
The Challenge of Neuropsychiatric Systemic Lupus Erythematosus: From Symptoms to Therapeutic Strategies

Abstract
1. Introduction
2. Clinical Manifestations
2.1. Focal CNS Syndromes
2.2. Focal Peripheral Nervous System Syndromes
2.3. Diffuse CNS Syndromes
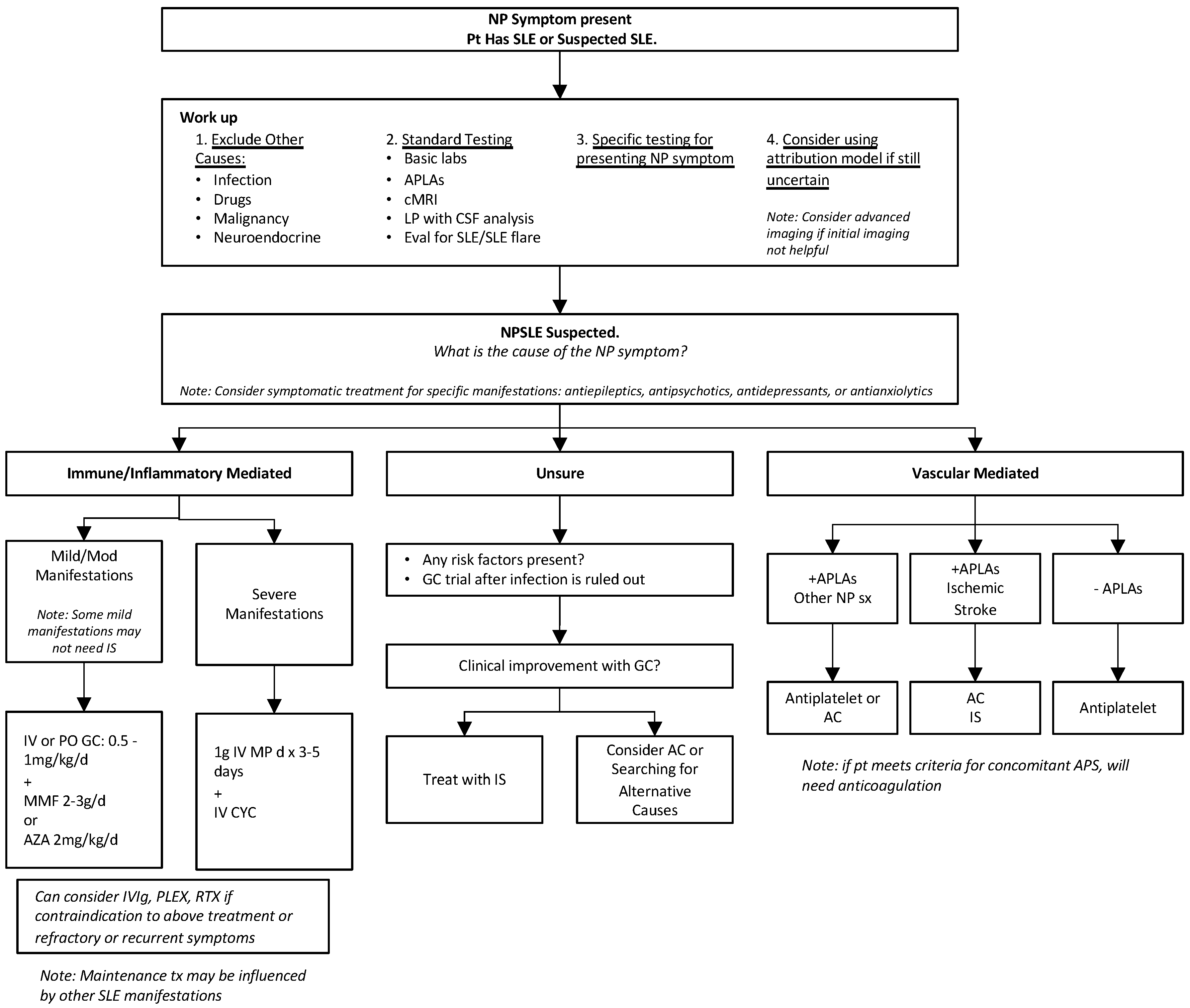
3. Diagnosis
3.1. Diagnosing SLE
Assessing for SLE Flares
3.2. Diagnosing Antiphospholipid Syndrome
3.3. Diagnosing NPSLE
3.3.1. Laboratory Testing
3.3.2. Imaging
3.3.3. Other Testing
3.4. Attribution Models
- The temporal relationship of NP events to the diagnosis of SLE (score: 0–3);
- Identification of minor or common NP events [19] (score 0–3);
- Recognition of confounding factors according to ACR case definitions (score > 1 factor = 0; 0 factors = 2);
- Favoring factors (derived from EULAR recommendations for NPSLE) (score of 0 = none, up to 2 if >1 factor).
