranin as a cerebrospinal fluid biomarker for synaptic loss in symptomatic Alzheimer disease. JAMA Neurol. 2015, 72, 1275–1280. [Google Scholar] [CrossRef] [Green Version]
- Kvartsberg, H.; Duits, F.H.; Ingelsson, M.; Andreasen, N.; Öhrfelt, A.; Andersson, K.; Brinkmalm, G.; Lannfelt, L.; Minthon, L.; Hansson, O.; et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 1180–1190. [Google Scholar] [CrossRef]
- Wellington, H.; Paterson, R.W.; Portelius, E.; Törnqvist, U.; Magdalinou, N.; Fox, N.C.; Blennow, K.; Schott, J.M.; Zetterberg, H. Increased CSF neurogranin concentration is specific to Alzheimer disease. Neurology 2016, 86, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Portelius, E.; Olsson, B.; Höglund, K.; Cullen, N.C.; Kvartsberg, H.; Andreasson, U.; Zetterberg, H.; Sandelius, Å.; Shaw, L.M.; Lee, V.M.Y.; et al. cerebrospinal fluid neurogranin concentration in neurodegeneration: Relation to clinical phenotypes and neuropathology. Acta Neuropathol. 2018, 136, 363–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkmalm, A.; Brinkmalm, G.; Honer, W.G.; Frölich, L.; Hausner, L.; Minthon, L.; Hansson, O.; Wallin, A.; Zetterberg, H.; Blennow, K.; et al. SNAP-25 is a promising novel cerebrospinal fluid biomarker for synapse degeneration in Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonucci, F.; Corradini, I.; Fossati, G.; Tomasoni, R.; Menna, E.; Matteoli, M. SNAP-25, a known presynaptic protein with emerging postsynaptic functions. Front. Synaptic Neurosci. 2016, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Halbgebauer, S.; Steinacker, P.; Hengge, S.; Oeckl, P.; Rumeileh, S.A.; Anderl-Straub, S.; Lombardi, J.; Von Arnim, C.A.F.; Giese, A.; Ludolph, A.C.; et al. CSF levels of SNAP-25 are increased early in Creutzfeldt-Jakob and Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2022, 93, 1059–1065. [Google Scholar] [CrossRef]
- Nilsson, J.; Ashton, N.J.; Benedet, A.L.; Montoliu-Gaya, L.; Gobom, J.; Pascoal, T.A.; Chamoun, M.; Portelius, E.; Jeromin, A.; Mendes, M.; et al. Quantification of SNAP-25 with Mass Spectrometry and Simoa: A Method Comparison in Alzheimer’s Disease. Alzheimer’s Res. Ther. 2022, 14, 78. [Google Scholar] [CrossRef]
- Milà-Alomà, M.; Brinkmalm, A.; Ashton, N.J.; Kvartsberg, H.; Shekari, M.; Operto, G.; Salvadó, G.; Falcon, C.; Gispert, J.D.; Vilor-Tejedor, N.; et al. CSF Synaptic Biomarkers in the Preclinical Stage of Alzheimer Disease and Their Association with MRI and PET: A Cross-sectional Study. Neurology 2021, 97, e2065–e2078. [Google Scholar] [CrossRef]
- Sutphen, C.L.; McCue, L.; Herries, E.M.; Xiong, C.; Ladenson, J.H.; Holtzman, D.M.; Fagan, A.M.; ADNI. Longitudinal decreases in multiple cerebrospinal fluid biomarkers of neuronal injury in symptomatic late onset Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 869–879. [Google Scholar] [CrossRef]
- Zhang, H.; Therriault, J.; Kang, M.S.; Ng, K.P.; Pascoal, T.A.; Rosa-Neto, P.; Gauthier, S.; Alzheimer’s Disease Neuroimaging Initiative. Cerebrospinal fluid synaptosomal-associated protein 25 is a key player in synaptic degeneration in mild cognitive impairment and Alzheimer’s disease. Alzheimer’s Res. Ther. 2018, 10, 80. [Google Scholar] [CrossRef] [Green Version]
- Neve, R.L.; Finch, E.A.; Bird, E.D.; Benowitz, L.I. Growth-associated protein GAP-43 is expressed selectively in associative regions of the adult human brain. Proc. Natl. Acad. Sci. USA 1988, 85, 3638–3642. [Google Scholar] [CrossRef] [Green Version]
