

Cutaneous Melanoma and Glioblastoma Multiforme Association—Case Presentation and Literature Review



 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction—Literature Review
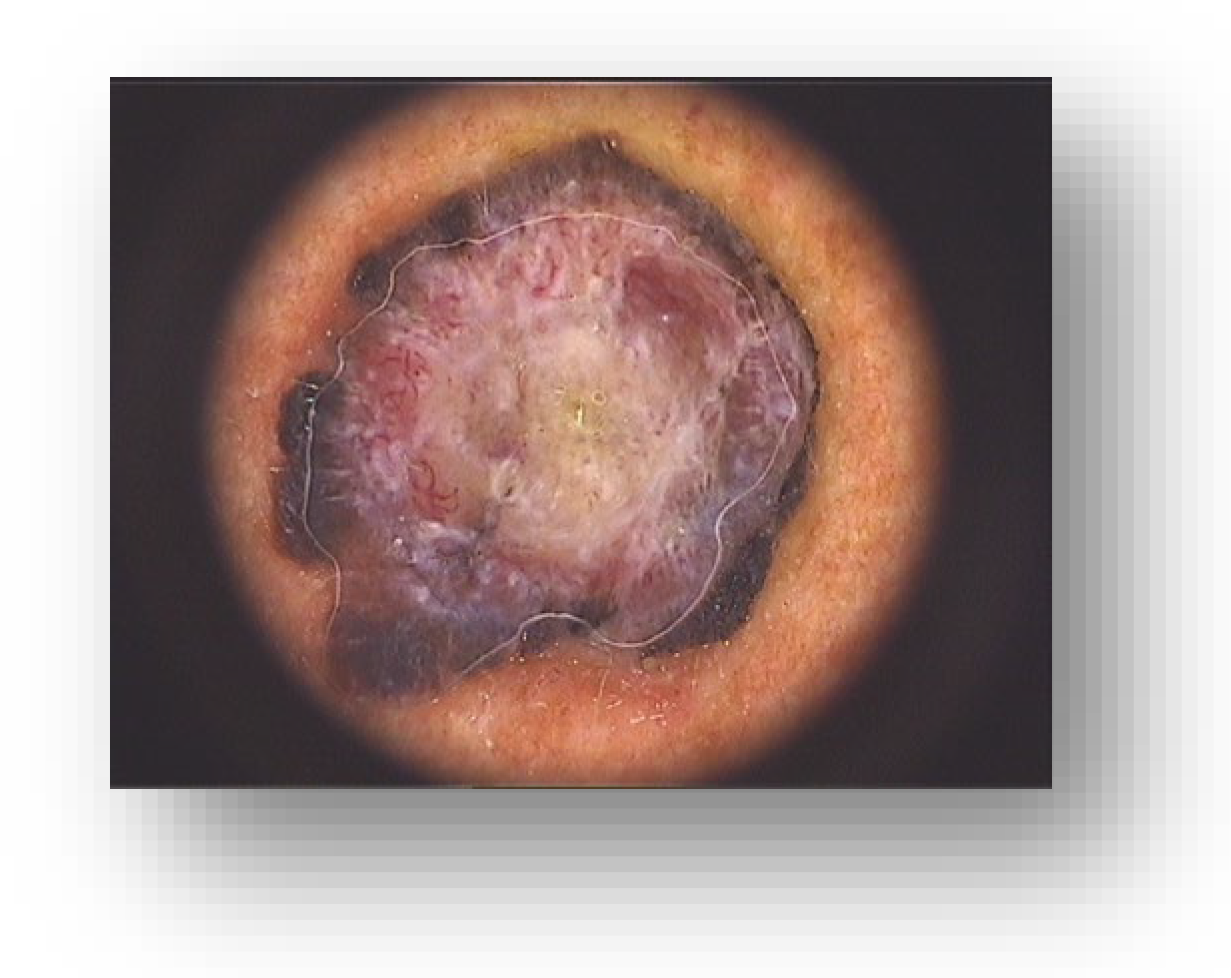
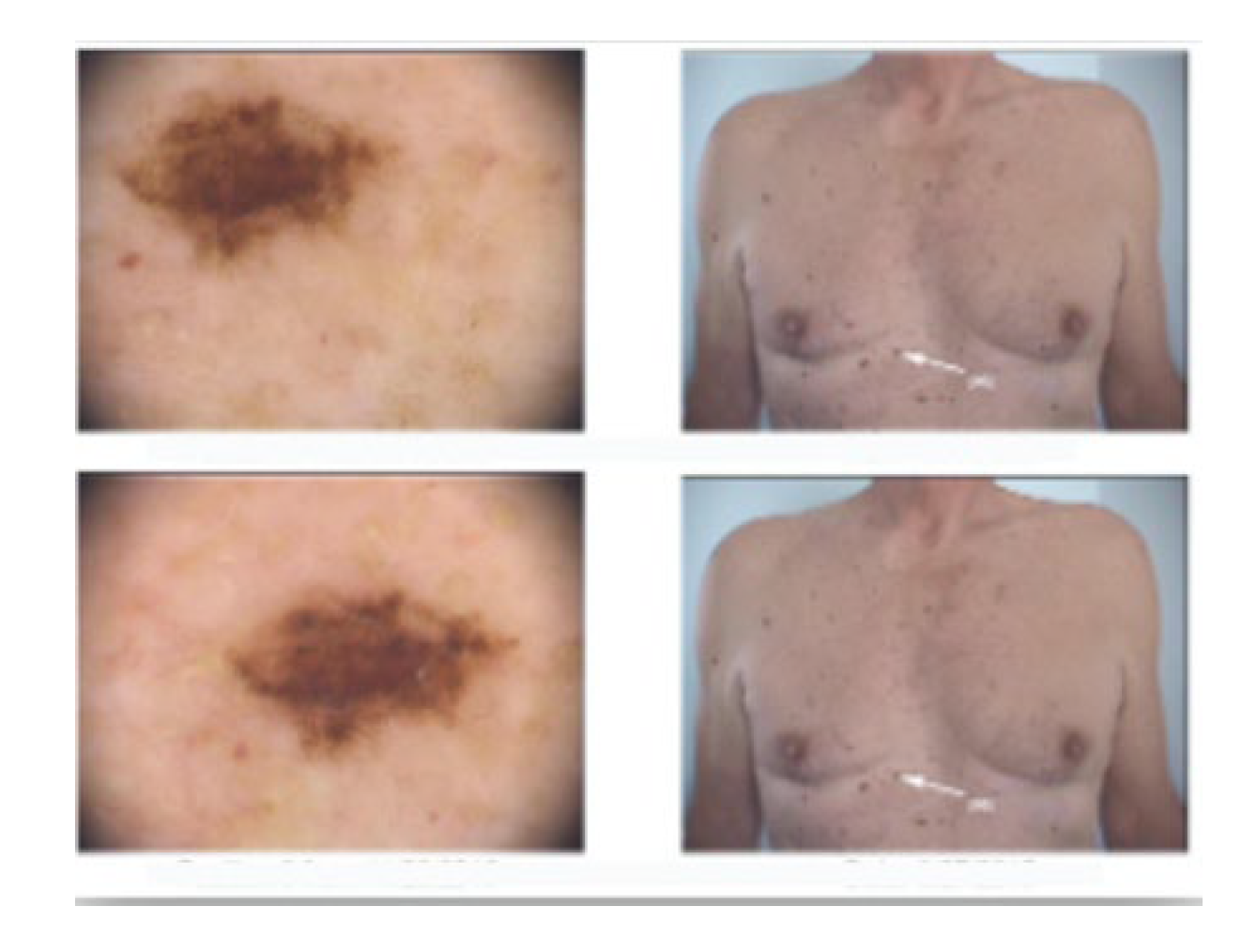
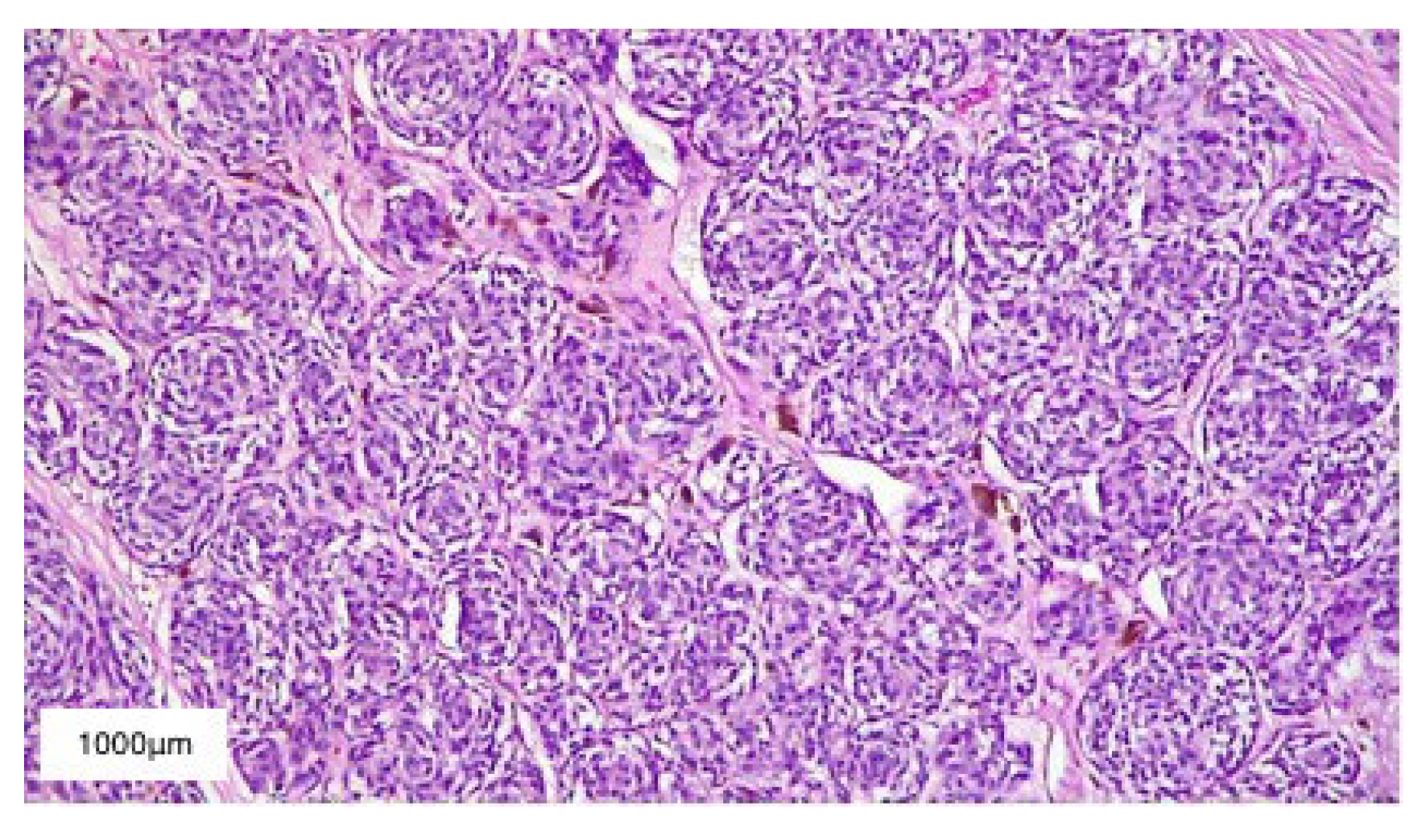
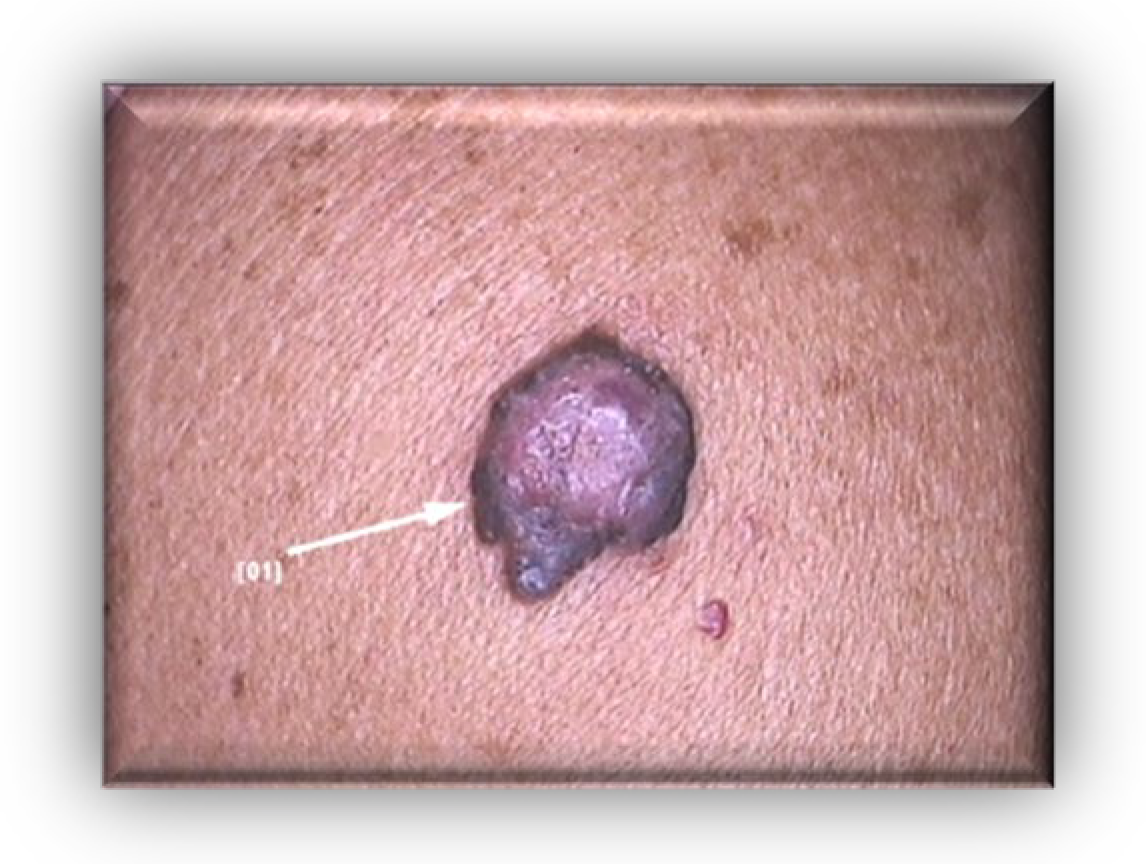
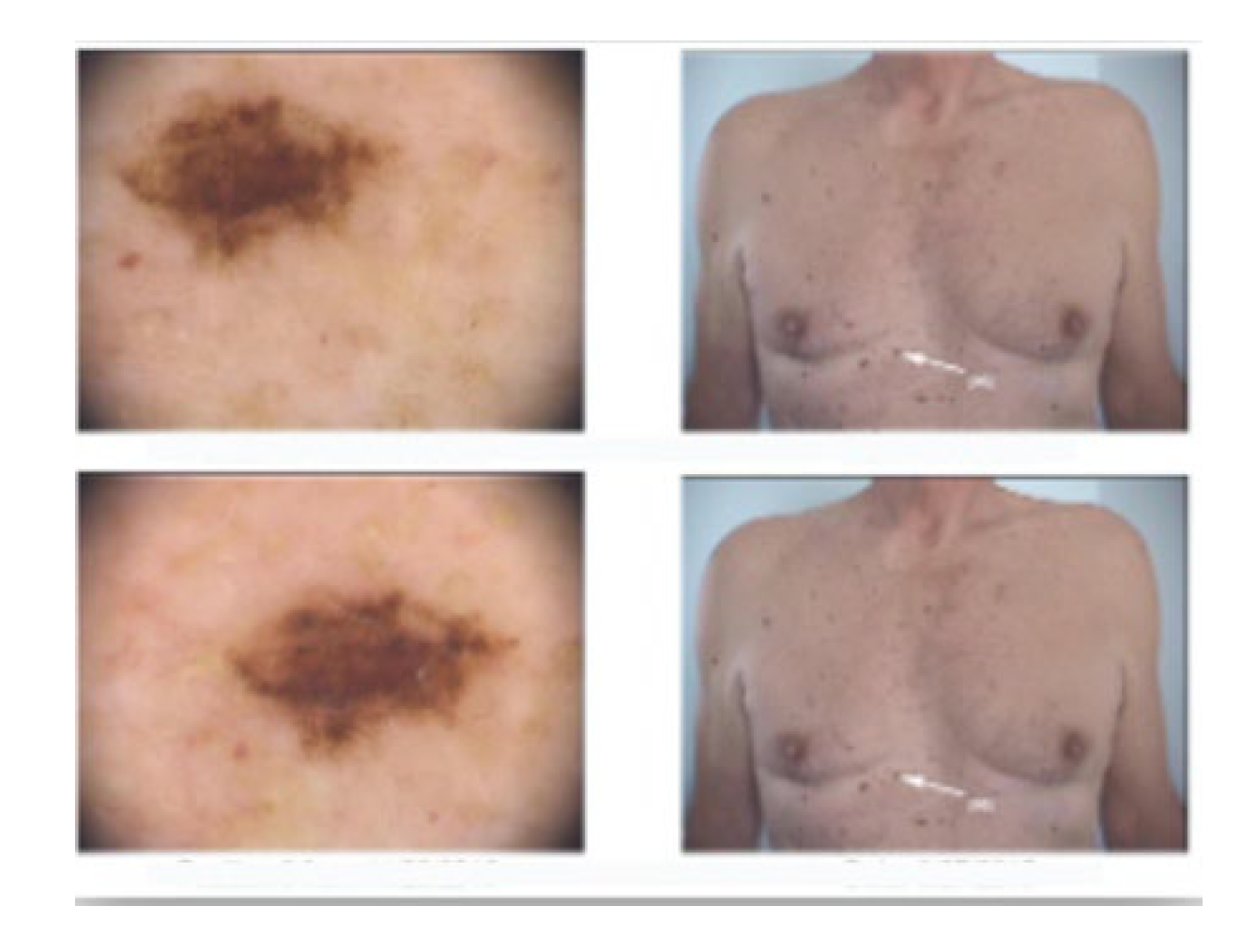
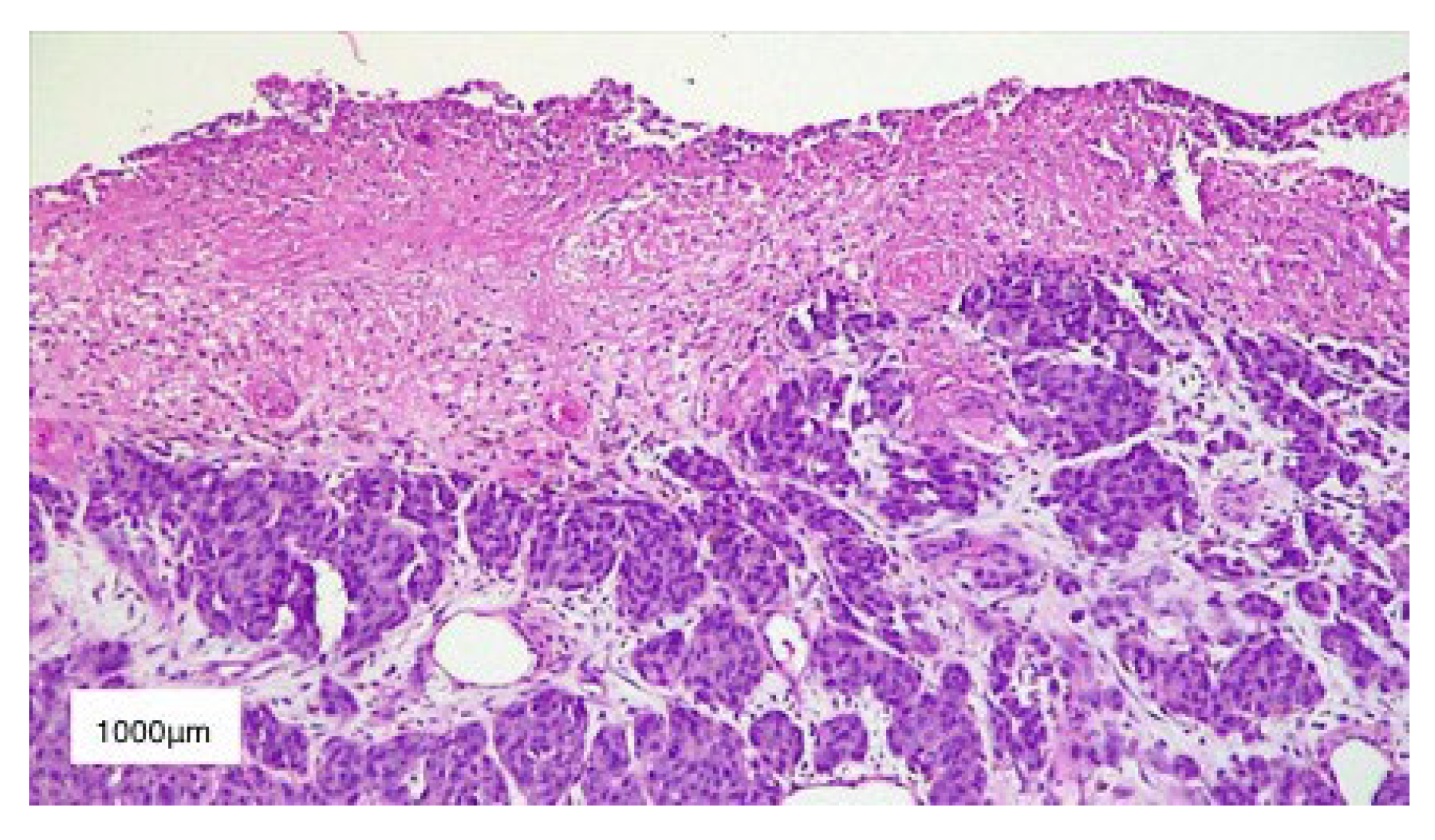
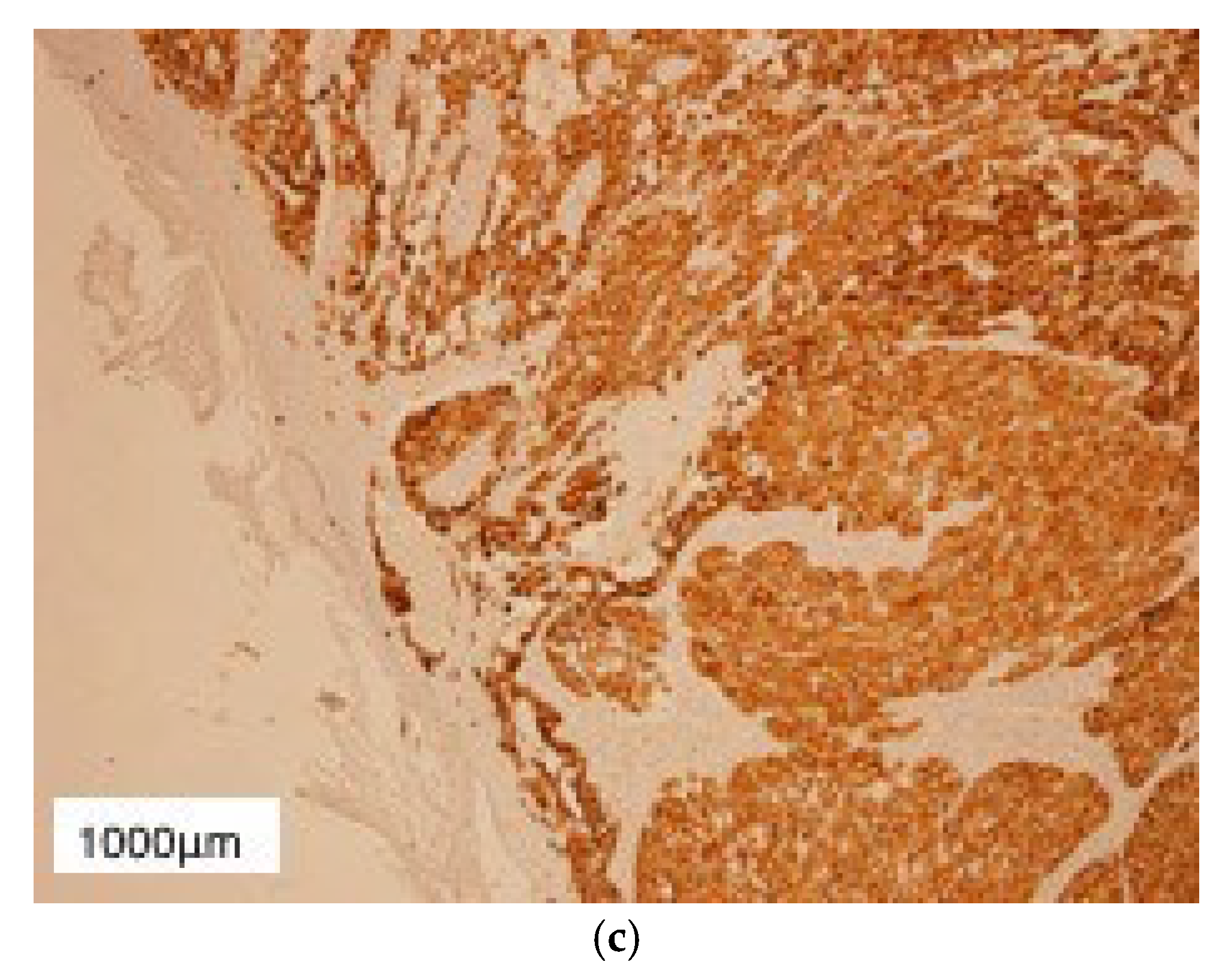
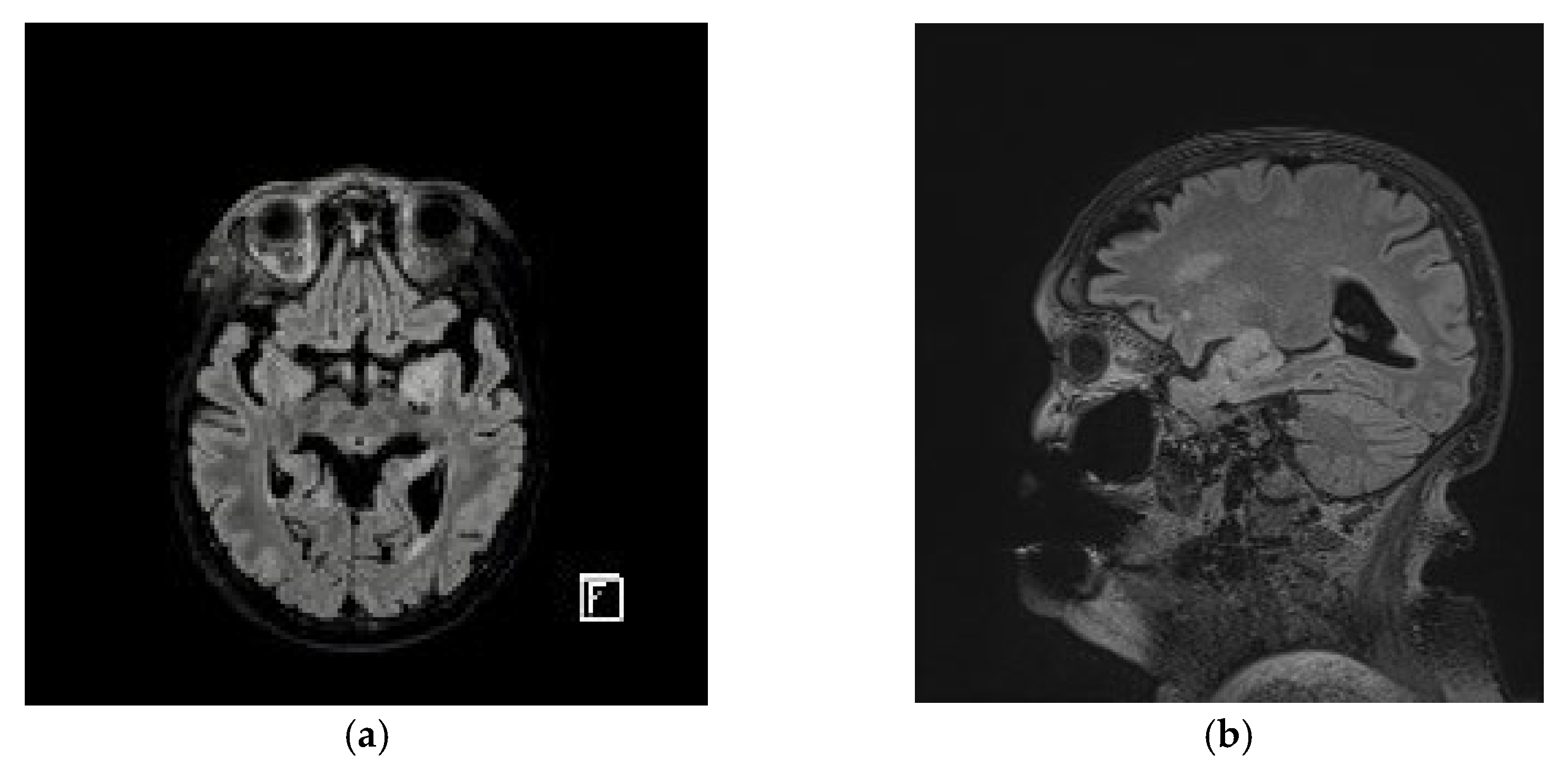
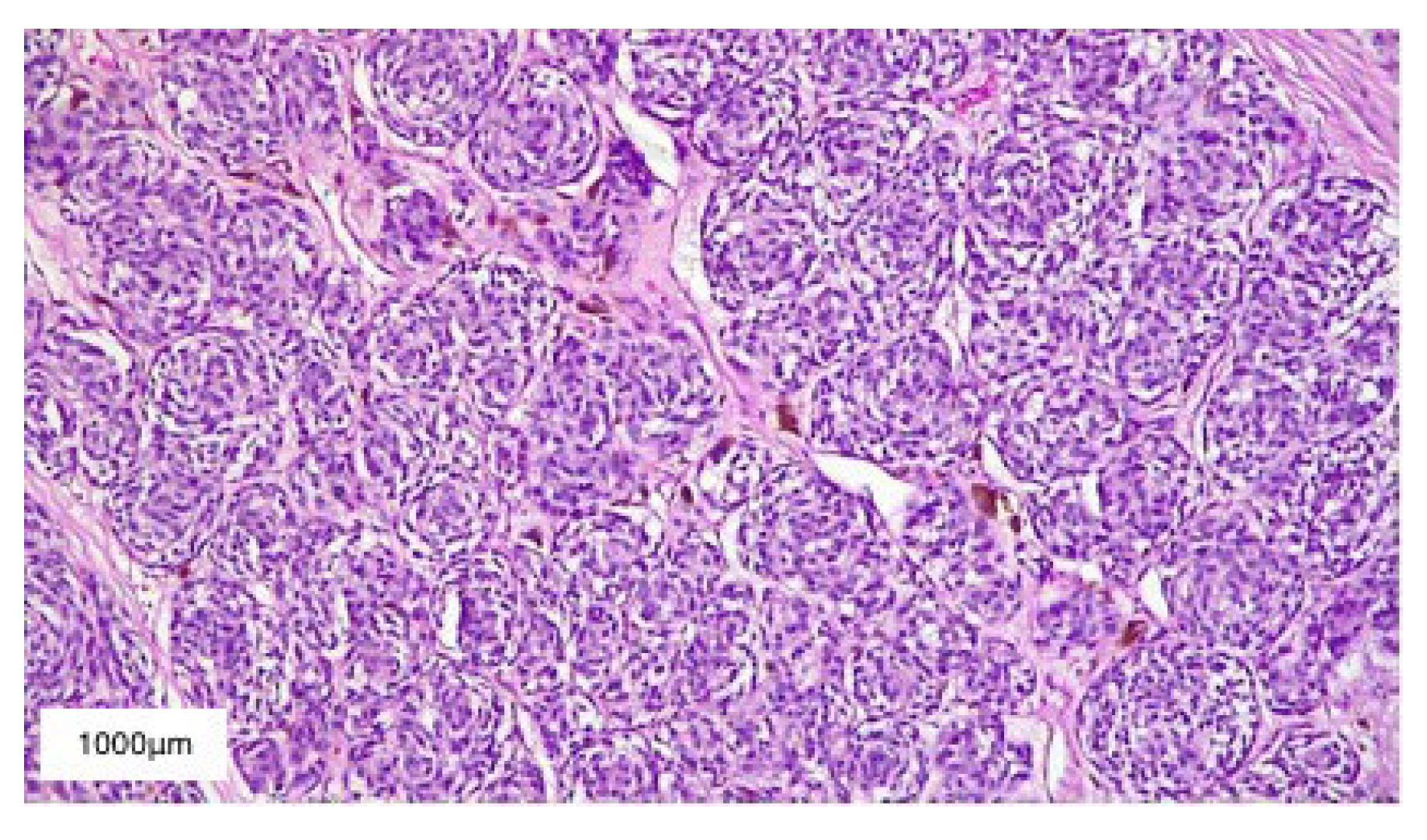
2. Case Presentation
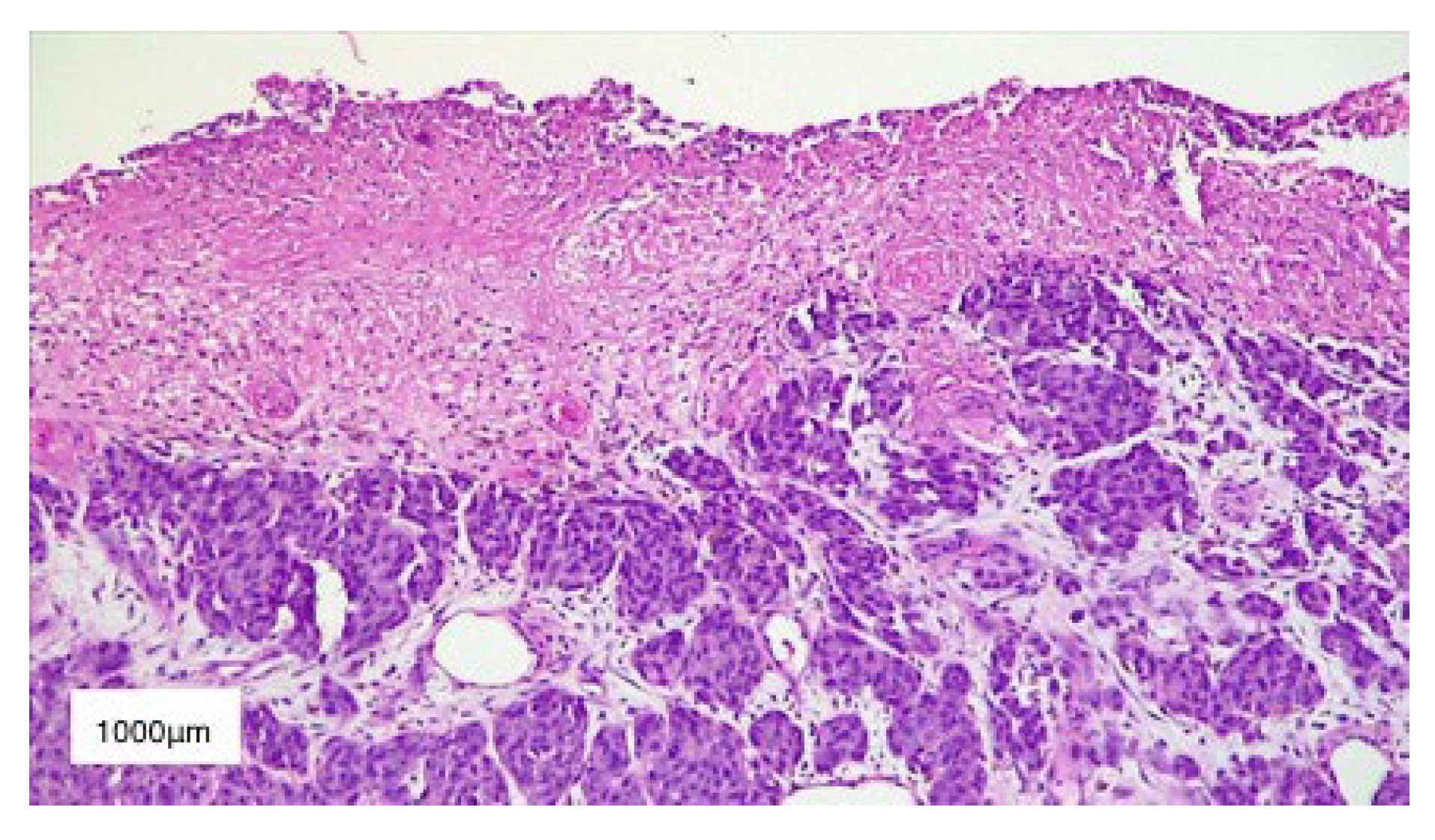
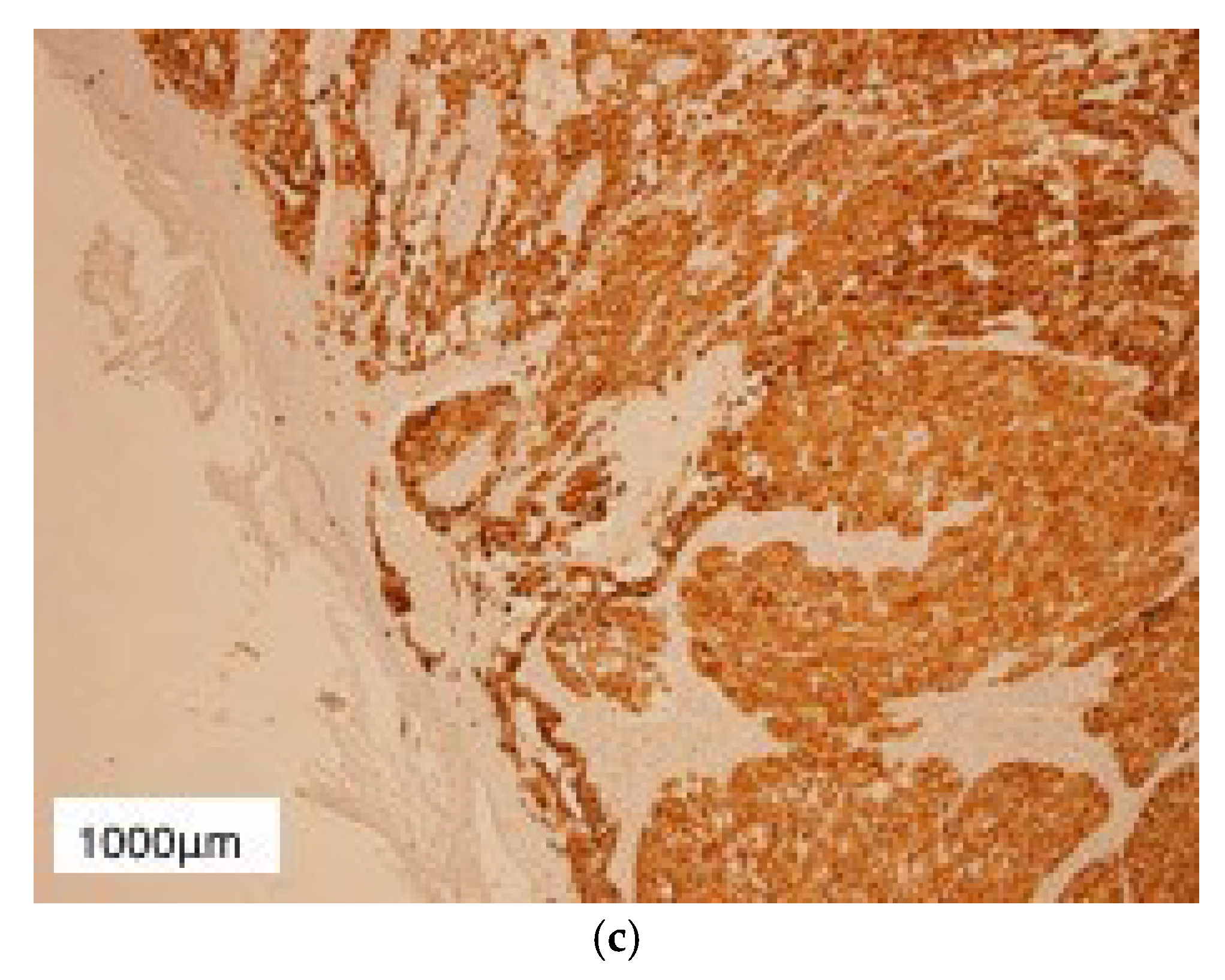
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bolognia, J.L.; Schaffer, J.V.; Cerroni, L. Dermatology, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2018; Chapter 113; pp. 1920–1933. [Google Scholar]
- Kaufman, D.K.; Kimmel, D.W.; Parisi, J.E.; Michels, V.V. A familial syndrome with cutaneous malignant melanoma and cerebral astrocytoma. Neurology 1993, 43, 1728–1731. [Google Scholar] [CrossRef] [PubMed]
- Takita, J.; Hayashi, Y.; Nakajima, T.; Adachi, J.; Tanaka, T.; Yamaguchi, N.; Ogawa, Y.; Hanada, R.; Yamamoto, K.; Yokota, J. The p16 (CDKN2A) gene is involved in the growth of neuroblastoma cells and its expression is associated with prognosis of neuroblastoma patients. Oncogene 1998, 17, 3137–3143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayward, N.K. Genetics of melanoma predisposition. Oncogene 2003, 22, 3053–3062. [Google Scholar] [CrossRef] [Green Version]
