Abstract
Wilson disease and alpha1-antitrypsin deficiency are two rare genetic diseases that may impact predominantly the liver and/or the brain, and the liver and/or the lung, respectively. The early diagnosis of these diseases is important in order to initiate a specific treatment, when available, ideally before irreversible organ damage, but also to initiate family screening. This review focuses on the non-invasive diagnostic tests available for clinicians in both diseases. These tests are crucial at diagnosis to reduce the potential diagnostic delay and assess organ involvement. They also play a pivotal role during follow-up to monitor disease progression and evaluate treatment efficacy of current or emerging therapies.
1. Introduction
Wilson disease (WD) and alpha1-antitrypsin deficiency (AATD) are two rare genetic diseases affecting predominantly the liver and/or the brain, and the liver and/or the lung, respectively [1,2]. The early diagnosis of these diseases is important in order to initiate a specific treatment, when available, ideally before irreversible organ damage, but also to initiate family screening. The diagnosis of AATD is usually relatively straightforward, as it relies on the measurement of a serum A1-antitrypsin (AAT) level that is below normal values, whereas the diagnosis of WD can be challenging, as no single test can confirm or exclude the disease.
Liver histology is not routinely required for establishing the diagnosis of AATD or WD but can help by identifying characteristics consistent with either disease, while at the same time excluding other causes, and also by staging/grading the hepatic involvement. In WD, histological findings from liver biopsies are often non-specific [3,4]. Mild steatosis and mild inflammation are often seen at the early stages. Later, an auto-immune hepatitis-like pattern of injury may be present, with parenchymal mononuclear inflammatory infiltrate and interface hepatitis. Portal and periportal fibrosis progresses to portal–portal bridging and finally mixed macro-/micronodular cirrhosis. In cases of acute liver failure (ALF) due to WD, multi-lobular (sub-massive) necrosis may be identified, almost always superimposed upon cirrhosis. Copper is heterogeneously deposited in the liver in WD, varying from lobule to lobule in early disease and from nodule to nodule in cirrhosis, which may lead to negative histochemical staining results for copper deposition, especially in small samples. Quantitative copper analysis of the liver biopsy can help, particularly in difficult cases: high intrahepatic copper (>250 µg/g dry weight) is in favor of the diagnosis of WD in the absence of chronic cholestasis, whereas in untreated patients, normal hepatic copper content (<50 µg/g dry weight) almost always excludes this diagnosis [4]. In patients with severe AATD, while the intrahepatic histological features are variable and mostly non-specific, the most characteristic feature seen is the presence of PAS-positive diastase-resistant inclusions in the cytoplasm of periportal hepatocytes. Other, non-specific, features are the presence of steatosis (sometimes with features of steatohepatitis), lymphocyte-predominant chronic inflammatory cell infiltrate (mostly portal and lobular inflammation, sometimes mild-to-moderate interface hepatitis or centrilobular inflammation), portal fibrosis, bridging fibrosis, and finally cirrhosis [5,6]. Liver biopsy may be helpful for some patients but is associated with morbidity in 0.5% patients (bleeding or pneumothorax) and mortality in 0.02%, and cannot, therefore, be recommended in all patients [7]. In children, it often requires general anesthesia and one overnight stay to observe the absence of bleeding [8].
2. Materials and Methods
In this review, we present the non-invasive tests available or in development for clinicians to establish a diagnosis and assess organ involvement in WD and AATD. PubMed/MEDLINE was searched for reports in English language using the following key words: Wilson disease, alpha1-antitrypsin, diagnosis, histology, imaging, genetic. We considered original articles, reviews, meta-analyses, and book chapters published in the last 30 years.
3. Results
3.1. Wilson Disease
WD is a rare autosomal recessive inherited copper overload disease caused by ATP7B mutations (coding for ATPase 7B) responsible for impaired biliary copper excretion. It results in the accumulation of copper in various tissues and organs, predominantly in the liver, brain and cornea [9]. Making a diagnosis can be challenging given that the disease presentations vary widely and need a combination of diagnostic tests [10]. For these reasons, delayed diagnoses are common. A composite scoring system referred to as the Leipzig score combines the assessment of clinical and mostly non-invasive laboratory or morphological findings (Kayser–Fleischer rings, neurological symptoms or abnormalities on brain MRI, serum ceruloplasmin, Coombs-negative hemolytic anemia, hepatic and urinary copper levels) with genetic data to aid in the diagnosis of WD (Table 1) [11].

Table 1.
Leipzig score (a scoring system for the diagnosis of Wilson disease).
3.1.1. Clinical Features at Onset
- WD generally presents in children and young adults between the second and fourth decade of life [12]. However, younger (<5 years) and older ages (>40 years) have been extensively reported at WD onset [13]. Late-onset WD has been reported for both hepatic or neuropsychiatric forms [13,14,15]. Age per se cannot eliminate the diagnosis of WD, even if the index of suspicion should be higher in children and young adults, and a wider screen should be performed at initial presentation in these patients [16]. It also has to be kept in mind that less than 10% of the WD patients were diagnosed after 45 years [17].
- WD patients can present with a wide range of hepatic, neurological, and psychiatric manifestations, combined in various and unpredictable ways [12]. All patterns of liver disease have been described in WD, in both pediatric and adult populations, including asymptomatic abnormalities in liver biochemistry, hepatic steatosis or hepatomegaly on imaging, acute or chronic hepatitis, cirrhosis and acute liver failure [18]. Dysarthria is the most frequent neurological feature reported in large cohorts and is followed by dystonia, tremor, abnormal gait, parkinsonism, and chorea [13]. Dysarthria, dystonia, tremor, and parkinsonism can be the sole initial manifestations of WD. Psychiatric manifestations vary from mood disorders (including depression and bipolar disorders), to personality disorders (including antisocial behavior and sexual disinhibition), and, finally, cognitive impairment [12].
- A Coombs-negative hemolytic anemia and ophthalmological findings such as Kayser–Fleischer ring (KFR) and sunflower cataract are classical non-hepatic/non-neuropsychiatric manifestations seen in WD. Non-immune hemolytic anemia occurs in 4–10% of cases and is highly suggestive of WD when associated with unexplained liver disease or movement disorders [16,19,20] but may also precede other symptoms [21]. The presentation of an unexplained Coombs-negative hemolytic anemia in a child or young adult should always be considered as a case of WD unless proved otherwise. In patients with severe liver failure, marked hemolysis is frequently seen (in around 50–80% of cases [22,23]) and may result from increased non-ceruloplasmin-bound copper released from impaired hepatocytes [20]. KFR consists of a ring-shaped copper deposit in the internal corneal layer of the Descemet’s membrane, seen on slit lamp examination in 90% of neurological presentations, 36–62% of hepatic presentations, and is highly suggestive of WD [24]. KFR can be visible as a yellowish-green or golden-brown discoloration at the periphery of each cornea, distinct from the underlying iris. KFR confirms the presence of excess free copper in the bloodstream but is not pathognomonic for WD, as it may occur in any disorder with impaired biliary copper excretion such as chronic cholestasis. Sunflower cataract is a rare but also characteristic sign, where copper deposits are situated under the lens capsule. Notably, sunflower cataracts do not impact visual acuity and can be reversible after anti-copper treatment [25].
- In non-diagnosed or undertreated women of child-bearing age, infertility or repeated miscarriages are often seen [4,13]. Other rare non-hepatic/non-neuropsychiatric manifestations may be present at the time of diagnosis or develop later, and include renal abnormalities (Fanconi syndrome, nephrolithiasis), cardiac abnormalities (cardiomyopathy, arrythmia), skeletal abnormalities (osteopenia/osteoporosis), pancreatitis, and hypo-parathyroidism.
3.1.2. Laboratory Tests of Copper Metabolism (Table 2)
- The classical triad of “low ceruloplasmin, low serum copper and increased 24-h urinary copper levels” is usually associated with the diagnosis of WD but may be absent in some cases confirmed by genetic testing, and is present in 16% of healthy heterozygous carriers [13]. Some conditions may be responsible for false positive or false negative results and may lead to an incorrect interpretation of copper balance.
- Ceruloplasmin
- A very low ceruloplasmin level (<0.10 g/L; informative cut-off in Leipzig score) is highly suggestive of WD; intermediate concentrations (0.10–0.20 g/L) are less specific, and up to 15% of patients with neurological presentations have normal concentrations (>0.20 g/L) [11,16]. However, a serum ceruloplasmin concentration (measured immunologically) below 0.14 g/L is reported to have a better positive predictive values than a cut-off of 0.20 g/L for the diagnosis of WD [26]. Ceruloplasmin levels have been found to be reduced in other chronic liver diseases, mostly as a consequence of cirrhosis and impaired liver function, but also in cases of malabsorption, malnutrition, marked renal protein loss, acquired copper deficiency, or other rare, inherited metabolic disorders mimicking WD (Menkes disease, aceruloplasminemia, congenital disorders of glycosylation, Nieman Pick C, MEDNIK syndrome) [27]. Moreover, up to 20% of heterozygous ATP7B carriers have a serum caeruloplasmin level of 0.15–0.19 g/L [28]. Conversely, ceruloplasmin is increased by estrogen, pregnancy and contraceptive pills. Being an acute-phase response protein, it also increases during inflammation or infections. Lastly, there are methodological concerns about the widely used immunological assay for ceruloplasmin, which also detects apo-ceruloplasmin and may overestimate results.