4. Pathogenesis
- Autoimmune/inflammatory pathway: Pro-inflammatory mediators or autoantibodies against neuronal cells cause damage due to intrathecal immune complex formation and disruption of the blood-brain barrier. Manifestations with optic neuritis, transverse myelitis, peripheral neuropathy (mono multiplex), recurrent seizures, and diffuse manifestations may be caused by this pathway.
- Vascular/ischemic/thrombotic pathway: Autoantibodies mediate vascular injury, causing cerebral microangiopathy, vascular occlusion, and hemorrhaging. Complement activation and deposition, accelerated atherosclerosis, coagulopathy, and immune complex deposition all contribute to damage in this pathway as well. Manifestations with positive APLAs, including cerebrovascular disease, chorea, seizures, myelopathy, and cognitive dysfunction, may be caused by this pathway.
5. Treatment
5.1. General Lupus Management
5.2. NPSLE Treatment
5.2.1. Immunosuppressive Treatment
5.2.2. Ischemic Treatment
5.2.3. Symptomatic Treatment
5.2.4. Therapeutic Challenges
6. Juvenile-Onset NPSLE
7. Future Directions
8. Conclusions
Funding
Conflicts of Interest
References
- Hanly, J.G.; Urowitz, M.B.; Gordon, C.; Bae, S.C.; Romero-Diaz, J.; Sanchez-Guerrero, J.; Bernatsky, S.; Clarke, A.E.; Wallace, D.J.; Isenberg, D.A.; et al. Neuropsychiatric events in systemic lupus erythematosus: A longitudinal analysis of outcomes in an international inception cohort using a multistate model approach. Ann. Rheum. Dis. 2020, 79, 356–362. [Google Scholar] [CrossRef] [PubMed]
- The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum. 1999, 42, 599–608. [CrossRef]
- Camones-Huerta, J.; Arias-Osorio, C.; Rodriguez-Hurtado, D.; Aguilar-Olano, J. Neuropsychiatric Manifestations in Systemic Lupus Erythematosus Patients at a Tertiary Hospital in Peru. Eur. J. Rheumatol. 2023, 10, 143–147. [Google Scholar] [PubMed]
- Bertsias, G.K.; Ioannidis, J.P.; Aringer, M.; Bollen, E.; Bombardieri, S.; Bruce, I.N.; Cervera, R.; Dalakas, M.; Doria, A.; Hanly, J.G.; et al. EULAR recommendations for the management of systemic lupus erythematosus with neuropsychiatric manifestations: Report of a task force of the EULAR standing committee for clinical affairs. Ann. Rheum. Dis. 2010, 69, 2074–2082. [Google Scholar] [CrossRef] [PubMed]
- Govoni, M.; Bortoluzzi, A.; Padovan, M.; Silvagni, E.; Borrelli, M.; Donelli, F.; Ceruti, S.; Trotta, F. The diagnosis and clinical management of the neuropsychiatric manifestations of lupus. J. Autoimmun. 2016, 74, 41–72. [Google Scholar] [CrossRef] [PubMed]
- Unterman, A.; Nolte, J.E.; Boaz, M.; Abady, M.; Shoenfeld, Y.; Zandman-Goddard, G. Neuropsychiatric syndromes in systemic lupus erythematosus: A meta-analysis. Semin. Arthritis Rheum. 2011, 41, 1–11. [Google Scholar] [CrossRef]
- Pamuk, O.N.; Raza, A.A.; Hasni, S. Neuropsychiatric lupus in late- and early-onset systemic lupus erythematosus patients: A systematic review and meta-analysis. Rheumatology 2024, 63, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Carrión-Barberà, I.; Salman-Monte, T.C.; Vílchez-Oya, F.; Monfort, J. Neuropsychiatric involvement in systemic lupus erythematosus: A review. Autoimmun. Rev. 2021, 20, 102780. [Google Scholar] [CrossRef] [PubMed]