- Soura, E.; Eliades, P.J.; Shannon, K.; Stratigos, A.J.; Tsao, H. Hereditary melanoma: Update on syndromes and management: Genetics of familial atypical multiple mole melanoma syndrome. J. Am. Acad. Dermatol. 2016, 74, 395–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laud, K.; Marian, C.; Avril, M.F.; Barrois, M.; Chompret, A.; Goldstein, A.M.; Tucker, M.A.; Clark, P.A.; Peters, G.; Chaudru, V.; et al. Comprehensive analysis of CDKN2A (p16INK4A/p14ARF) and CDKN2B genes in 53 melanoma index cases considered to be at heightened risk of melanoma. J. Med. Genet. 2006, 43, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Cen, L.; Carlson, B.L.; Schroeder, M.A.; Ostrem, J.L.; Kitange, G.J.; Mladek, A.C.; Fink, S.R.; Decker, P.A.; Wu, W.; Kim, J.S.; et al. p16-Cdk4-Rb axis controls sensitivity to a cyclin-dependent kinase inhibitor PD0332991 in glioblastoma xenograft cells. Neuro Oncol. 2012, 14, 870–881. [Google Scholar] [CrossRef] [Green Version]
- Petronzelli, F.; Sollima, D.; Coppola, G.; Martini-Neri, M.E.; Neri, G.; Genuardi, M. CDKN2A germline splicing mutation affecting both p16(ink4) and p14(arf) RNA processing in a melanoma/neurofibroma kindred. Genes Chromosomes Cancer 2001, 31, 398–401. [Google Scholar] [CrossRef]
- Knappskog, S.; Geisler, J.; Arnesen, T.; Lillehaug, J.R.; Lønning, P.E. A novel type of deletion in the CDKN2A gene identified in a melanoma-prone family. Genes Chromosomes Cancer 2007, 45, 1155–1163, Erratum in: Genes Chromosomes Cancer. 2007, 46, 716. [Google Scholar] [CrossRef]