- Andersson, A.; Remnestål, J.; Nellgård, B.; Vunk, H.; Kotol, D.; Edfors, F.; Uhlén, M.; Schwenk, J.M.; Ilag, L.L.; Zetterberg, H.; et al. Development of parallel reaction monitoring assays for cerebrospinal fluid proteins associated with Alzheimer’s disease. Clin. Chim. Acta 2019, 494, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Remnestål, J.; Just, D.; Mitsios, N.; Fredolini, C.; Mulder, J.; Schwenk, J.M.; Uhlén, M.; Kultima, K.; Ingelsson, M.; Kilander, L.; et al. CSF profiling of the human brain enriched proteome reveals associations of neuromodulin and neurogranin to Alzheimer’s disease. Proteom. Clin. Appl. 2016, 10, 1242–1253. [Google Scholar] [CrossRef] [PubMed]
- Lan, G.; Cai, Y.; Li, A.; Liu, Z.; Ma, S.; Guo, T.; Alzheimer’s Disease Neuroimaging Initiative. Association of presynaptic loss with Alzheimer’s disease and cognitive decline. Ann. Neurol. 2022, 92, 1001–1015. [Google Scholar] [CrossRef]
- Sandelius, Å.; Sandgren, S.; Axelsson, M.; Malmeström, C.; Novakova, L.; Kostanjevecki, V.; Vandijck, M.; Blennow, K.; Zetterberg, H.; Lycke, J. Cerebrospinal fluid growth-associated protein 43 in multiple sclerosis. Sci. Rep. 2019, 9, 17309. [Google Scholar] [CrossRef] [Green Version]
- Rot, U.; Sandelius, Å.; Emeršič, A.; Zetterberg, H.; Blennow, K. Cerebrospinal fluid GAP-43 in early multiple sclerosis. Mult. Scler. J. Exp. Transl. Clin. 2018, 4, 2055217318792931. [Google Scholar] [CrossRef] [Green Version]
- Constantinides, V.C.; Paraskevas, G.P.; Emmanouilidou, E.; Petropoulou, O.; Bougea, A.; Vekrellis, K.; Evdokimidis, I.; Stamboulis, E.; Kapaki, E. CSF biomarkers β-amyloid, Tau proteins and a-synuclein in the differential diagnosis of Parkinson-plus syndromes. J. Neurol. Sci. 2017, 382, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Kapaki, E.; Paraskevas, G.P.; Emmanouilidou, E.; Vekrellis, K. The diagnostic value of CSF α-synuclein in the differential diagnosis of dementia with Lewy bodies vs. normal subjects and patients with Alzheimer’s disease. PLoS ONE 2013, 8, e81654. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; Candelise, N.; Baiardi, S.; Capellari, S.; Giannini, G.; Orrù, C.D.; Antelmi, E.; Mammana, A.; Hughson, A.G.; Calandra-Buonaura, G.; et al. Ultrasensitive RT-QuIC assay with high sensitivity and specificity for Lewy body-associated synucleinopathies. Acta Neuropathol. 2020, 140, 49–62. [Google Scholar] [CrossRef]
- Quadalti, C.; Calandra-Buonaura, G.; Baiardi, S.; Mastrangelo, A.; Rossi, M.; Zenesini, C.; Giannini, G.; Candelise, N.; Sambati, L.; Polischi, B.; et al. Neurofilament light chain and α-synuclein RT-QuIC as differential diagnostic biomarkers in parkinsonisms and related syndromes. NPJ Park. Dis. 2021, 7, 93. [Google Scholar] [CrossRef]
- Poggiolini, I.; Gupta, V.; Lawton, M.; Lee, S.; El-Turabi, A.; Querejeta-Coma, A.; Trenkwalder, C.; Sixel-Döring, F.; Foubert-Samier, A.; Pavy-Le Traon, A.; et al. Diagnostic value of cerebrospinal fluid alpha-synuclein seed quantification in synucleinopathies. Brain 2022, 145, 584–595. [Google Scholar] [CrossRef]