- 24 h urinary copper excretion
- The urinary copper concentration is a relatively simple and sensitive test for the diagnosis of WD but also for the monitoring of the effectiveness of WD treatment [4,29,30,31]. In untreated WD patients, the 24 h copper urinary excretion reflects the amount of non-ceruloplasmin-bound copper in the circulation [32]. Diagnostic cut-offs and ranges depend on the laboratory. In a study of 111 healthy adults from the UK, the mean copper output was 0.34 μmol/24 h (21 μg/24 h) [33]. The literature favors cut-offs >1.6 μmol/24 h (>100 μg/24 h) for the diagnosis of WD [15,29,30,34]. However, multiple studies have shown this cut-off to be too high, particularly in children and asymptomatic siblings. With a cutoff >0.64 μmol/24 h (>40 μg/24-h), urinary copper output has 79% sensitivity and 88% specificity for the diagnosis of Wilson disease in children [35]. Moreover, high urinary copper values could be seen in other types of liver disease (e.g., autoimmune hepatitis, NAFLD, cholestasis, and during acute hepatic failure of any origin). Heterozygotes may also have intermediate levels [36].
- The D-penicillamine challenge test (PCT), which consists of measuring the 24 h urinary copper excretion after a dose of D-penicillamine (usually 1 g/day, either once 1 g or twice 500 mg) has been proposed as a tool for WD diagnosis. In a preliminary study, the measurement of 24 h urine copper excretion after PCT was significantly higher in WD patients (three adults and seven children; mean ± standard deviation 37 ± 5.7 µmol/24 h [2330 ± 360 µg/24 h]) in comparison to 10 heterozygote adults (15.9 ± 3.3 µmol/24 h [1000 ± 210 µg/24 h]) and 26 normal adults (10.2 ± 3.7 µmol/24 h [640 ± 230 µg/24 h]) [36]. This was then validated mostly in children: a cut-off >25 µmol/24 h (1600 µg/24 h) had a good sensitivity and positive predictive value for symptomatic patients, but this cut-off was not sufficiently sensitive in asymptomatic patients with WD [37,38,39].
- Serum copper
- Most of the copper measured as serum copper is contained within ceruloplasmin, which explains why in WD patients serum copper is usually low. However, in cases of severe injury related to WD, high serum copper is frequently seen related to a massive release of hepatocellular copper. When considering the diagnosis of WD in the setting of acute liver failure, the interpretation of conventional copper testing is often misleading, as serum copper is high, 24 h urinary copper excretion very high, and ceruloplasmin levels can be either low, normal, or high. These tests are not as reliable as in chronic WD patients and are less sensitive and specific than other available tests such as hemoglobin, serum transaminases, alkaline phosphatase and total bilirubin [40]. In cases of acute liver failure, the presence of a nonimmune acute intravascular hemolysis with severe coagulopathy, relatively modest elevations of serum aminotransferases, normal or subnormal alkaline phosphatase and progression to renal failure is suggestive of the diagnosis of WD [4,41].
- Non-ceruloplasmin-bound copper (NCC)
- The proportion of non-ceruloplasmin-bound (“free”) copper with respect to total copper is increased in patients with WD. NCC can be calculated in µmol/L by subtracting the ceruloplasmin in g/L multiplied by 47 from the serum copper in µmol/L [42]. Variations in the sensitivity and specificity of ceruloplasmin assays between laboratories preclude the definition of an optimal cut-off at diagnosis and during follow-up [43]. Calculated NCC is not useful for WD diagnosis but may help to monitor response with chelation or zinc therapy [42,44].
- An alternative approach consists of measuring the labile fraction of copper bound to albumin and other peptides called exchangeable copper (CuEXC), which represents the majority of NCC. CuEXC is measured by adding EDTA (that binds to the complex ceruloplasmin–copper) to a serum sample and performing ultrafiltration prior to copper quantification [45]. Levels of CuEXC are higher in animal models of WD, in newly diagnosed symptomatic patients, in non-compliant patients, as well as in patients with extrahepatic involvement, and also correlate with neurological severity [46,47,48,49,50]. Expressed as the percentage of total serum copper, relative exchangeable copper (REC) is a valuable diagnostic test with a high sensitivity and specificity. With a cut-off of 18.5%, REC was able to discriminate WD patients from heterozygous ATP7B carriers, healthy controls and patients with other liver diseases [45,47,48,51]. CuEXC has also shown interesting results to monitor treatment response in animal models and WD patients, but an optimal cut-off has yet to be defined [46,48].
- Other techniques directly measuring NCC, such as copper speciation, have been reported recently and are currently under investigation to assess the treatment efficacy in clinical trials [52].
Table 2.
Classical features of diagnostic tests for WD.
Table 2.
Classical features of diagnostic tests for WD.
| Normal | Heterozygotes | Pre-Symptomatic WD | Chronic Hepatic WD | ALF-WD | Neurologic WD | |
|---|---|---|---|---|---|---|
| Ceruloplasmin (g/L) | normal (≥0.20) | usually normal 0.15–0.19 in 10–15% | ≤0.14 but may be normal | ≤0.14 but may be normal | usually ≤0.14 often 0.15–0.19 or ≥0.20 | ≤0.14 but may be normal |
| Total copper | normal | normal, sometimes decreased | usually decreased | usually decreased | normal or increased | usually decreased |
| Exchangeable copper | normal | normal | usually normal * | increased but may be normal | Increased * | increased |
| REC (%) | <18.5 | <18.5 | >18.5 sometimes 10–18.5% * | >18.5 | >18.5 * | >18.5 |
| 24 h urine copper (µg/24 h) | <40 | usually <40 sometimes 40–100 | usually >40 | usually >100 sometimes 40–100 | >100 (frequently >1000) | >100 |
| Intrahepatic copper (µg/g dry weight) | absent | absent | probably >50 (LB usually not performed) | usually >250 almost always >50 | usually >250 almost always >50 | usually >250 almost always >50 (LB usually not performed) |
| Nonimmune hemolytic anemia | absent | absent | absent | sometimes ≤10% | very frequent >60% | sometimes ≤10% |
| KF ring | absent | absent | <50% | 50% | 50% | >95% |
| Brain MRI abnormalities | absent | absent | <50% | 50% | 50% | >95% |
| Number of alleles with ATPase 7B disease-causing mutation | 0 | 1 | 2 | 2 | 2 | 2 |
| Ceruloplasmin (g/L) | normal (≥0.20) | usually normal 0.15–0.19 in 10–15% | ≤0.14 but may be normal | ≤0.14 but may be normal | usually ≤0.14 often 0.15–0.19 or ≥0.20 | ≤0.14 but may be normal |
| Total copper | normal | normal, sometimes decreased | usually decreased | usually decreased | normal or increased | usually decreased |
| Exchangeable copper | normal | normal | usually normal * | increased but may be normal | increased * | increased |
| REC (%) | <18.5 | <18.5 | >18.5 sometimes 10–18.5% * | >18.5 | >18.5 * | >18.5 |
| 24 h urine copper (µg/24 h) | <40 | usually <40 sometimes 40–100 | usually >40 | usually >100 sometimes 40–100 | >100 (frequently >1000) | >100 |
| Intrahepatic copper (µg/g dry weight) | absent | absent | probably >50 (LB usually not performed) | usually >250 almost always >50 | usually >250 almost always >50 | usually >250 almost always >50 (LB usually not performed) |
| Nonimmune hemolytic anemia | absent | absent | absent | sometimes ≤10% | very frequent >60% | sometimes ≤10% |
| KF ring | absent | absent | <50% | 50% | 50% | >95% |
| Brain MRI abnormalities | absent | absent | <50% | 50% | 50% | >95% |
| Number of alleles with ATPase 7B disease-causing mutation | 0 | 1 | 2 | 2 | 2 | 2 |
Abbreviations: WD, Wilson disease; ALF, acute liver failure; LB, liver biopsy; KF, Kayser–Fleischer; MRI, magnetic resonance imaging. * personal data.
3.1.3. Genetic Testing
ATP7B sequencing has an important role in confirming the clinical diagnosis in all patients suspected of having WD but should not delay the initiation of treatment. It is also helpful when initial investigations are inconclusive and for family screening. A great number of mutations have been reported that hamper the proper function of ATPase 7B. More than 900 deleterious variants in the ATP7B gene have been identified, with single-nucleotide missense and nonsense mutations being the most common, followed by small deletions, splice site mutations and small insertions [53]. In a genetic study of 181 patients from the UK, two mutations were identified in 98% of the participants when using a combination of Sanger sequencing (including coding regions, splice sites, and promoter regions) and multiplex ligation-dependent probe amplification (for the identification of deletions and duplications) [54]. The most common mutation among patients from Northern and Eastern Europe is His1069Gln, but its frequency varies significantly between countries: 86% of patients in Poland, 19% in UK, and 14% in France [54,55,56]. To date, most of the studies have not established a clear phenotype–genotype correlation regarding initial symptomatic manifestations [57]. Studies on modifier genes are ongoing in order to better understand differences in clinical expression despite similar mutations.