- Shaban, A.; Leira, E.C. Neurological Complications in Patients with Systemic Lupus Erythematosus. Curr. Neurol. Neurosci. Rep. 2019, 19, 97. [Google Scholar] [CrossRef]
- Barbhaiya, M.; Zuily, S.; Naden, R.; Hendry, A.; Manneville, F.; Amigo, M.C.; Amoura, Z.; Andrade, D.; Andreoli, L.; Artim-Esen, B.; et al. The 2023 ACR/EULAR Antiphospholipid Syndrome Classification Criteria. Arthritis Rheumatol. 2023, 75, 1687–1702. [Google Scholar] [CrossRef]
- Garcia, D.; Erkan, D. Diagnosis and Management of the Antiphospholipid Syndrome. N. Engl. J. Med. 2018, 378, 2010–2021. [Google Scholar] [CrossRef] [PubMed]
- Piga, M.; Chessa, E.; Peltz, M.T.; Floris, A.; Mathieu, A.; Cauli, A. Demyelinating syndrome in SLE encompasses different subtypes: Do we need new classification criteria? Pooled results from systematic literature review and monocentric cohort analysis. Autoimmun. Rev. 2017, 16, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Justiz-Vaillant, A.A.; Gopaul, D.; Soodeen, S.; Arozarena-Fundora, R.; Barbosa, O.A.; Unakal, C.; Thompson, R.; Pandit, B.; Umakanthan, S.; Akpaka, P.E. Neuropsychiatric Systemic Lupus Erythematosus: Molecules Involved in Its Imunopathogenesis, Clinical Features, and Treatment. Molecules 2024, 29, 747. [Google Scholar] [CrossRef] [PubMed]
- Bortoluzzi, A.; Piga, M.; Silvagni, E.; Chessa, E.; Mathieu, A.; Govoni, M. Peripheral nervous system involvement in systemic lupus erythematosus: A retrospective study on prevalence, associated factors and outcome. Lupus 2019, 28, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Constantin, A.; Năstase, D.; Tulbă, D.; Bălănescu, P.; Băicuș, C. Immunosuppressive therapy of systemic lupus erythematosus associated peripheral neuropathy: A systematic review. Lupus 2020, 29, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Mizrachi, M.; Anderson, E.; Carroll, K.R.; Tehrani, N.; Volpe, B.T.; Diamond, B. Cognitive dysfunction in SLE: An understudied clinical manifestation. J. Autoimmun. 2022, 132, 102911. [Google Scholar] [CrossRef] [PubMed]
- Hanly, J.G.; Li, Q.; Su, L.; Urowitz, M.B.; Gordon, C.; Bae, S.; Romero-Diaz, J.; Sanchez-Guerrero, J.; Bernatsky, S.; Clarke, A.E.; et al. Psychosis in Systemic Lupus Erythematosus: Results From an International Inception Cohort Study. Arthritis Rheumatol. 2019, 71, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Zandman-Goddard, G.; Chapman, J.; Shoenfeld, Y. Autoantibodies involved in neuropsychiatric SLE and antiphospholipid syndrome. Semin. Arthritis Rheum. 2007, 36, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Ainiala, H.; Hietaharju, A.; Loukkola, J.; Peltola, J.; Korpela, M.; Metsänoja, R.; Auvinen, A. Validity of the new American College of Rheumatology criteria for neuropsychiatric lupus syndromes: A population-based evaluation. Arthritis Rheum. 2001, 45, 419–423. [Google Scholar] [CrossRef]
- Hanly, J.G.; Su, L.; Farewell, V.; McCURDY, G.; Fougere, L.; Thompson, K. Prospective study of neuropsychiatric events in systemic lupus erythematosus. J. Rheumatol. 2009, 36, 1449–1459. [Google Scholar] [CrossRef]