- Goldman, J.S.; Farmer, J.M.; Wood, E.M.; Johnson, J.K.; Boxer, A.; Neuhaus, J.; Lomen-Hoerth, C.; Wilhelmsen, K.C.; Lee, V.M.; Grossman, M.; et al. Comparison of family histories in FTLD subtypes and related tauopathies. Neurology 2005, 65, 1817–1819. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, I.R.; Rademakers, R. The molecular genetics and neuropathology of frontotemporal lobar degeneration: Recent developments. Neurogenetics 2007, 8, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Xu, F.; Huang, S.; Li, X.Y.; Lin, J.; Feng, X.; Xie, S.; Wang, Z.; Li, X.; Zhu, J.; Lai, H.; et al. Identification of TARDBP Gly298Ser as a founder mutation for amyotrophic lateral sclerosis in Southern China. BMC Med. Genom. 2022, 15, 173. [Google Scholar] [CrossRef]
- Hasegawa, M.; Arai, T.; Nonaka, T.; Kametani, F.; Yoshida, M.; Hashizume, Y.; Beach, T.G.; Buratti, E.; Baralle, F.; Morita, M.; et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 2008, 64, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Steinacker, P.; Hendrich, C.; Sperfeld, A.D.; Jesse, S.; von Arnim, C.A.; Lehnert, S.; Pabst, A.; Uttner, I.; Tumani, H.; Lee, V.M.; et al. TDP-43 in cerebrospinal fluid of patients with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch. Neurol. 2008, 65, 1481–1487. [Google Scholar] [CrossRef] [Green Version]
- Kasai, T.; Tokuda, T.; Ishigami, N.; Sasayama, H.; Foulds, P.; Mitchell, D.J.; Mann, D.M.; Allsop, D.; Nakagawa, M. Increased TDP-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2009, 117, 55–62. [Google Scholar] [CrossRef]
- Junttila, A.; Kuvaja, M.; Hartikainen, P.; Siloaho, M.; Helisalmi, S.; Moilanen, V.; Kiviharju, A.; Jansson, L.; Tienari, P.J.; Remes, A.M.; et al. Cerebrospinal fluid TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis patients with and without the C9ORF72 hexanucleotide expansion. Dement. Geriatr. Cogn. Dis. Extra 2016, 6, 142–149. [Google Scholar] [CrossRef]
- Kapaki, E.; Boufidou, F.; Bourbouli, M.; Pyrgelis, E.S.; Constantinides, V.C.; Anastassopoulou, C.; Paraskevas, G.P. Cerebrospinal fluid biomarker profile in TDP-43-related genetic frontotemporal dementia. J. Pers. Med. 2022, 12, 1747. [Google Scholar] [CrossRef]
- Bourbouli, M.; Paraskevas, G.P.; Rentzos, M.; Mathioudakis, L.; Zouvelou, V.; Bougea, A.; Tychalas, A.; Kimiskidis, V.K.; Constantinides, V.; Zafeiris, S.; et al. Genotyping and plasma/cerebrospinal fluid profiling of a cohort of frontotemporal dementia-amyotrophic lateral sclerosis patients. Brain Sci. 2021, 11, 1239. [Google Scholar] [CrossRef] [PubMed]
- Bourbouli, M.; Rentzos, M.; Bougea, A.; Zouvelou, V.; Constantinides, V.C.; Zaganas, I.; Evdokimidis, I.; Kapaki, E.; Paraskevas, G.P. Cerebrospinal fluid TAR DNA-binding protein 43 combined with tau proteins as a candidate biomarker for amyotrophic lateral sclerosis and frontotemporal dementia spectrum disorders. Dement. Geriatr. Cogn. Disord. 2017, 44, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Foulds, P.G.; Davidson, Y.; Mishra, M.; Hobson, D.J.; Humphreys, K.M.; Taylor, M.; Johnson, N.; Weintraub, S.; Akiyama, H.; Arai, T.; et al. Plasma phosphorylated-TDP-43 protein levels correlate with brain pathology in frontotemporal lobar degeneration. Acta Neuropathol. 2009, 118, 647–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Songsrirote, K.; Li, Z.; Ashford, D.; Bateman, A.; Thomas-Oates, J. Development and application of mass spectrometric methods for the analysis of progranulin N-glycosylation. J. Proteom. 2010, 73, 1479–1490. [Google Scholar] [CrossRef]