3.1.4. Brain Magnetic Resonance Imaging (MRI)
Brain MRI is the most sensitive neuroimaging method for the diagnosis of neurological WD. More than 90% of WD patients with neurological disease and approximately half of the patients with hepatic symptoms have abnormalities on brain MRI. Symmetric hyperintense signal abnormality in the basal ganglia, thalamus, and/or brainstem in T2-weighted or FLAIR images is characteristic of WD [58]. The face of the giant panda sign, where hyperintense signal abnormality surrounds the red nucleus and substantia nigra, is highly specific for WD but only seen in a minority of patients. In addition, signs of diffuse tissue atrophy and hypointensities in T2/T2* and SWI images in the deep gray matter caused by abnormal iron accumulation are frequently present. In order to assess the severity of the radiological findings, Dusek et al. recently proposed a semiquantitative MRI scale that includes an acute toxicity subscore based on the distribution and severity of these hyperintense signal abnormalities and a chronic damage subscore based on brain atrophy and hypointense signal abnormalities on T2/T2*/SWI [59]. In a validation study that included 39 treatment-naïve patients, the acute toxicity subscore improved with treatment (p = 0.02) but did not correlate with the Unified Wilson Disease Rating Scale (UWDRS) score at baseline and after 2 years. The chronic damage subscore correlated with UWDRS at baseline (r = 0.59, p = 0.005) and 24 months (r = 0.68, p < 0.001). However, a recent report on six children with neurological WD has shown that brain MRI had poor prognostic value in distinguishing reversible from irreversible T2-hyperintense lesions after LT [60]. Therefore, brain changes found on MRI should not preclude LT in these patients because they failed to predict clinical outcomes after LT.
3.1.5. Liver Imaging
All patients with suspected WD should have a liver ultrasound (US) evaluation irrespective of their clinical presentation. Patients with exclusive neurological symptoms have a high rate of liver abnormalities on imaging, and morphological signs of liver cirrhosis are frequently found in these patients [61]. Hepatic steatosis is the most common finding, seen in 35–88% of WD patients. Cholelithiasis is not rare in WD and has been reported in 20–25% of patients [62,63]. Irregular liver edge, reversed portal vein flow, splenomegaly or ascites are signs that suggest the presence of cirrhosis. Computed tomography (CT) and/or MRI can also show signs of liver cirrhosis and portal hypertension (PHT). Multiple hyperechoic and hypoechoic nodular lesions with honeycomb pattern, a perihepatic fat layer, and the absence of caudate lobe hypertrophy in a cirrhotic liver are radiological features often seen in WD patients [62,64]. The occurrence of hepatobiliary malignancies (hepatocellular carcinoma and cholangiocarcinoma) during follow-up is a rare complication of WD [17,65]; however, a semestrial screening with abdominal US should be performed in all patients with cirrhosis. Interestingly, in cases of liver tumor, a higher rate of cholangiocarcinoma than expected is observed, which advocates for a frequent histological assessment [65].
3.1.6. Liver Stiffness Measurement (LSM)
Data regarding LSM are relatively scarce in WD, particularly in newly diagnosed patients [66,67,68,69,70]. Paternostro et al. analyzed the results of LSM measured by transient elastography (TE) in 188 WD patients who had concomitant liver biopsy, 44 of whom had recent WD diagnosis before treatment [67]. LSM was higher in cirrhotic compared to non-cirrhotic patients (11.3 vs. 6.1 kPa, p < 0.001). This was even more pronounced in recently diagnosed patients (35.2 kPa vs. 6.4 kPa, p < 0.001). The accuracy of an LSM cutoff ≥9.9 kPa for the diagnosis of cirrhosis was better in recently diagnosed (predictive positive value (PPV): 74%, negative predictive value (NPV): 100%) vs. previously diagnosed (PPV: 53%, NPV: 82%) patients. In most WD patients, LSM measured by TE remained stable over time (median follow-up of 46 (24–66) months), while a significant proportion (30.8%) of cirrhotic patients showed improvements of LSM under WD therapy. LSM only worsened in 5.6% of recently diagnosed patients and in 8.5% of previously diagnosed patients. In another recent study, a value >10.45 kPa of liver stiffness measured by two-dimensional real-time shear wave elastography (2D-SWE) was associated with an increased risk of hypersplenism [71].
3.1.7. New Non-Invasive Tests
- ATP7B peptides
- Direct quantification of ATP7B peptides on dried blood spots using immunoaffinity-enriched mass spectrometry has recently been proposed as a novel diagnostic test for WD [72]. This technique identified WD patients with a sensitivity of 92% and specificity of 98% in a retrospective cohort of 264 patients and 150 healthy controls. ATP7B peptide concentrations were below diagnostic cut-offs in 101/107 (94%) with Cp < 10 mg/dL, 70/77 (91%) with Cp 10–20 mg/dL, and 14/16 (88%) with normal serum ceruloplasmin (>0.2 g/L). This suggests that a combination of ATP7B peptide quantification and serum ceruloplasmin is likely to improve diagnostic accuracy. Further studies are needed to validate this new technique and confirm these encouraging preliminary findings.
- Anterior segment optical coherence tomography (AS-OCT)
- AS-OCT could be used for the detection of KFR. In a study of 29 patients with WD, 15 had normal slit-lamp evaluation but abnormal AS-OCT (p < 0.001), suggesting that AS-OCT is a more accurate diagnostic tool that could detect significantly more cases of KFR as compared to the slit-lamp examination [73]. This technique could allow more easy recognition of KFR by non-expert ophthalmologists, as well as non-ophthalmologists [74,75,76]. It also can be useful to quantify the KF rings that can be followed over time in patients undergoing treatment to assess its effectiveness.
- Brain magnetic resonance spectroscopy (MRS)
- Brain MRS could potentially serve as a method for detecting early neurological involvement, particularly in pediatric patients with WD [77]. MRS demonstrates that N-acetyl-aspartate (NAA) to creatine (Cr) ratios are consistently reduced in the basal ganglia of patients with neurological involvement and that myo-inositol is increased in patients with portosystemic shunting. In a recent case–control prospective study in a pediatric population, the levels of NAA, Cr and choline (Cho), as well as ratios of NAA/Cho, NAA/Cr, and Cho/ Cr in brain tissue as determined by MRS were significantly lower in 26 WD patients compared to 26 healthy individuals [78]. A more severe decrease in NAA, NAA/Cr, and NAA/Cho was observed in WD patients with mixed neurological and hepatic involvement compared with those with liver-only involvement. MRS was more sensitive than conventional MRI in detecting early changes in brains in children with WD before structural changes became visible on MRI.
- Copper absorption test
- Dynamic assessment of copper metabolism and fluxes in the body can be evaluated with radioactive copper tracer, 64Cu. Abnormal copper metabolism with increased hepatic 64Cu retention, reduced fecal excretion of the radiotracer, and altered 64Cu blood kinetics was found in WD patients and in animal models [79,80]. 64Cu incorporation into ceruloplasmin has demonstrated excellent diagnostic accuracy in patients with WD compared to heterozygous control subjects [81]. Interestingly, dynamic positron emission tomography (PET) analysis with 64Cu tracer was used in a recent animal study involving gene therapy [80]. This study found that gene therapy restored physiological copper metabolism in WD mice, confirming the mechanism of action, and demonstrated the translational potential of 64Cu-chloride PET to explore gene therapy pharmacodynamics in a minimally invasive and sensitive manner in WD patients.
3.2. A1-Antitrypsin Deficiency
AATD is a rare inherited autosomal recessive disease predisposing to liver damage in both adults and children, and lung involvement in adulthood [82,83]. It results from a misfolding and polymerization of a mutant alpha1-antitrypsin (AAT) protein within the endoplasmic reticulum of hepatocytes. There are more than 500 different alleles described for the protease inhibitor (Pi) allele in the SERPINA1 gene on the long arm of chromosome 14 [84]. The most common pathogenic mutation leading to this misfolded protein is Glu342Lys (Z allele), and a homozygous mutation of the Z allele leads to PiZZ genotype, which predisposes to liver fibrosis and emphysema [85,86]. Prevalence of the Z allele is 1 in 25 persons, and the PiZZ genotype concerns approximately 1 in 2000 persons in European populations [87]. The S allele leads to milder deficiency and less severe phenotypes [86]. Biochemical and genetic tests can confirm this underdiagnosed disease, and different non-invasive tools are available to assess liver and lung involvement.
3.2.1. Circumstances for Diagnosis
- Lung disease results from loss of function due to a low level of serine protease inhibitor capable of inhibiting neutrophil elastase in the lungs, which can lead to the development of early-onset panlobular basal emphysema. AATD-related lung disease only affects adults and should be considered in all cases of chronic obstructive pulmonary disease (COPD), emphysema, bronchiectasis, or asthma with unsuitable response to bronchodilators.
- Liver damage results from the degradation of accumulated AAT polymers in hepatocytes, through autophagy and endoplasmic reticulum pathways, leading to a proinflammatory and fibrogenic response [86,88]. Liver disease may present in infants as neonatal cholestasis with jaundice, pale stools, and elevated serum conjugated bilirubin; this situation requires the early intervention of a pediatric hepatologist [89]. Later during childhood and adulthood, AATD should be considered in patients with chronic elevation of transaminases or, in case of diagnosis of cirrhosis, PHT [90,91,92]. Adults patients with cirrhosis may also present with hepatocellular carcinoma. Insofar as AATD is now a recognized cofactor in chronic liver diseases such as alcoholic and non-alcoholic fatty liver disease, even in heterozygous state, the diagnosis should be sought in all chronic liver disease [93,94,95,96,97].
- Other rare conditions have been reported in ZZ patients, such as neutrophilic panniculitis, ANCA-vasculitis or chronic kidney disease [86,98].
- Since AATD is a genetic disease transmitted in an autosomal recessive mode, family screening can be proposed to first-degree relatives, and be the mode of diagnosis [82].
3.2.2. Diagnostic Tests
- The measurement of serum AAT level is the first test to identify AATD [99]. A concentration < 1.1 g/L is considered low, especially in the absence of inflammatory syndrome, and the most severe phenotypes are usually characterized by AAT concentration < 0.57 g/L. When serum AAT is <1.1 g/L, phenotyping or genotyping tests should be performed in an experienced laboratory (Figure 1) [86,99].