- Palazzo, L.; Lindblom, J.; Cetrez, N.; Ala, H.; Parodis, I. Determinants of neuropsychiatric flares in patients with systemic lupus erythematosus: Results from five phase III trials of belimumab. Rheumatology 2024, 63, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.; Orbai, A.M.; Alarcón, G.S.; Gordon, C.; Merrill, J.T.; Fortin, P.R.; Bruce, I.N.; Isenberg, D.; Wallace, D.J.; Nived, O.; et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012, 64, 2677–2686. [Google Scholar] [CrossRef] [PubMed]
- Aringer, M.; Costenbader, K.; Daikh, D.; Brinks, R.; Mosca, M.; Ramsey-Goldman, R.; Smolen, J.S.; Wofsy, D.; Boumpas, D.T.; Kamen, D.L.; et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Ann. Rheum. Dis. 2019, 78, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- Nikolopoulos, D.; Fanouriakis, A.; Bertsias, G. Treatment of neuropsychiatric systemic lupus erythematosus: Clinical challenges and future perspectives. Expert Rev. Clin. Immunol. 2021, 17, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Inglese, F.; Jaarsma-Coes, M.G.; Steup-Beekman, G.M.; Monahan, R.; Huizinga, T.; van Buchem, M.A.; Ronen, I.; de Bresser, J. Neuropsychiatric systemic lupus erythematosus is associated with a distinct type and shape of cerebral white matter hyperintensities. Rheumatology 2022, 61, 2663–2671. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, G.A.; Rocca, M.A.; Preziosa, P.; Bozzolo, E.P.; Pagani, E.; Canti, V.; Moiola, L.; Rovere-Querini, P.; Manfredi, A.A.; Filippi, M. Quantitative MRI adds to neuropsychiatric lupus diagnostics. Rheumatology 2021, 60, 3278–3288. [Google Scholar] [CrossRef] [PubMed]
- Ota, Y.; Srinivasan, A.; Capizzano, A.A.; Bapuraj, J.R.; Kim, J.; Kurokawa, R.; Baba, A.; Moritani, T. Central Nervous System Systemic Lupus Erythematosus: Pathophysiologic, Clinical, and Imaging Features. Radiographics 2022, 42, 212–232. [Google Scholar] [CrossRef] [PubMed]
- Castellino, G.; Govoni, M.; Giacuzzo, S.; Trotta, F. Optimizing clinical monitoring of central nervous system involvement in SLE. Autoimmun. Rev. 2008, 7, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.; Huang, M.W.; Putterman, C. Advances in the diagnosis, pathogenesis and treatment of neuropsychiatric systemic lupus erythematosus. Curr. Opin. Rheumatol. 2020, 32, 152–158. [Google Scholar] [CrossRef]
- Bortoluzzi, A.; Scirè, C.A.; Bombardieri, S.; Caniatti, L.; Conti, F.; De Vita, S.; Doria, A.; Ferraccioli, G.; Gremese, E.; Mansutti, E.; et al. Development and validation of a new algorithm for attribution of neuropsychiatric events in systemic lupus erythematosus. Rheumatology 2015, 54, 891–898. [Google Scholar] [CrossRef]
- Fanouriakis, A.; Pamfil, C.; Rednic, S.; Sidiropoulos, P.; Bertsias, G.; Boumpas, D.T. Is it primary neuropsychiatric systemic lupus erythematosus? Performance of existing attribution models using physician judgment as the gold standard. Clin. Exp. Rheumatol. 2016, 34, 910–917. [Google Scholar] [PubMed]
- Govoni, M.; Hanly, J.G. The management of neuropsychiatric lupus in the 21st century: Still so many unmet needs? Rheumatology 2020, 59 (Suppl. 5), v52–v62. [Google Scholar] [CrossRef] [PubMed]