- Chitramuthu, B.P.; Bennett Hugh, P.J.; Bateman, A. Progranulin: A new avenue towards the understanding and treatment of neurodegenerative disease. Brain 2017, 140, 3081–3104. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Kukar, T.; Rademakers, R. Lysosomal dysfunction and other pathomechanisms in FTLD: Evidence from progranulin genetics and biology. Adv. Exp. Med. Biol. 2021, 1281, 219–242. [Google Scholar] [CrossRef]
- Van Damme, P.; van Hoecke, A.; Lambrechts, D.; Vanacker, P.; Bogaert, E.; van Swieten, J.; Carmeliet, P.; Van Den Bosch, L.; Robberecht, W. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J. Cell Biol. 2008, 181, 37–41. [Google Scholar] [CrossRef] [Green Version]
- Petkau, T.L.; Leavitt, B.R. Progranulin in neurodegenerative disease. Trends Neurosci. 2014, 37, 388–398. [Google Scholar] [CrossRef]
- Tanaka, Y.; Chambers, J.K.; Matsuwaki, T.; Yamanouchi, K.; Nishihara, M. Possible involvement of lysosomal dysfunction in pathological changes of the brain in aged progranulin-deficient mice. Acta Neuropathol. Commun. 2014, 2, 78. [Google Scholar] [CrossRef]
- Lui, H.; Zhang, J.; Makinson, S.R.; Cahill, M.K.; Kelley, K.W.; Huang, H.Y.; Shang, Y.; Oldham, M.C.; Martens, L.H.; Gao, F.; et al. Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell 2016, 165, 921–935. [Google Scholar] [CrossRef] [Green Version]
- Baker, M.; Mackenzie, I.R.; Pickering-Brown, S.M.; Gass, J.; Rademakers, R.; Lindholm, C.; Snowden, J.; Adamson, J.; Sadovnick, A.D.; Rollinson, S.; et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006, 442, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Rademakers, R.; Neumann, M.; Mackenzie, I.R. Advances in understanding the molecular basis of frontotemporal dementia. Nat. Rev. Neurol. 2012, 8, 423–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, D.C.; Lehmann, M.; Yokoyama, J.S.; Karydas, A.; Lee, J.J.; Coppola, G.; Grinberg, L.T.; Geschwind, D.; Seeley, W.W.; Miller, B.L.; et al. Progranulin mutations as risk factors for Alzheimer disease. JAMA Neurol. 2013, 70, 774–778. [Google Scholar] [CrossRef] [PubMed]
- Sieben, A.; van Langenhove, T.; Engelborghs, S.; Martin, J.J.; Boon, P.; Cras, P.; de Deyn, P.P.; Santens, P.; van Broeckhoven, C.; Cruts, M. The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol. 2012, 124, 353–372. [Google Scholar] [CrossRef] [Green Version]
- Meeter, L.H.; Patzke, H.; Loewen, G.; Dopper, E.G.; Pijnenburg, Y.A.; van Minkelen, R.; van Swieten, J.C. Progranulin Levels in Plasma and Cerebrospinal Fluid in Granulin Mutation Carriers. Dement. Geriatr. Cogn. Dis. Extra 2016, 6, 330–340. [Google Scholar] [CrossRef]
- Galimberti, D.; Bonsi, R.; Fenoglio, C.; Serpente, M.; Cioffi, S.M.; Fumagalli, G.; Arighi, A.; Ghezzi, L.; Arcaro, M.; Mercurio, M.; et al. Inflammatory molecules in Frontotemporal Dementia: Cerebrospinal fluid signature of progranulin mutation carriers. Brain Behav. Immun. 2015, 49, 182–187. [Google Scholar] [CrossRef]
- Wilke, C.; Gillardon, F.; Deuschle, C.; Hobert, M.A.; Jansen, I.E.; Metzger, F.G.; Heutink, P.; Gasser, T.; Maetzler, W.; Blauwendraat, C.; et al. Cerebrospinal Fluid Progranulin, but Not Serum Progranulin, Is Reduced in GRN-Negative Frontotemporal Dementia. Neurodegener. Dis. 2017, 17, 83–88. [Google Scholar] [CrossRef]