- Loo, J.C.; Liu, L.; Hao, A.; Gao, L.; Agatep, R.; Shennan, M.; Summers, A.; Goldstein, A.M.; Tucker, M.A.; Deters, C.; et al. Germline splicing mutations of CDKN2A predispose to melanoma. Oncogene 2003, 22, 6387–6394. [Google Scholar] [CrossRef] [Green Version]
- Harland, M.; Mistry, S.; Bishop, D.T.; Bishop, J.A. A deep intronic mutation in CDKN2A is associated with disease in a subset of melanoma pedigrees. Hum. Mol. Genet. 2001, 10, 2679–2686. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Choi, B.Y.; Lee, M.H.; Bode, A.M.; Dong, Z. Implications of Genetic and Epigenetic Alterations of CDKN2A (p16(INK4a)) in Cancer. EBioMedicine 2016, 8, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, A.M. Familial melanoma, pancreatic cancer and germline CDKN2A mutations. Hum. Mutat. 2004, 23, 630. [Google Scholar] [CrossRef]
- Castejón-Griñán, M.; Herraiz, C.; Olivares, C.; Jiménez-Cervantes, C.; García-Borrón, J.C. cAMP-independent non-pigmentary actions of variant melanocortin 1 receptor: AKT-mediated activation of protective responses to oxidative DNA damage. Oncogene 2018, 37, 3631–3646. [Google Scholar] [CrossRef] [PubMed]
- Harding, R.M.; Healy, E.; Ray, A.J.; Ellis, N.S.; Flanagan, N.; Todd, C.; Dixon, C.; Sajantila, A.; Jackson, I.J.; Birch-Machin, M.A.; et al. Evidence for variable selective pressures at MC1R. Am. J. Hum. Genet. 2000, 66, 1351–1361. [Google Scholar] [CrossRef] [Green Version]