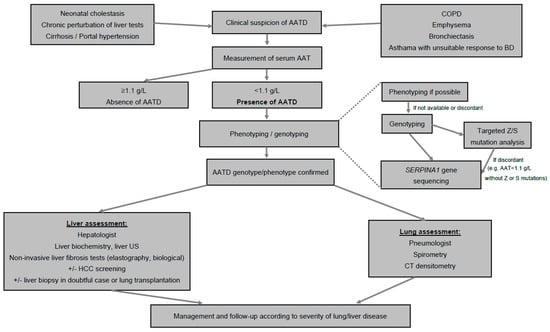 Figure 1. Algorithm for diagnosis of AAT. Abbreviations: AAT, alpha1-antitrypsin; AATD, alpha1-antitrypsin deficiency; COPD, chronic obstructive pulmonary disease; BD, bronchodilators; US, ultrasound; HCC, hepatocellular carcinoma; CT, computed tomography.
Figure 1. Algorithm for diagnosis of AAT. Abbreviations: AAT, alpha1-antitrypsin; AATD, alpha1-antitrypsin deficiency; COPD, chronic obstructive pulmonary disease; BD, bronchodilators; US, ultrasound; HCC, hepatocellular carcinoma; CT, computed tomography. - Phenotyping is performed by iso-electric focusing, which allows one to identify normal (M) and most frequent pathological alleles (Z, S, Mmalton) as well as some rarer ones, according to their electrophoretic mobility, and, therefore, to infer the genotype.
- Sometimes, genotyping of genomic DNA by PCR targeting the most frequent mutant alleles (Z, S) replaces phenotyping, which is controversial because it is a genetic test, and may sometimes wrongly reassure in the absence of Z or S mutation, whereas the AAT level is low due to the presence of other, non-detected (non Z and non S) mutations.
- In doubtful cases, genotyping should be performed by SERPINA1 gene sequencing in order to confirm the genotype. New approaches are emerging based on blood spot that allow the identification of the Z protein and even a panel of the most frequent phenotypes [100].
- All patients diagnosed with AATD should then be assessed for liver and lung involvement (Figure 1).
3.2.3. Assessment of Liver Disease
- AATD individuals should be referred for clinical evaluation, liver biochemistry, liver ultrasound, and non-invasive fibrosis assessment at the time of diagnosis to detect potential complications and for correct risk stratification [101]. Assessing liver disease severity can be challenging for physicians because fibrosis may develop without severe abnormalities of liver biochemistry, particularly in the case of severe AATD, and because signs of PHT may appear without severe fibrosis [102,103,104,105]. To date, there is no specific treatment available to treat specifically hepatic involvement, and liver transplantation is currently the sole option in cases of decompensated cirrhosis [106,107]. Emerging therapies that may prevent the progression of liver disease are currently under investigation [108,109].
- Clinical evaluation should pay attention to signs of PHT and liver failure.
- Simple blood tests are required, such as for plasma levels of liver enzymes (aspartate amino-transferase (AST), alanine amino-transferase (ALT), gamma-glutamyl-transferase (GGT), alkaline phosphatase (ALP), bilirubin), markers of liver function (prothrombin time (PT) and/or international normalized ratio (INR), albumin, ammoniemia, factor V), and hemogram to identify hypersplenism.
- Abdominal ultrasound (US) should be performed in all patients with AATD to assess liver morphology and search for signs of PHT and focal lesions. In cases with obese patients, CT and/or MRI can be helpful to identify signs of liver cirrhosis and portal hypertension. Screening for hepatocellular carcinoma should be performed every 6 months using US and measurement of serum alpha-fetoprotein in patients with cirrhosis.
- As in all chronic liver diseases, assessment of liver fibrosis severity is a key point in severe AATD. In ZZ patients, classical hepatic risk factors (male sex, age > 50 years, obesity and presence of diabetes) and repeated elevated liver enzymes were associated with significant liver fibrosis [103,110]. Liver biopsy is not necessary for the diagnosis of AATD. It remains the gold standard test for the determination of fibrosis, but is rarely performed in AATD due to its invasive nature, and because non-invasive techniques have been developed over the last years, although there are still limited data available for AATD patients. Liver biopsy remains valuable in discordant situations, when other, concomitant liver disease is associated, and before lung transplantation to assess precisely the liver status [5,102].
- TE is the most widely available technique used routinely in several chronic liver disease to assess liver fibrosis in adults and children by measuring LSM and to estimate steatosis by measuring the controlled attenuation parameter (CAP) [102,111,112,113]. Preliminary studies have found that TE is an easy and repeatable tool to screen for the presence of significant liver fibrosis in AATD adults, and LSM was ≥7.1 kPA (suggesting the presence of significant liver fibrosis) in 18.0–23.6% of adult ZZ patients [94,114]. Clark et al. studied the results of non-invasive tests in comparison to liver histology (METAVIR score) in a population of 94 ZZ adult patients and reported that the best LSM cut-off to identify F ≥ 2 (35.1% patients) and F ≥ 3 (6.4% patients) was 5.45 kPa and 8.45 kPa, respectively [93]. In children, Shneider et al. evaluated TE in a population of cholestatic children including AATD, and showed that this technique was feasible and correlated with liver parameters and PHT. Median LSM was significantly higher in children with PHT (23.8 kPa) compared to patients without PHT (7.8 kPa). In another study, TE had good accuracy in a population of 50 children with AATD, with higher median LSM values in patients with PHT (15.8 kPa) vs. patients without PHT (4.8 kPa), and a threshold of 7.1 kPa (sensitivity 100%, specificity 85%) to detect PHT (personal data). In addition, mean CAP values, an established surrogate of liver steatosis, were found to be higher in Pi*ZZ carriers than in non-carriers, and Pi*ZZ carriers had more severe steatosis than non-carriers, whereas the median body mass index as well as the frequency of obesity and diabetes were similar in both groups [101]. Other techniques, such as acoustic radiation force impulse (ARFI) quantification, two-dimensional shear wave elastography (2D-SWE), and magnetic resonance elastography (MRE), have been assessed in small cohorts of Pi*ZZ individuals and remain to be comprehensively validated [115,116]. MRE, which is a less routinely available technique, seems to be particularly useful in difficult cases such as those involving obesity and allows steatosis quantification.
- In addition to elastography methods, or when these are not available, simple liver biology tests can be used to calculate indirect non-invasive biological fibrosis scores, such as the AST to platelet ratio index (APRI) and the Fibrosis-4 test (FIB-4). Although data are scarce, these non-invasive markers of fibrosis seem to have good accuracy to detect fibrosis in adults and perform as well as transient elastography, whereas they are not as accurate to determine F ≥ 3 [93]. When compared with liver histology, APRI > 0.43 and 0.63 were associated with fibrosis ≥ F2 and ≥F3, respectively, with high specificity (87 and 94%) but low sensitivity (59 and 57%) in adults. In children, recent data suggest that a value > 0.67 (sensitivity 94% and specificity 95%) was associated with significant PHT (personal data). The accuracy of FIB-4 seems to be poorer in adults when compared with liver histology with lower sensitivity (74 and 71%) and specificity (64 and 78%) for cut-offs >1.43 and 1.90, respectively for F ≥ 2 and F ≥ 3 in adults [93]. Other non-invasive laboratory tests (Fibrotest®, Fibrometer®, ELF®) have not been studied in AATD. Similarly to the management of other liver diseases, the use of a combination of a laboratory test and an elastography method to identify and monitor liver fibrosis could be proposed, and in the event of discordant results, a biopsy might be considered [102]. However, such a strategy remains to be specifically validated in AATD patients.
3.2.4. Assessment of Lung Disease
- The severity of lung disease is variable, from asymptomatic to severe, and depends on genotypes and behavior such as smoking. Lung involvement could be treated with AAT augmentation therapy and may require lung transplantation in the most severe patients [86]. Therefore, it is crucial to assess severity at diagnosis and to monitor it [82]. The clinical manifestations of AATD-related lung disease are similar to those of other causes of emphysema, which contributes to underdiagnosis and late diagnosis of the disease. It is characterized by basal panlobular emphysema at an early age, with generally reversible airflow obstruction, usually chronic bronchitis, and bronchiectasis.
- Simple clinical evaluations using the 6 min walk test and health-related quality of life questionnaires should be regularly performed [82]. Pulmonary function tests including spirometry with the forced expiratory volume in the first second (FEV1) and the diffusing capacity of the lungs for carbon monoxide (DLCO) are performed to assess severity during follow-up [82].
- CT densitometry is an imaging method used to assess lung volume and emphysema, and in AATD this provides essential information regarding the presence, distribution and morphology of emphysema but also offers the option to quantify the extent of emphysema. These data, combined with the results of pulmonary function tests, have implications for treatment decisions such as initiation of AAT therapy, or suitability for surgical or endoscopic interventions for reducing lung volume [117]. In addition, the course of the lung density seems to be one of the most specific and sensitive surrogate end-points for evaluation of the therapeutic benefit of augmentation therapy and represents a paradigm imaging biomarker [82,118].
4. Conclusions
Non-invasive diagnostic tests are crucial for the diagnosis of rare genetic diseases such as WD and AATD to reduce the diagnostic delay and assess organ involvement. These tests also play a pivotal role during the follow-up of patients by monitoring the effectiveness and tolerability of current treatments and new emerging therapies, such as gene therapy and new molecules, which should be available soon.