- Morenas-Rodríguez, E.; Cervera-Carles, L.; Vilaplana, E.; Alcolea, D.; Carmona-Iragui, M.; Dols-Icardo, O.; Ribosa-Nogué, R.; Muñoz-Llahuna, L.; Sala, I.; Belén Sánchez-Saudinós, M.; et al. Progranulin Protein Levels in Cerebrospinal Fluid in Primary Neurodegenerative Dementias. J. Alzheimer’s Dis. 2016, 50, 539–546. [Google Scholar] [CrossRef]
- Pawlitzki, M.; Sweeney-Reed, C.M.; Bittner, D.; Lux, A.; Vielhaber, S.; Schreiber, S.; Paul, F.; Neumann, J. CSF-progranulin and neurofilament light chain levels in patients with radiologically isolated syndrome-sign of inflammation. Front. Neurol. 2018, 9, 1075. [Google Scholar] [CrossRef] [Green Version]
- De Riz, M.; Galimberti, D.; Fenoglio, C.; Piccio, L.M.; Scalabrini, D.; Venturelli, E.; Pietroboni, A.; Piola, M.; Naismith, R.T.; Parks, B.J.; et al. Cerebrospinal fluid progranulin levels in patients with different multiple sclerosis subtypes. Neurosci. Lett. 2010, 469, 234–236. [Google Scholar] [CrossRef] [Green Version]
- Vercellino, M.; Grifoni, S.; Romagnolo, A.; Masera, S.; Mattioda, A.; Trebini, C.; Chiavazza, C.; Caligiana, L.; Capello, E.; Mancardi, G.L.; et al. Progranulin expression in brain tissue and cerebrospinal fluid levels in multiple sclerosis. Mult. Scler. 2011, 17, 1194–1201. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Preeti, K.; Fernandes, V.; Khatri, D.K.; Singh, S.B. Role of MicroRNAs, Aptamers in Neuroinflammation and Neurodegenerative Disorders. Cell Mol. Neurobiol. 2022, 42, 2075–2095. [Google Scholar] [CrossRef] [PubMed]
- Selmaj, K.W.; Mycko, M.P.; Furlan, R.; Rejdak, K. Fluid phase biomarkers in multiple sclerosis. Curr. Opin. Neurol. 2022, 35, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Mandolesi, G.; Rizzo, F.R.; Balletta, S.; Stampanoni Bassi, M.; Gilio, L.; Guadalupi, L.; Nencini, M.; Moscatelli, A.; Ryan, C.P.; Licursi, V.; et al. The microRNA let-7b-5p Is Negatively Associated with Inflammation and Disease Severity in Multiple Sclerosis. Cells 2021, 10, 330. [Google Scholar] [CrossRef] [PubMed]
- De Vito, F.; Musella, A.; Fresegna, D.; Rizzo, F.R.; Gentile, A.; Stampanoni Bassi, M.; Gilio, L.; Buttari, F.; Procaccini, C.; Colamatteo, A.; et al. MiR-142-3p regulates synaptopathy-driven disease progression in multiple sclerosis. Neuropathol. Appl. Neurobiol. 2022, 48, e12765. [Google Scholar] [CrossRef]
- Su, Y.; Li, Z.; Rang, X.; Wang, Y.; Fu, J. Integrated Analysis and Identification of CSF-Derived Risk miRNAs and Pivotal Genes in Multiple Sclerosis. J. Mol. Neurosci. 2022, 72, 1916–1928. [Google Scholar] [CrossRef]
- Ahlbrecht, J.; Martino, F.; Pul, R.; Skripuletz, T.; Sühs, K.W.; Schauerte, C.; Yildiz, Ö.; Trebst, C.; Tasto, L.; Thum, S.; et al. Deregulation of microRNA-181c in cerebrospinal fluid of patients with clinically isolated syndrome is associated with early conversion to relapsing-remitting multiple sclerosis. Mult. Scler. 2016, 22, 1202–1214. [Google Scholar] [CrossRef]
- Bergman, P.; Piket, E.; Khademi, M.; James, T.; Brundin, L.; Olsson, T.; Piehl, F.; Jagodic, M. Circulating miR-150 in CSF is a novel candidate biomarker for multiple sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e219. [Google Scholar] [CrossRef] [Green Version]
- Perdaens, O.; Dang, H.A.; D’Auria, L.; van Pesch, V. CSF microRNAs discriminate MS activity and share similarity to other neuroinflammatory disorders. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e673. [Google Scholar] [CrossRef] [Green Version]