- Box, N.F.; Wyeth, J.R.; O’Gorman, L.E.; Martin, N.G.; Sturm, R.A. Characterization of melanocyte stimulating hormone receptor variant alleles in twins with red hair. Hum. Mol. Genet. 1997, 6, 1891–1897. [Google Scholar] [CrossRef] [Green Version]
- Palmer, J.S.; Duffy, D.L.; Box, N.F.; Aitken, J.F.; O’Gorman, L.E.; Green, A.C.; Hayward, N.K.; Martin, N.G.; Sturm, R.A. Melanocortin-1 receptor polymorphisms and risk of melanoma: Is the association explained solely by pigmentation phenotype? Am. J. Hum. Genet. 2000, 66, 176–186. [Google Scholar] [CrossRef] [Green Version]
- Box, N.F.; Duffy, D.L.; Chen, W.; Stark, M.; Martin, N.G.; Sturm, R.A.; Hayward, N.K. MC1R genotype modifies risk of melanoma in families segregating CDKN2A mutations. Am. J. Hum. Genet. 2001, 69, 765–773. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Wan, L.; Hacker, E.; Dai, X.; Lenna, S.; Jimenez-Cervantes, C.; Wang, Y.; Leslie, N.R.; Xu, G.X.; Widlund, H.R.; et al. MC1R is a potent regulator of PTEN after UV exposure in melanocytes. Mol. Cell. 2013, 51, 409–422. [Google Scholar] [CrossRef] [Green Version]
- Georgescu, M.M. PTEN Tumor Suppressor Network in PI3K-Akt Pathway Control. Genes Cancer 2010, 1, 1170–1177. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, C.; Zhang, W.; Wang, G.; Yao, K.; Wang, Z.; Li, G.; Qian, Z.; Li, Y.; Jiang, T.; et al. ATRX, IDH1-R132H and Ki-67 immunohistochemistry as a classification scheme for astrocytic tumors. Oncoscience 2016, 3, 258–265. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Zhu, P.; Zhang, C.; Li, Q.; Wang, Z.; Li, G.; Wang, G.; Yang, P.; Li, J.; Han, B.; et al. Detection of ATRX and IDH1-R132H immunohistochemistry in the progression of 211 paired gliomas. Oncotarget 2016, 7, 16384–16395. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.S.; Choe, G.; Nam, K.H.; Seo, A.N.; Yun, S.; Kim, K.J.; Cho, H.J.; Park, S.H. Immunohistochemical classification of primary and secondary glioblastomas. Korean J. Pathol. 2013, 47, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Crespo, I.; Vital, A.L.; Gonzalez-Tablas, M.; Patino Mdel, C.; Otero, A.; Lopes, M.C.; de Oliveira, C.; Domingues, P.; Orfao, A.; Tabernero, M.D. Molecular and Genomic Alterations in Glioblastoma Multiforme. Am. J. Pathol. 2015, 185, 1820–1833. [Google Scholar] [CrossRef] [Green Version]
- Ogura, R.; Tsukamoto, Y.; Natsumeda, M.; Isogawa, M.; Aoki, H.; Kobayashi, T.; Yoshida, S.; Okamoto, K.; Takahashi, H.; Fujii, Y.; et al. Immunohistochemical profiles of IDH1, MGMT and P53: Practical significance for prognostication of patients with diffuse gliomas. Neuropathology 2015, 35, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Gershenwald, J.E.; Scolyer, R.A.; Hess, K.R.; Sondak, V.K.; Long, G.V.; Ross, M.I.; Lazar, A.J.; Faries, M.B.; Kirkwood, J.M.; McArthur, G.A.; et al. Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2017, 67, 472–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbe, C.; Peris, K.; Hauschild, A.; Saiag, P.; Middleton, M.; Bastholt, L.; Grob, J.J.; Malvehy, J.; Newton-Bishop, J.; Stratigos, A.J.; et al. Diagnosis and treatment of melanoma. European consensus-based interdisciplinary guideline—Update 2016. Eur. J. Cancer 2016, 63, 201–217. [Google Scholar] [CrossRef]
- Cohen, A.; Holmen, S.; Colman, H. IDH1 and IDH2 Mutations in Gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345. [Google Scholar] [CrossRef] [Green Version]
- Horbinsky, C. What do we know about IDH1/2 mutations so far, and how do we use it? Acta Neuropathol. 2013, 125, 621–636. [Google Scholar] [CrossRef] [Green Version]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011, 19, 17–30. [Google Scholar] [CrossRef] [Green Version]
- National Comprehensive Cancer Network. Clinical Practice Guidelines in Oncology—Central Nervous System Cancers; Version I.2018; NCCN.Org: Bethesda, MD, USA, 2018. [Google Scholar]
- Frigerio, S.; Disciglio, V.; Manoukian, S.; Peissel, B.; Della Torre, G.; Maurichi, A.; Collini, P.; Pasini, B.; Gotti, G.; Ferrari, A.; et al. A large de novo 9p21.3 deletion in a girl affected by astrocytoma and multiple melanoma. BMC Med. Genet. 2014, 15, 59. [Google Scholar] [CrossRef] [Green Version]
- Braam, K.I.; Overbeek, A.; Kaspers, G.J.; Ronckers, C.M.; Schouten-van Meeteren, A.Y.; Van Dulmen-Den Broeder, E.; Veening, M.A. Malignant melanoma as second malignant neoplasm in long-term childhood cancer survivors: A systematic review. Pediatr. Blood Cancer 2012, 58, 665–674. [Google Scholar] [CrossRef]