Author Contributions
Conceptualization, O.G.; methodology, O.G.; writing—original draft preparation, O.G. and M.R.; writing—review and editing, O.G., J.D., E.C.-B. and M.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Philip Robinson (DRS, Hospices Civils de Lyon) for help in manuscript preparation.
Conflicts of Interest
O.G. received personal fees as speaker from Echosens. M.R. received personal fees as consultant from Takeda, Grifols, and as speaker from CSL Behring.
References
- Ala, A.; Walker, A.P.; Ashkan, K.; Dooley, J.S.; Schilsky, M.L. Wilson’s disease. Lancet 2007, 369, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Fromme, M.; Schneider, C.V.; Trautwein, C.; Brunetti-Pierri, N.; Strnad, P. Alpha-1 antitrypsin deficiency: A re-surfacing adult liver disorder. J. Hepatol. 2021, 76, 946–958. [Google Scholar] [CrossRef]
- Langner, C.; Denk, H. Wilson disease. Virchows Arch. 2004, 445, 111–118. [Google Scholar] [CrossRef]
- Schilsky, M.L.; Roberts, E.A.; Bronstein, J.M.; Dhawan, A.; Hamilton, J.P.; Rivard, A.M.; Washington, M.K.; Weiss, K.H.; Zimbrean, P.C. A Multidisciplinary Approach to the Diagnosis and Management of Wilson Disease: Executive Summary of the 2022 Practice Guidance on Wilson Disease from the American Association for the Study of Liver Diseases. Hepatology 2022. [Google Scholar] [CrossRef]
- Morer, L.; Choudat, L.; Dauriat, G.; Durand, F.; Cazals-Hatem, D.; Thabut, G.; Brugière, O.; Castier, Y.; Mal, H. Liver Involvement in Patients with PiZZ-Emphysema, Candidates for Lung Transplantation. Am. J. Transplant. 2017, 17, 1389–1395. [Google Scholar] [CrossRef]
- Dawwas, M.F.; Davies, S.E.; Griffiths, W.J.H.; Lomas, D.A.; Alexander, G.J. Prevalence and risk factors for liver involvement in individuals with PiZZ-related lung disease. Am. J. Respir. Crit. Care Med. 2013, 187, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Atwell, T.D.; Spanbauer, J.C.; McMenomy, B.P.; Stockland, A.H.; Hesley, G.K.; Schleck, C.D.; Harmsen, W.S.; Welch, T.J. The Timing and Presentation of Major Hemorrhage After 18,947 Image-Guided Percutaneous Biopsies. Am. J. Roentgenol. 2015, 205, 190–195. [Google Scholar] [CrossRef]
- Dezsőfi, A.; Baumann, U.; Dhawan, A.; Durmaz, O.; Fischler, B.; Hadzic, N.; Hierro, L.; Lacaille, F.; McLin, V.A.; Nobili, V.; et al. Liver biopsy in children: Position paper of the ESPGHAN Hepatology Committee. J. Pediatr. Gastroenterol. Nutr. 2015, 60, 408–420. [Google Scholar] [CrossRef]
- Członkowska, A.; Litwin, T.; Dusek, P.; Ferenci, P.; Lutsenko, S.; Medici, V.; Rybakowski, J.K.; Weiss, K.H.; Schilsky, M.L. Wilson disease. Nat. Rev. Dis. Prim. 2018, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Schilsky, M.L. Wilson Disease: Diagnosis, Treatment, and Follow-up. Clin. Liver Dis. 2017, 21, 755–767. [Google Scholar] [CrossRef]
- Ferenci, P.; Caca, K.; Loudianos, G.; Mieli-Vergani, G.; Tanner, S.; Sternlieb, I.; Schilsky, M.; Cox, D.; Berr, F. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003, 23, 139–142. [Google Scholar] [CrossRef]
- Schroeder, S.M.; Matsukuma, K.E.; Medici, V. Wilson disease and the differential diagnosis of its hepatic manifestations: A narrative review of clinical, laboratory, and liver histological features. Ann. Transl. Med. 2021, 9, 1394. [Google Scholar] [CrossRef]
- Poujois, A.; Woimant, F. Challenges in the diagnosis of Wilson disease. Ann. Transl. Med. 2019, 7 (Suppl. 2), S67. [Google Scholar] [CrossRef] [PubMed]
- Shribman, S.; Webb, G.; Taylor, R.; Warner, T.T.; Duckworth, A.; Gimson, A.; Shenoy, A.; Griffiths, W. Liver transplantation for late-onset presentations of acute liver failure in Wilson’s disease: The UK experience over 2 decades. JHEP Rep. 2020, 2, 100096. [Google Scholar] [CrossRef]
- Ferenci, P.; Członkowska, A.; Merle, U.; Ferenc, S.; Gromadzka, G.; Yurdaydin, C.; Vogel, W.; Bruha, R.; Schmidt, H.T.; Stremmel, W. Late-onset Wilson’s disease. Gastroenterology 2007, 132, 1294–1298. [Google Scholar] [CrossRef] [PubMed]
- Shribman, S.; Marjot, T.; Sharif, A.; Vimalesvaran, S.; Ala, A.; Alexander, G.; Dhawan, A.; Dooley, J.; Gillett, G.T.; Kelly, D.; et al. Investigation and management of Wilson’s disease: A practical guide from the British Association for the Study of the Liver. Lancet Gastroenterol. Hepatol. 2022, 7, 560–575. [Google Scholar] [CrossRef]
- Beinhardt, S.; Leiss, W.; Stättermayer, A.F.; Graziadei, I.; Zoller, H.; Stauber, R.; Maieron, A.; Datz, C.; Steindl-Munda, P.; Hofer, H.; et al. Long-term Outcomes of Patients With Wilson Disease in a Large Austrian Cohort. Clin. Gastroenterol. Hepatol. 2014, 12, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Boga, S.; Ala, A.; Schilsky, M.L. Hepatic features of Wilson disease. In Handbook of Clinical Neurology; Członkowska, A., Schilsky, M.L., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; Volume 142, pp. 91–99. [Google Scholar] [CrossRef]
- Walshe, J.M. The acute haemolytic syndrome in Wilson’s disease—A review of 22 patients. QJM Int. J. Med. 2013, 106, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Dzieżyc-Jaworska, K.; Litwin, T.; Członkowska, A. Clinical manifestations of Wilson disease in organs other than the liver and brain. Ann. Transl. Med. 2019, 7 (Suppl. 2), S62. [Google Scholar] [CrossRef] [PubMed]
- Balkema, S.; Hamaker, M.E.; Visser, H.P.J.; Heine, G.D.N.; Beuers, U. Haemolytic anaemia as a first sign of Wilson’s disease. Neth. J. Med. 2008, 66, 344–347. [Google Scholar] [PubMed]
- Fang, W.-Y.; Abuduxikuer, K.; Shi, P.; Qiu, Y.-L.; Zhao, J.; Li, Y.-C.; Zhang, X.-Y.; Wang, N.-L.; Xie, X.-B.; Lu, Y.; et al. Pediatric Wilson disease presenting as acute liver failure: Prognostic indices. World J. Clin. Cases 2021, 9, 3273–3286. [Google Scholar] [CrossRef]
- Guillaud, O.; Dumortier, J.; Sobesky, R.; Debray, D.; Wolf, P.; Vanlemmens, C.; Durand, F.; Calmus, Y.; Duvoux, C.; Dharancy, S.; et al. Long term results of liver transplantation for Wilson’s disease: Experience in France. J. Hepatol. 2014, 60, 579–589. [Google Scholar] [CrossRef]
- Chevalier, K.; Mauget-Faÿsse, M.; Vasseur, V.; Azar, G.; Obadia, M.A.; Poujois, A. Eye Involvement in Wilson’s Disease: A Review of the Literature. J. Clin. Med. 2022, 11, 2528. [Google Scholar] [CrossRef] [PubMed]
- Langwińska-Wośko, E.; Litwin, T.; Dzieżyc, K.; Członkowska, A. The sunflower cataract in Wilson’s disease: Pathognomonic sign or rare finding? Acta Neurol. Belg. 2016, 116, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Mak, C.M.; Lam, C.-W.; Tam, S. Diagnostic accuracy of serum ceruloplasmin in Wilson disease: Determination of sensitivity and specificity by ROC curve analysis among ATP7B-genotyped subjects. Clin. Chem. 2008, 54, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, A.; Bhatt, M. Wilson disease. Curr. Opin. Neurol. 2020, 33, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.A.; Schilsky, M.L. American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: An update. Hepatology 2008, 47, 2089–2111. [Google Scholar] [CrossRef]
- Socha, P.; Janczyk, W.; Dhawan, A.; Baumann, U.; D’Antiga, L.; Tanner, S.; Iorio, R.; Vajro, P.; Houwen, R.; Fischler, B.; et al. Wilson’s Disease in Children: A Position Paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J. Pediatr. Gastroenterol. Nutr. 2018, 66, 334–344. [Google Scholar] [CrossRef]
- European Association for Study of Liver. EASL Clinical Practice Guidelines: Wilson’s disease. J. Hepatol. 2012, 56, 671–685. [Google Scholar] [CrossRef]