- Zheleznyakova, G.Y.; Piket, E.; Needhamsen, M.; Hagemann-Jensen, M.; Ekman, D.; Han, Y.; James, T.; Khademi, M.; Al Nimer, F.; Scicluna, P.; et al. Small noncoding RNA profiling across cellular and biofluid compartments and their implications for multiple sclerosis immunopathology. Proc. Natl. Acad. Sci. USA 2021, 118, e2011574118. [Google Scholar] [CrossRef]
- Sala Frigerio, C.; Lau, P.; Salta, E.; Tournoy, J.; Bossers, K.; Vandenberghe, R.; Wallin, A.; Bjerke, M.; Zetterberg, H.; Blennow, K.; et al. Reduced expression of hsa-miR-27a-3p in CSF of patients with Alzheimer disease. Neurology 2013, 81, 2103–2106. [Google Scholar] [CrossRef]
- Burgos, K.; Malenica, I.; Metpally, R.; Courtright, A.; Rakela, B.; Beach, T.; Shill, H.; Adler, C.; Sabbagh, M.; Villa, S.; et al. Profiles of extracellular miRNA in cerebrospinal fluid and serum from patients with Alzheimer’s and Parkinson’s diseases correlate with disease status and features of pathology. PLoS ONE 2014, 9, e94839. [Google Scholar] [CrossRef] [Green Version]
- Kiko, T.; Nakagawa, K.; Tsuduki, T.; Furukawa, K.; Arai, H.; Miyazawa, T. MicroRNAs in plasma and cerebrospinal fluid as potential markers for Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 39, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Jäkel, L.; Bruinsma, I.B.; Claassen, J.A.; Kuiperij, H.B.; Verbeek, M.M. MicroRNA-29a is a candidate biomarker for Alzheimer’s disease in cell-free cerebrospinal fluid. Mol. Neurobiol. 2016, 53, 2894–2899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sørensen, S.S.; Nygaard, A.B.; Christensen, T. miRNA expression profiles in cerebrospinal fluid and blood of patients with Alzheimer’s disease and other types of dementia—An exploratory study. Transl. Neurodegener. 2016, 5, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starhof, C.; Hejl, A.M.; Heegaard, N.H.H.; Carlsen, A.L.; Burton, M.; Lilje, B.; Winge, K. The biomarker potential of cell-free microrna from cerebrospinal fluid in parkinsonian syndromes. Mov. Disord. 2019, 34, 246–254. [Google Scholar] [CrossRef] [PubMed]
- De Felice, B.; Annunziata, A.; Fiorentino, G.; Borra, M.; Biffali, E.; Coppola, C.; Cotrufo, R.; Brettschneider, J.; Giordana, M.L.; Dalmay, T.; et al. miR-338-3p is over-expressed in blood, CFS, serum and spinal cord from sporadic amyotrophic lateral sclerosis patients. Neurogenetics 2014, 15, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Chandrananda, D.; Thorne, N.P.; Bahlo, M. High-resolution characterization of sequence signatures due to non-random cleavage of cell-free DNA. BMC Med. Genom. 2015, 8, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, W.; Gu, W.; Nagpal, S.; Gephart, M.H.; Quake, S.R. Brain tumor mutations detected in cerebral spinal fluid. Clin. Chem. 2015, 61, 514–522. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Springer, S.; Zhang, M.; McMahon, K.W.; Kinde, I.; Dobbyn, L.; Ptak, J.; Brem, H.; Chaichana, K.; Gallia, G.L.; et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc. Natl. Acad. Sci. USA 2015, 112, 9704–9709. [Google Scholar] [CrossRef] [Green Version]