- Pasmant, E.; Laurendeau, I.; Héron, D.; Vidaud, M.; Vidaud, D.; Bièche, I. Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: Identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res. 2007, 67, 3963–3969. [Google Scholar] [CrossRef] [Green Version]
- Chan, A.K.; Han, S.J.; Choy, W.; Beleford, D.; Aghi, M.K.; Berger, M.S.; Shieh, J.T.; Bollen, A.W.; Perry, A.; Phillips, J.J.; et al. Familial melanoma-astrocytoma syndrome: Synchronous diffuse astrocytoma and pleomorphic xanthoastrocytoma in a patient with germline CDKN2A/B deletion and a significant family history. Clin. Neuropathol. 2017, 36, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiondella, L.; Cavallieri, F.; Napoli, M.; Froio, E.; Serra, S.; Borsari, S.; Giaccherini, L.; Ghadirpour, R.; Cozzi, S.; Pascarella, R.; et al. Glioblastoma and malignant melanoma: Serendipitous or anticipated association? Neuropathology 2021, 41, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Malmström, A.; Grønberg, B.H.; Marosi, C.; Stupp, R.; Frappaz, D.; Schultz, H.; Abacioglu, U.; Tavelin, B.; Lhermitte, B.; Hegi, M.E.; et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: The Nordic randomised, phase 3 trial. Lancet Oncol. 