- Pfeiffenberger, J.; Lohse, C.M.; Gotthardt, D.; Rupp, C.; Weiler, M.; Teufel, U.; Weiss, K.H.; Gauss, A. Long-term evaluation of urinary copper excretion and non-caeruloplasmin associated copper in Wilson disease patients under medical treatment. J. Inherit. Metab. Dis. 2019, 42, 371–380. [Google Scholar] [CrossRef]
- Walshe, J.M. The pattern of urinary copper excretion and its response to treatment in patients with Wilson’s disease. QJM Int. J. Med. 2011, 104, 775–778. [Google Scholar] [CrossRef]
- Sieniawska, C.E.; Jung, L.C.; Olufadi, R.; Walker, V. Twenty-four-hour urinary trace element excretion: Reference intervals and interpretive issues. Ann. Clin. Biochem. 2012, 49, 341–351. [Google Scholar] [CrossRef]
- Merle, U.; Schaefer, M.; Ferenci, P.; Stremmel, W. Clinical presentation, diagnosis and long-term outcome of Wilson’s disease: A cohort study. Gut 2007, 56, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Ryan, A.; Nevitt, S.J.; Tuohy, O.; Cook, P. Biomarkers for diagnosis of Wilson’s disease. Cochrane Database Syst. Rev. 2019, 2019, CD012267. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.-B.; Blackwell, R.Q. Studies on levels of penicillamine-induced cupriuresis in heterozygotes of Wilson’s disease. Metabolism 1967, 16, 507–513. [Google Scholar] [CrossRef]
- da Costa, C.M.; Baldwin, D.; Portmann, B.; Lolin, Y.; Mowat, A.P.; Mieli-Vergani, G. Value of urinary copper excretion after penicillamine challenge in the diagnosis of Wilson’s disease. Hepatology 1992, 15, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Koppikar, S.; Taylor, R.M.; Carragher, F.; Schlenck, B.; Heinz-Erian, P.; Kronenberg, F.; Ferenci, P.; Tanner, S.; Siebert, U.; et al. Re-evaluation of the penicillamine challenge test in the diagnosis of Wilson’s disease in children. J. Hepatol. 2007, 47, 270–276. [Google Scholar] [CrossRef]
- Steindl, P.; Ferenci, P.; Dienes, H.P.; Grimm, G.; Pabinger, I.; Madl, C.; Dobersberger, T.M.; Herneth, A.; Dragosics, B.; Meryn, S.; et al. Wilson’s disease in patients presenting with liver disease: A diagnostic challenge. Gastroenterology 1997, 113, 212–218. [Google Scholar] [CrossRef]
- Korman, J.D.; Volenberg, I.; Balko, J.; Webster, J.; Schiodt, F.V.; Squires, R.H., Jr.; Fontana, R.J.; Lee, W.M.; Schilsky, M.L.; Pediatric and Adult Acute Liver Failure Study Groups. Screening for Wilson disease in acute liver failure: A comparison of currently available diagnostic tests. Hepatology 2008, 48, 1167–1174. [Google Scholar] [CrossRef]
- Yasmin, A.; Rukunuzzaman, M.; Karim, A.S.M.B.; Alam, R.; Hossen, K.; Sonia, Z.F. Ratio of aspartate aminotransferase to alanine aminotransferase and alkaline phosphatase to total bilirubin in Wilsonian acute liver failure in children. Indian J. Gastroenterol. 2022, 41, 224–230. [Google Scholar] [CrossRef]
- Zulkufli, N.S.; Sthaneshwar, P.; Chan, W.-K. Calculated parameters for the diagnosis of Wilson disease. Singap. Med. J. 2022, 1, 20. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.; Yacoubian, C.; Beetham, R.; Catchpole, A.; Bullock, D. The role of calculated non-caeruloplasmin-bound copper in Wilson’s disease. Ann. Clin. Biochem. 2017, 54, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Chanpong, A.; Dhawan, A. Long-Term Urinary Copper Excretion on Chelation Therapy in Children with Wilson Disease. J. Craniofacial Surg. 2021, 72, 210–215. [Google Scholar] [CrossRef] [PubMed]
- El Balkhi, S.; Trocello, J.-M.; Poupon, J.; Chappuis, P.; Massicot, F.; Girardot-Tinant, N.; Woimant, F. Relative exchangeable copper: A new highly sensitive and highly specific biomarker for Wilson’s disease diagnosis. Clin. Chim. Acta 2011, 412, 2254–2260. [Google Scholar] [CrossRef]
- Heissat, S.; Harel, A.; Um, K.; Brunet, A.-S.; Hervieu, V.; Guillaud, O.; Dumortier, J.; Lachaux, A.; Mintz, E.; Bost, M. Evaluation of the accuracy of exchangeable copper and relative exchangeable copper (REC) in a mouse model of Wilson’s disease. J. Trace Elem. Med. Biol. 2018, 50, 652–657. [Google Scholar] [CrossRef]
- Guillaud, O.; Brunet, A.-S.; Mallet, I.; Dumortier, J.; Pelosse, M.; Heissat, S.; Rivet, C.; Lachaux, A.; Bost, M. Relative exchangeable copper: A valuable tool for the diagnosis of Wilson disease. Liver Int. 2018, 38, 350–357. [Google Scholar] [CrossRef]
- Ngwanou, D.H.; Couchonnal, E.; Parant, F.; Belmalih, A.; Guillaud, O.; Dumortier, J.; Bost, M.; Lachaux, A. Long-Term Urinary Copper Excretion and Exchangeable copper in children with Wilson Disease under chelation therapy. J. Pediatr. Gastroenterol. Nutr. 2022, 75, 75–80. [Google Scholar] [CrossRef]
- Jacquelet, E.; Poujois, A.; Pheulpin, M.-C.; Demain, A.; Tinant, N.; Gastellier, N.; Woimant, F. Adherence to treatment, a challenge even in treatable metabolic rare diseases: A cross sectional study of Wilson’s disease. J. Inherit. Metab. Dis. 2021, 44, 1481–1488. [Google Scholar] [CrossRef]
- Poujois, A.; Trocello, J.-M.; Djebrani-Oussedik, N.; Poupon, J.; Collet, C.; Girardot-Tinant, N.; Sobesky, R.; Habès, D.; Debray, D.; Vanlemmens, C.; et al. Exchangeable copper: A reflection of the neurological severity in Wilson’s disease. Eur. J. Neurol. 2017, 24, 154–160. [Google Scholar] [CrossRef]
- Guillaud, O.; Woimant, F.; Couchonnal, E.; Dumortier, J.; Laurencin, C.; Lion-François, L.; Belmalih, A.; Bost, M.; Morvan, E.; Oussedik-Djebrani, N.; et al. Maintenance therapy simplification using a single daily dose: A preliminary real-life feasibility study in patients with Wilson disease. Clin. Res. Hepatol. Gastroenterol. 2022, 46, 101978. [Google Scholar] [CrossRef]
- Solovyev, N.; Ala, A.; Schilsky, M.; Mills, C.; Willis, K.; Harrington, C.F. Biomedical copper speciation in relation to Wilson’s disease using strong anion exchange chromatography coupled to triple quadrupole inductively coupled plasma mass spectrometry. Anal. Chim. Acta 2020, 1098, 27–36. [Google Scholar] [CrossRef]
- Espinós, C.; Ferenci, P. Are the new genetic tools for diagnosis of Wilson disease helpful in clinical practice? JHEP Rep. 2020, 2, 100114. [Google Scholar] [CrossRef] [PubMed]
- Coffey, A.J.; Durkie, M.; Hague, S.; McLay, K.; Emmerson, J.; Lo, C.; Klaffke, S.; Joyce, C.J.; Dhawan, A.; Hadzic, N.; et al. A genetic study of Wilson’s disease in the United Kingdom. Brain 2013, 136, 1476–1487. [Google Scholar] [CrossRef] [PubMed]
- Kluska, A.; Kulecka, M.; Litwin, T.; Dziezyc, K.; Balabas, A.; Piatkowska, M.; Paziewska, A.; Dabrowska, M.; Mikula, M.; Diana, K.; et al. Whole-exome sequencing identifies novel pathogenic variants across the ATP7B gene and some modifiers of Wilson’s disease phenotype. Liver Int. 2019, 39, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Couchonnal, E.; Bouchard, S.; Sandahl, T.D.; Pagan, C.; Lion-François, L.; Guillaud, O.; Habes, D.; Debray, D.; Lamireau, T.; Broué, P.; et al. ATP7B variant spectrum in a French pediatric Wilson disease cohort. Eur. J. Med. Genet. 2021, 64, 104305. [Google Scholar] [CrossRef] [PubMed]
- Ferenci, P.; Stremmel, W.; Członkowska, A.; Szalay, F.; Viveiros, A.; Stättermayer, A.F.; Bruha, R.; Houwen, R.; Pop, T.L.; Stauber, R.; et al. Age and Sex but Not ATP7B Genotype Effectively Influence the Clinical Phenotype of Wilson Disease. Hepatology 2019, 69, 1464–1476. [Google Scholar] [CrossRef] [PubMed]
- Shribman, S.; Poujois, A.; Bandmann, O.; Czlonkowska, A.; Warner, T.T. Wilson’s disease: Update on pathogenesis, biomarkers and treatments. J. Neurol. Neurosurg. Psychiatry 2021, 92, 1053–1061. [Google Scholar] [CrossRef]