- García-Romero, N.; Carrión-Navarro, J.; Areal-Hidalgo, P.; Ortiz de Mendivil, A.; Asensi-Puig, A.; Madurga, R.; Núñez-Torres, R.; González-Neira, A.; Belda-Iniesta, C.; González-Rumayor, V.; et al. BRAF V600E detection in liquid biopsies from pediatric central nervous system tumors. Cancers 2019, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Lee, J.; Jung, E.S.; Kim, M.H.; Kim, I.B.; Son, H.; Kim, S.; Kim, S.; Park, Y.M.; Mook-Jung, I.; et al. Brain somatic mutations observed in Alzheimer’s disease associated with aging and dysregulation of tau phosphorylation. Nat. Commun. 2019, 10, 3090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pai, M.C.; Kuo, Y.M.; Wang, I.F.; Chiang, P.M.; Tsai, K.J. The role of methylated circulating nucleic acids as a potential biomarker in Alzheimer’s disease. Mol. Neurobiol. 2019, 56, 2440–2449. [Google Scholar] [CrossRef] [PubMed]
- Bahado-Singh, R.O.; Radhakrishna, U.; Gordevičius, J.; Aydas, B.; Yilmaz, A.; Jafar, F.; Imam, K.; Maddens, M.; Challapalli, K.; Metpally, R.P.; et al. Artificial intelligence and circulating cell-free DNA methylation profiling: Mechanism and detection of Alzheimer’s disease. Cells 2022, 11, 1744. [Google Scholar] [CrossRef] [PubMed]
- Lehmann-Werman, R.; Neiman, D.; Zemmour, H.; Moss, J.; Magenheim, J.; Vaknin-Dembinsky, A.; Rubertsson, S.; Nellgård, B.; Blennow, K.; Zetterberg, H.; et al. Identification of tissue-specific cell death using methylation patterns of circulating dna. Proc. Natl. Acad. Sci. USA 2016, 113, E1826–E1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendioroz, M.; Martínez-Merino, L.; Blanco-Luquin, I.; Urdánoz, A.; Roldán, M.; Jericó, I. Liquid biopsy: A new source of candidate biomarkers in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2018, 5, 763–768. [Google Scholar] [CrossRef] [Green Version]
- Caggiano, C.; Celona, B.; Garton, F.; Mefford, J.; Black, B.L.; Henderson, R.; Lomen-Hoerth, C.; Dahl, A.; Zaitlen, N. Comprehensive cell type decomposition of circulating cell-free DNA with CelFiE. Nat. Commun. 2021, 12, 2717. [Google Scholar] [CrossRef]
- Grazioli, S.; Pugin, J. Mitochondrial damage-associated molecular patterns: From inflammatory signaling to human diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef] [Green Version]
- Lowes, H.; Pyle, A.; Duddy, M.; Hudson, G. Cell-free mitochondrial DNA in progressive multiple sclerosis. Mitochondrion 2019, 46, 307–312. [Google Scholar] [CrossRef]
- Leurs, C.E.; Podlesniy, P.; Trullas, R.; Balk, L.; Steenwijk, M.D.; Malekzadeh, A.; Piehl, F.; Uitdehaag, B.M.; Killestein, J.; van Horssen, J.; et al. Cerebrospinal fluid mtDNA concentration is elevated in multiple sclerosis disease and responds to treatment. Mult. Scler. 2018, 24, 472–480. [Google Scholar] [CrossRef]
- Podlesniy, P.; Figueiro-Silva, J.; Llado, A.; Antonell, A.; Sanchez-Valle, R.; Alcolea, D.; Lleo, A.; Molinuevo, J.L.; Serra, N.; Trullas, R. Low cerebrospinal fluid concentration of mitochondrial DNA in preclinical Alzheimer disease. Ann. Neurol. 2013, 74, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Pyle, A.; Brennan, R.; Kurzawa-Akanbi, M.; Yarnall, A.; Thouin, A.; Mollenhauer, B.; Burn, D.; Chinnery, P.F.; Hudson, G. Reduced cerebrospinal fluid mitochondrial DNA is a biomarker for early-stage Parkinson’s disease. Ann. Neurol. 2015, 78, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Lowes, H.; Pyle, A.; Santibanez-Koref, M.; Hudson, G. Circulating cell-free mitochondrial DNA levels in Parkinson’s disease are influenced by treatment. Mol. Neurodegener. 2020, 15, 10. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Biomarker | Specific Biological Process | Neurodegenerative Diseases | Neuroinflammatory Diseases | Methodology (Most Used) |