- Sarwar, S.; Mohamed, A.S.; Rogers, S.; Sarmast, S.T.; Kataria, S.; Mohamed, K.H.; Khalid, M.Z.; Saeeduddin, M.O.; Shiza, S.T.; Ahmad, S.; et al. Neuropsychiatric Systemic Lupus Erythematosus: A 2021 Update on Diagnosis, Management, and Current Challenges. Cureus 2021, 13, e17969. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, N.; Stock, A.D.; Putterman, C. Neuropsychiatric lupus: New mechanistic insights and future treatment directions. Nat. Rev. Rheumatol. 2019, 15, 137–152. [Google Scholar] [CrossRef]
- Lindblom, J.; Mohan, C.; Parodis, I. Biomarkers in Neuropsychiatric Systemic Lupus Erythematosus: A Systematic Literature Review of the Last Decade. Brain Sci. 2022, 12, 192. [Google Scholar] [CrossRef]
- Sato, S.; Temmoku, J.; Fujita, Y.; Yashiro-Furuya, M.; Matsuoka, N.; Asano, T.; Kobayashi, H.; Watanabe, H.; Migita, K. Autoantibodies associated with neuropsychiatric systemic lupus erythematosus: The quest for symptom-specific biomarkers. Fukushima J. Med. Sci. 2020, 66, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.; Rijnink, E.C.; Nabuurs, R.J.A.; Steup-Beekman, G.M.; Versluis, M.J.; Emmer, B.J.; Zandbergen, M.; van Buchem, M.A.; Allaart, C.F.; Wolterbeek, R.; et al. Brain histopathology in patients with systemic lupus erythematosus: Identification of lesions associated with clinical neuropsychiatric lupus syndromes and the role of complement. Rheumatology 2017, 56, 77–86. [Google Scholar] [CrossRef]
- Magro-Checa, C.; Zirkzee, E.J.; Huizinga, T.W.; Steup-Beekman, G.M. Management of Neuropsychiatric Systemic Lupus Erythematosus: Current Approaches and Future Perspectives. Drugs 2016, 76, 459–483. [Google Scholar] [CrossRef]
- Barile-Fabris, L.; Ariza-Andraca, R.; Olguin-Ortega, L.; Jara, L.J.; Fraga-Mouret, A.; Miranda-Limon, J.M.; de la Mata, J.F.; Clark, P.; Vargas, F.; Alocer-Varela, J. Controlled clinical trial of IV cyclophosphamide versus IV methylprednisolone in severe neurological manifestations in systemic lupus erythematosus. Ann. Rheum. Dis. 2005, 64, 620–625. [Google Scholar] [CrossRef]
- Papachristos, D.; Oon, S.; Hanly, J.; Nikpour, M. Management of inflammatory neurologic and psychiatric manifestations of systemic lupus erythematosus: A systematic review. Semin. Arthritis Rheum. 2021, 51, 49–71. [Google Scholar] [CrossRef]
- Monahan, R.C.; de Voorde, L.J.J.B.-V.; Fronczek, R.; de Bresser, J.; Eikenboom, J.; Kloppenburg, M.; Middelkoop, H.A.M.; Terwindt, G.M.; van der Wee, N.J.A.; Huizinga, T.W.J.; et al. Clinical outcome in patients with suspected inflammatory neuropsychiatric lupus treated with immunosuppression: An observational cohort study. Lupus Sci. Med. 2023, 10, e000850. [Google Scholar] [CrossRef] [PubMed]
- Bortoluzzi, A.; Fanouriakis, A.; Silvagni, E.; Appenzeller, S.; Carli, L.; Carrara, G.; Cauli, A.; Conti, F.; Costallat, L.T.L.; De Marchi, G.; et al. Therapeutic strategies and outcomes in neuropsychiatric systemic lupus erythematosus: An international multicentre retrospective study. Rheumatology 2024, keae119. [Google Scholar] [CrossRef] [PubMed]