- Dusek, P.; Smolinski, L.; Redzia-Ogrodnik, B.; Golebiowski, M.; Skowronska, M.; Poujois, A.; Laurencin, C.; Jastrzebska-Kurkowska, I.; Litwin, T.; Członkowska, A. Semiquantitative Scale for Assessing Brain MRI Abnormalities in Wilson Disease: A Validation Study. Mov. Disord. 2020, 35, 994–1001. [Google Scholar] [CrossRef]
- Pinto, C.; Malaquias, M.J.; Miranda, H.P.; Temudo, T.; Silva, E.; Ramos, C.; Magalhães, M. Brain MRI in the Decision for Liver Transplantation in Pediatric Neurological Wilson’s Disease. Mov. Disord. Clin. Pract. 2022, 9, 941–948. [Google Scholar] [CrossRef]
- Przybyłkowski, A.; Gromadzka, G.; Chabik, G.; Wierzchowska, A.; Litwin, T.; Członkowska, A. Liver cirrhosis in patients newly diagnosed with neurological phenotype of Wilson’s disease. Funct. Neurol. 2014, 29, 23–29. [Google Scholar]
- Akpinar, E.; Akhan, O. Liver imaging findings of Wilson’s disease. Eur. J. Radiol. 2007, 61, 25–32. [Google Scholar] [CrossRef]
- Cançado, E.L.; de Souza Rocha, M.; Barbosa, E.R.; Scaff, M.; Cerri, G.G.; Magalhães, A.; Canelas, H.M. Abdominal ultrasonography in hepatolenticular degeneration. A study of 33 patients. Arq. Neuropsiquiatr. 1987, 45, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhao, X.; Zhan, Q.; Chai, W.; Mahyoub, R.; Yang, Y.; Shen, B.; Chen, K. Unique CT imaging findings of liver in Wilson’s disease. Abdom. Imaging 2011, 36, 69–73. [Google Scholar] [CrossRef]
- Pfeiffenberger, J.; Mogler, C.; Gotthardt, D.N.; Schulze-Bergkamen, H.; Litwin, T.; Reuner, U.; Hefter, H.; Huster, D.; Schemmer, P.; Członkowska, A.; et al. Hepatobiliary malignancies in Wilson disease. Liver Int. 2015, 35, 1615–1622. [Google Scholar] [CrossRef]
- Sobesky, R.; Guillaud, O.; Bouzbib, C.; Sogni, P.; Poujois, A.; Woimant, F.; Duclos-Vallée, J.C.; Bourlière, M.; de Lédinghen, V.; Ganne-Carrié, N.; et al. Non-invasive diagnosis and follow-up of rare genetic liver diseases. Clin. Res. Hepatol. Gastroenterol. 2022, 46, 101768. [Google Scholar] [CrossRef]
- Paternostro, R.; Pfeiffenberger, J.; Ferenci, P.; Stättermayer, A.F.; Stauber, R.E.; Wrba, F.; Longerich, T.; Lackner, K.; Trauner, M.; Ferlitsch, A.; et al. Non-invasive diagnosis of cirrhosis and long-term disease monitoring by transient elastography in patients with Wilson disease. Liver Int. 2020, 40, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Stefanescu, A.C.; Pop, T.L.; Stefanescu, H.; Miu, N. Transient elastography of the liver in children with Wilson’s disease: Preliminary results. J. Clin. Ultrasound 2016, 44, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Karlas, T.; Hempel, M.; Tröltzsch, M.; Huster, D.; Günther, P.; Tenckhoff, H.; Mössner, J.; Berg, T.; Keim, V.; Wiegand, J. Non-invasive evaluation of hepatic manifestation in Wilson disease with transient elastography, ARFI, and different fibrosis scores. Scand. J. Gastroenterol. 2012, 47, 1353–1361. [Google Scholar] [CrossRef]
- Sini, M.; Sorbello, O.; Civolani, A.; Liggi, M.; Demelia, L. Non-invasive assessment of hepatic fibrosis in a series of patients with Wilson’s Disease. Dig. Liver Dis. 2012, 44, 487–491. [Google Scholar] [CrossRef]
- Wang, J.; Hu, M.; Zhu, Q.; Sun, L. Liver stiffness assessed by real-time two-dimensional shear wave elastography predicts hypersplenism in patients with Wilson’s disease: A prospective study. BMC Med. Imaging 2022, 22, 25. [Google Scholar] [CrossRef]
- Collins, C.J.; Yi, F.; Dayuha, R.; Duong, P.; Horslen, S.; Camarata, M.; Coskun, A.K.; Houwen, R.H.; Pop, T.L.; Zoller, H.; et al. Direct Measurement of ATP7B Peptides Is Highly Effective in the Diagnosis of Wilson Disease. Gastroenterology 2021, 160, 2367–2382. [Google Scholar] [CrossRef]
- Broniek-Kowalik, K.; Dzieżyc, K.; Litwin, T.; Członkowska, A.; Szaflik, J.P. Anterior segment optical coherence tomography (AS-OCT) as a new method of detecting copper deposits forming the Kayser-Fleischer ring in patients with Wilson disease. Acta Ophthalmol. 2019, 97, 757–760. [Google Scholar] [CrossRef]
- Sridhar, M.S. Advantages of Anterior Segment Optical Coherence Tomography Evaluation of the Kayser-Fleischer Ring in Wilson Disease. Cornea 2017, 36, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, M.S.; Rangaraju, A.; Anbarasu, K.; Reddy, S.P.; Daga, S.; Jayalakshmi, S.; Shaik, B. Evaluation of Kayser-Fleischer ring in Wilson disease by anterior segment optical coherence tomography. Indian J. Ophthalmol. 2017, 65, 354–357. [Google Scholar] [CrossRef]
- Sridhar, M.S.; Pineda, R. Anterior segment optical coherence tomography to look for Kayser-Fleischer rings. Pract. Neurol. 2017, 17, 222–223. [Google Scholar] [CrossRef]
- Moini, M.; To, U.; Schilsky, M.L. Recent advances in Wilson disease. Transl. Gastroenterol. Hepatol. 2021, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Basha, M.M.A.; Refaat, R.; Ahmed, A.F.; Yousef, H.Y.; Alsowey, A.M.; Metwally, M.I.; Aly, S.A.; Hussien, H.M.; El-Saadany, H.F.; AlGhobashy, A.A.; et al. Brain magnetic resonance spectroscopy (MRS) as a diagnostic tool for detecting early neurological changes in children with Wilson’s disease. Eur. J. Radiol. 2019, 111, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Osborn, S.B.; Walshe, J.M. Studies with Radioactive Copper (64Cu AND 67Cu) in Relation to the Natural History of Wilson’s Disease. Lancet 1967, 289, 346–350. [Google Scholar] [CrossRef]
- Murillo, O.; Collantes, M.; Gazquez, C.; Moreno, D.; Hernandez-Alcoceba, R.; Barberia, M.; Ecay, M.; Tamarit, B.; Douar, A.; Ferrer, V.; et al. High value of 64Cu as a tool to evaluate the restoration of physiological copper excretion after gene therapy in Wilson’s disease. Mol. Ther. Methods Clin. Dev. 2022, 26, 98–106. [Google Scholar] [CrossRef]
- Członkowska, A.; Rodo, M.; Wierzchowska-Ciok, A.; Smolinski, L.; Litwin, T. Accuracy of the radioactive copper incorporation test in the diagnosis of Wilson disease. Liver Int. 2018, 38, 1860–1866. [Google Scholar] [CrossRef]
- Miravitlles, M.; Dirksen, A.; Ferrarotti, I.; Koblizek, V.; Lange, P.; Mahadeva, R.; McElvaney, N.G.; Parr, D.; Piitulainen, E.; Roche, N.; et al. European Respiratory Society statement: Diagnosis and treatment of pulmonary disease in α1-antitrypsin deficiency. Eur. Respir. J. 2017, 50, 1700610. [Google Scholar] [CrossRef]
- Greene, C.M.; Marciniak, S.J.; Teckman, J.; Ferrarotti, I.; Brantly, M.L.; Lomas, D.A.; Stoller, J.K.; McElvaney, N.G. α1-Antitrypsin deficiency. Nat. Rev. Dis. Prim. 2016, 2, 16051. [Google Scholar] [CrossRef]
- Seixas, S.; Marques, P.I. Known Mutations at the Cause of Alpha-1 Antitrypsin Deficiency an Updated Overview of SERPINA1 Variation Spectrum. Appl. Clin. Genet. 2021, 14, 173–194. [Google Scholar] [CrossRef]
- Torres-Durán, M.; López-Campos, J.L.; Rodríguez-Hermosa, J.L.; Esquinas, C.; Martínez-González, C.; Hernández-Pérez, J.M.; Rodríguez, C.; Bustamante, A.; Casas-Maldonado, F.; Barrecheguren, M.; et al. Demographic and clinical characteristics of patients with α1-antitrypsin deficiency genotypes PI*ZZ and PI*SZ in the Spanish registry of EARCO. ERJ Open Res. 2022, 8, 00213-2022. [Google Scholar] [CrossRef]
- Strnad, P.; McElvaney, N.G.; Lomas, D.A. Alpha1-Antitrypsin Deficiency. N. Engl. J. Med. 2020, 382, 1443–1455. [Google Scholar] [CrossRef]
- Blanco, I.; Bueno, P.; Diego, I.; Pérez-Holanda, S.; Casas-Maldonado, F.; Esquinas, C.; Miravitlles, M. Alpha-1 antitrypsin Pi*Z gene frequency and Pi*ZZ genotype numbers worldwide: An update. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; McAllister, S.L.; Teckman, J.H. Alpha-1 antitrypsin deficiency liver disease. Transl. Gastroenterol. Hepatol. 2021, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.; Lacaille, F.; Berthiller, J.; Joly, P.; Dumortier, J.; Aumar, M.; Bridoux-Henno, L.; Jacquemin, E.; Lamireau, T.; Broué, P.; et al. Liver disease related to alpha1-antitrypsin deficiency in French children: The DEFI-ALPHA cohort. Liver Int. 2019, 39, 1136–1146. [Google Scholar] [CrossRef]