|---|---|---|---|---|
| KFLC | Intrathecal immunoglobulin synthesis | N/A | MS, CIS | Nephelometry |
| YKL-40 | Glial activation | AD, ALS, CJD, FTD | RRMS, CIS, NMO | Elisa |
| sTREM2 | Glial activation | AD, PD, FTD, CJD | MS | Elisa |
| sCD163 | Activated microglia and tissue macrophages | Not studied | MS | Elisa |
| CXCL13 | B-cell activation and recruitment to CNS | Not studied | RRMS, CIS, NMO | Elisa |
| IL-6 | Th2 and Th17-related inflammation | Not studied | NMO | Elisa |
| MCP-1/CCL2 | Glial activation, monocyte recruitment | AD | MS (reduced) | Elisa |
| GFAP | Glial activation (astrocytes) | AD | SPMS > RPMS, NMO | Elisa |
| Biomarker | Specific Biological Process | Neurodegenerative Diseases | Neuroinflammatory Diseases | Methodology (Most Used) |
|---|---|---|---|---|
| NF-L | Axonal dysfunction | AD, FTD, VaD, CJD, ALS, PSP, MSA, CBD | RRMS, SPMS, PPMS, CIS, NMO, MOGAD | ELISA |
| VILIP-1 | Axonal dysfunction | AD | Not studied | ELISA |
| UCH-L1 | Axonal dysfunction | AD | No difference | ELISA |
| Neurogranin | Synaptic degeneration | AD | Not studied | ELISA, mass spectrometry |
| SNAP-25 | Synaptic degeneration | AD | not studied | ELISA, mass spectrometry, SiMoA |
| GAP-43 | Synaptic degeneration | AD | contradictory results | ELISA |
| Protein Metabolism/Aggregation | ||||
|---|---|---|---|---|
| Biomarker | Specific Biological Process | Neurodegenerative Diseases | Neuroinflammatory Diseases | Methodology (Most Used) |
| a-synuclein | a-Syn pathology | AD, PD, CBD, MSA, DLB | Not applicable | ELISA, RT-QuIC, PMCA |
| TDP-43 | TDP-43 metabolism | ALS, FTD | Not applicable | ELISA |
| Progranulin | Glial activation inhibitor | AD, FTD | MS | ELISA |
| Molecular biomarkers | ||||
| miRNA | Various molecular targets implicated in molecular networks | AD (miR-27a-3p miR-101 miR-29a, miR-29b, miR- 34a, miR-125b, miR-29c-3p, miR15a-5p, let-7i-5p, miR-146a) PD (miR-7-5p, miR-331-5p, miR-145-5p), MSA (miR-9-3p and miR-106b-5p), PSP (miR-106b-5p), ALS (miR-338-3P) | MS (miR-181c; miR-150; miR-328; miR-34c-5p; miR-142-3p; miR-let-7b-5p and miR-15a-3p/124-5p/149-3p/29c-3p/33a-3p/34c-5p/297) | RT-qPCR, NGS |
| Cell-free DNA and mitochondrial DNA | Cell death | AD, PD | MS | RT-qPCR, ddPCR, fluorometric analysis, sequencing for mutations, methylation sequencing (qMSP) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapaki, E.; Vakrakou, A.G.; Boufidou, F. Novel CSF Biomarkers Tracking Autoimmune Inflammatory and Neurodegenerative Aspects of CNS Diseases. Diagnostics 2023, 13, 73. https://doi.org/10.3390/diagnostics13010073
Kapaki E, Vakrakou AG, Boufidou F. Novel CSF Biomarkers Tracking Autoimmune Inflammatory and Neurodegenerative Aspects of CNS Diseases. Diagnostics. 2023; 13(1):73. https://doi.org/10.3390/diagnostics13010073
Chicago/Turabian StyleKapaki, Elisabeth, Aigli G. Vakrakou, and Fotini Boufidou. 2023. "Novel CSF Biomarkers Tracking Autoimmune Inflammatory and Neurodegenerative Aspects of CNS Diseases" Diagnostics 13, no. 1: 73. https://doi.org/10.3390/diagnostics13010073
APA StyleKapaki, E., Vakrakou, A. G., & Boufidou, F. (2023). Novel CSF Biomarkers Tracking Autoimmune Inflammatory and Neurodegenerative Aspects of CNS Diseases. Diagnostics, 13(1), 73. https://doi.org/10.3390/diagnostics13010073