2012, 13, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma [Internet]; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; Chapter 8. Available online: https://www.ncbi.nlm.nih.gov/books/NBK470003/ (accessed on 12 February 2022). [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orzan, O.A.; Giurcăneanu, C.; Dima, B.; Dima, M.B.; Ion, A.; Bălăceanu, B.; Nițipir, C.; Tudose, I.; Nicolae, C.A.; Dorobanțu, A.M. Cutaneous Melanoma and Glioblastoma Multiforme Association—Case Presentation and Literature Review. Diagnostics 2023, 13, 1046. https://doi.org/10.3390/diagnostics13061046
Orzan OA, Giurcăneanu C, Dima B, Dima MB, Ion A, Bălăceanu B, Nițipir C, Tudose I, Nicolae CA, Dorobanțu AM. Cutaneous Melanoma and Glioblastoma Multiforme Association—Case Presentation and Literature Review. Diagnostics. 2023; 13(6):1046. https://doi.org/10.3390/diagnostics13061046
Chicago/Turabian StyleOrzan, Olguța Anca, Călin Giurcăneanu, Bogdan Dima, Monica Beatrice Dima, Ana Ion, Beatrice Bălăceanu, Cornelia Nițipir, Irina Tudose, Cătălina Andreea Nicolae, and Alexandra Maria Dorobanțu. 2023. "Cutaneous Melanoma and Glioblastoma Multiforme Association—Case Presentation and Literature Review" Diagnostics 13, no. 6: 1046. https://doi.org/10.3390/diagnostics13061046
APA StyleOrzan, O. A., Giurcăneanu, C., Dima, B., Dima, M. B., Ion, A., Bălăceanu, B., Nițipir, C., Tudose, I., Nicolae, C. A., & Dorobanțu, A. M. (2023). Cutaneous Melanoma and Glioblastoma Multiforme Association—Case Presentation and Literature Review. Diagnostics, 13(6), 1046. https://doi.org/10.3390/diagnostics13061046