- Patel, D.; Teckman, J. Liver disease with unknown etiology—Have you ruled out alpha-1 antitrypsin deficiency? Ther. Adv. Chronic Dis. 2021, 12, 2040622321995684. [Google Scholar] [CrossRef] [PubMed]
- Antoury, C.; Lopez, R.; Zein, N.; Stoller, J.; Alkhouri, N. Alpha-1 Antitrypsin Deficiency and the Risk of Hepatocellular Carcinoma in Patients With End-Stage Liver Disease. World J. Hepatol. 2015, 7, 1427–1432. [Google Scholar] [CrossRef]
- Manne, V.; Kowdley, K.V. Alpha1-Antitrypsin Deficiency: A Cause of Chronic Liver Disease. Clin. Liver Dis. 2020, 24, 483–492. [Google Scholar] [CrossRef]
- Clark, V.C.; Marek, G.; Liu, C.; Collinsworth, A.; Shuster, J.; Kurtz, T.; Nolte, J.; Brantly, M. Clinical and histologic features of adults with alpha-1 antitrypsin deficiency in a non-cirrhotic cohort. J. Hepatol. 2018, 69, 1357–1364. [Google Scholar] [CrossRef]
- Hamesch, K.; Mandorfer, M.; Pereira, V.M.; Moeller, L.S.; Pons, M.; Dolman, G.E.; Reichert, M.C.; Schneider, C.V.; Woditsch, V.; Voss, J.; et al. Liver Fibrosis and Metabolic Alterations in Adults With alpha-1-antitrypsin Deficiency Caused by the Pi*ZZ Mutation. Gastroenterology 2019, 157, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Strnad, P.; Buch, S.; Hamesch, K.; Fischer, J.; Rosendahl, J.; Schmelz, R.; Brueckner, S.; Brosch, M.; Heimes, C.V.; Woditsch, V.; et al. Heterozygous carriage of the alpha1-antitrypsin Pi*Z variant increases the risk to develop liver cirrhosis. Gut 2019, 68, 1099–1107. [Google Scholar] [CrossRef]
- Meister, F.; Lurje, G.; Neumann, U.P.; Trautwein, C.; Strnad, P. Heterozygosity for the Alpha-1-Antitrypsin Z Allele in Cirrhosis Is Associated With More Advanced Disease. Liver Transplant. 2019, 25, 342–343. [Google Scholar] [CrossRef] [PubMed]
- Goltz, D.; Hittetiya, K.; Vossing, L.M.; Kirfel, J.; Spengler, U.; Fischer, H.-P. Alpha(1)-antitrypsin PiMZ heterozygosity has an independent aggravating effect on liver fibrosis in alcoholic liver disease. Virchows Arch. 2014, 465, 539–546. [Google Scholar] [CrossRef]
- Deshayes, S.; Silva, N.M.; Khoy, K.; Mariotte, D.; Mauff, B.L.; Mornex, J.-F.; Pison, C.; Cuvelier, A.; Balduyck, M.; Pujazon, M.-C.; et al. Prevalence of Anti-Neutrophil Cytoplasmic Antibodies and Associated Vasculitis in COPD Associated with Alpha-1 Antitrypsin Deficiency: An Ancillary Study to a Prospective Study on 180 French Patients. Chest 2020, 158, 1919–1922. [Google Scholar] [CrossRef]
- Franciosi, A.N.; Carroll, T.P.; McElvaney, N.G. Pitfalls and caveats in α1-antitrypsin deficiency testing: A guide for clinicians. Lancet Respir. Med. 2019, 7, 1059–1067. [Google Scholar] [CrossRef]
- Ottaviani, S.; Barzon, V.; Buxens, A.; Gorrini, M.; Larruskain, A.; El Hamss, R.; Balderacchi, A.M.; Corsico, A.G.; Ferrarotti, I. Molecular diagnosis of alpha1-antitrypsin deficiency: A new method based on Luminex technology. J. Clin. Lab. Anal. 2020, 34, e23279. [Google Scholar] [CrossRef]
- Hamesch, K.; Strnad, P. Non-Invasive Assessment and Management of Liver Involvement in Adults With Alpha-1 Antitrypsin Deficiency. Chronic Obstr. Pulm. Dis. 2020, 7, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Tapper, E.B.; Lok, A.S.-F. Use of Liver Imaging and Biopsy in Clinical Practice. N. Engl. J. Med. 2017, 377, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Tanash, H.A.; Piitulainen, E. Liver disease in adults with severe alpha-1-antitrypsin deficiency. J. Gastroenterol. 2019, 54, 541–548. [Google Scholar] [CrossRef]
- Clark, V.C.; Dhanasekaran, R.; Brantly, M.; Rouhani, F.; Schreck, P.; Nelson, D.R. Liver Test Results Do Not Identify Liver Disease in Adults With α1-Antitrypsin Deficiency. Clin. Gastroenterol. Hepatol. 2012, 10, 1278–1283. [Google Scholar] [CrossRef]
- Bernspång, E.; Carlson, J.; Piitulainen, E. The liver in 30-year-old individuals with alpha1-antitrypsin deficiency. Scand. J. Gastroenterol. 2009, 44, 1349–1355. [Google Scholar] [CrossRef]
- Guillaud, O.; Jacquemin, E.; Couchonnal, E.; Vanlemmens, C.; Francoz, C.; Chouik, Y.; Conti, F.; Duvoux, C.; Hilleret, M.-N.; Kamar, N.; et al. Long term results of liver transplantation for alpha-1 antitrypsin deficiency. Dig. Liver Dis. 2021, 53, 606–611. [Google Scholar] [CrossRef]
- Carey, E.J.; Iyer, V.N.; Nelson, D.R.; Nguyen, J.H.; Krowka, M.J. Outcomes for recipients of liver transplantation for alpha-1-antitrypsin deficiency-related cirrhosis. Liver Transplant. 2013, 19, 1370–1376. [Google Scholar] [CrossRef]
- Lomas, D.A.; Hurst, J.R.; Gooptu, B. Update on alpha-1 antitrypsin deficiency: New therapies. J. Hepatol. 2016, 65, 413–424. [Google Scholar] [CrossRef]
- Suri, A.; Patel, D.; Teckman, J. Alpha-1-Antitrypsin Deficiency. Clin. Liver Dis. 2022, 19, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Fromme, M.; Schneider, C.V.; Pereira, V.; Hamesch, K.; Pons, M.; Reichert, M.C.; Benini, F.; Ellis, P.; Thorhauge, K.H.; Mandorfer, M.; et al. Hepatobiliary phenotypes of adults with alpha-1 antitrypsin deficiency. Gut 2022, 71, 415–423. [Google Scholar] [CrossRef]
- Banc-Husu, A.M.; Bass, L.M. Transient Elastography in Pediatric Liver Disease. J. Pediatr. Gastroenterol. Nutr. 2021, 73, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.-T.; Xiang, L.-L.; Qi, F.; Zhang, Y.-J.; Chen, Y.; Zhou, X.-Q. Accuracy of controlled attenuation parameter (CAP) and liver stiffness measurement (LSM) for assessing steatosis and fibrosis in non-alcoholic fatty liver disease: A systematic review and meta-analysis. eClinicalMedicine 2022, 51, 101547. [Google Scholar] [CrossRef] [PubMed]
- Karlas, T.; Petroff, D.; Sasso, M.; Fan, J.-G.; Mi, Y.-Q.; de Lédinghen, V.; Kumar, M.; Lupsor-Platon, M.; Han, K.-H.; Cardoso, A.C.; et al. Individual patient data meta-analysis of controlled attenuation parameter (CAP) technology for assessing steatosis. J. Hepatol. 2017, 66, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Guillaud, O.; Dumortier, J.; Traclet, J.; Restier, L.; Joly, P.; Chapuis-Cellier, C.; Lachaux, A.; Mornex, J.F. Assessment of liver fibrosis by transient elastography (Fibroscan®) in patients with A1AT deficiency. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.G.; Nguyen, P.; Bettencourt, R.; Dulai, P.S.; Haufe, W.; Hooker, J.; Minocha, J.; Valasek, M.A.; Aryafar, H.; Brenner, D.A.; et al. Magnetic resonance elastography identifies fibrosis in adults with alpha-1 antitrypsin deficiency liver disease: A prospective study. Aliment. Pharmacol. Ther. 2016, 44, 287–299. [Google Scholar] [CrossRef]
- Reiter, R.; Wetzel, M.; Hamesch, K.; Strnad, P.; Asbach, P.; Haas, M.; Siegmund, B.; Trautwein, C.; Hamm, B.; Klatt, D.; et al. Comparison of non-invasive assessment of liver fibrosis in patients with alpha1-antitrypsin deficiency using magnetic resonance elastography (MRE), acoustic radiation force impulse (ARFI) Quantification, and 2D-shear wave elastography (2D-SWE). PLoS ONE 2018, 13, e0196486. [Google Scholar] [CrossRef]
- Huang, Y.-C.T.; Wencker, M.; Driehuys, B. Imaging in alpha-1 antitrypsin deficiency: A window into the disease. Ther. Adv. Chronic Dis. 2021, 12, 20406223211024523. [Google Scholar] [CrossRef]
- Barjaktarevic, I.; Campos, M. Management of lung disease in alpha-1 antitrypsin deficiency: What we do and what we do not know. Ther. Adv. Chronic Dis. 2021, 12, 20406223211010172. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).