- Furie, R.; Rovin, B.H.; Houssiau, F.; Malvar, A.; Teng, Y.O.; Contreras, G.; Amoura, Z.; Yu, X.; Mok, C.-C.; Santiago, M.B.; et al. Two-Year, Randomized, Controlled Trial of Belimumab in Lupus Nephritis. N. Engl. J. Med. 2020, 383, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Morand, E.F.; Furie, R.; Tanaka, Y.; Bruce, I.N.; Askanase, A.D.; Richez, C.; Bae, S.C.; Brohawn, P.Z.; Pineda, L.; Berglind, A.; et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N. Engl. J. Med. 2020, 382, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.; Naqibuddin, M.; Sampedro, M.; Omdal, R.; Carson, K.A. Memantine in systemic lupus erythematosus: A randomized, double-blind placebo-controlled trial. Semin. Arthritis Rheum. 2011, 41, 194–202. [Google Scholar] [CrossRef]
- Kısaarslan, A.P.; Çiçek, S.; Batu, E.D.; Şahin, S.; Gürgöze, M.K.; Çetinkaya, S.B.; Ekinci, M.K.; Atmış, B.; Barut, K.; Adrovic, A.; et al. Neuropsychiatric involvement in juvenile-onset systemic lupus erythematosus: A multicenter study. Jt. Bone Spine 2023, 90, 105559. [Google Scholar] [CrossRef] [PubMed]
- Nikolaidou, A.; Beis, I.; Dragoumi, P.; Zafeiriou, D. Neuropsychiatric manifestations associated with Juvenile Systemic Lupus Erythematosus: An overview focusing on early diagnosis. Brain Dev. 2024, 46, 125–134. [Google Scholar] [CrossRef]
- Labouret, M.; Trebossen, V.; Ntorkou, A.; Bartoli, S.; Aubart, M.; Auvin, S.; Bader-Meunier, B.; Baudouin, V.; Corseri, O.; Dingulu, G.; et al. Juvenile neuropsychiatric systemic lupus erythematosus: A specific clinical phenotype and proposal of a probability score. Lupus 2024, 33, 328–339. [Google Scholar] [CrossRef]

{kind=link}
| Focal | Diffuse | ||
|---|---|---|---|
| Central NS | Helpful Diagnostics to Consider | Central NS | Helpful Diagnostics to Consider |
| Aseptic Meningitis | LP and MRI | Acute Confusional state | LP and MRI to exclude infection Neuropsych testing |
| Cerebrovascular Disease | EKG, echocardiogram, carotid doppler MRA and LP (if concern for CNS vasculitis) | Anxiety | |
| Demyelinating Syndromes | LP and MRI Consider testing for MS | Cognitive dysfunction | Neuropsych testing |
| Headaches | Mood disorder | Psychiatric evaluation | |
| Movement Disorders: Chorea | Psychosis | Psychiatric evaluation | |
| Myelopathy | Gad-enhanced MRI and LP and CSF analysis | ||
| Seizure Disorder | MRI, EEG, LP Important to exclude structural brain disease and inflammatory or metabolic conditions, infections | ||
| Peripheral NS | Helpful Diagnostics to Consider | ||
| AIDP | EMG and NCV, LP | ||
| Autonomic Disorder | |||
| Mononeuropathy (Single or Multiplex) | EMG and NCV | ||
| Myasthenia Gravis | CT exclude thyroid disease, specific ab testing (AchR, MuSK, LRP4) | ||
| Plexopathy | |||
| Polyneuropathy | EMG and NCV | ||
| Cranial Neuropathies | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, V. The Challenge of Neuropsychiatric Systemic Lupus Erythematosus: From Symptoms to Therapeutic Strategies. Diagnostics 2024, 14, 1186. https://doi.org/10.3390/diagnostics14111186
Patel V. The Challenge of Neuropsychiatric Systemic Lupus Erythematosus: From Symptoms to Therapeutic Strategies. Diagnostics. 2024; 14(11):1186. https://doi.org/10.3390/diagnostics14111186
Chicago/Turabian StylePatel, Veena. 2024. "The Challenge of Neuropsychiatric Systemic Lupus Erythematosus: From Symptoms to Therapeutic Strategies" Diagnostics 14, no. 11: 1186. https://doi.org/10.3390/diagnostics14111186
APA StylePatel, V. (2024). The Challenge of Neuropsychiatric Systemic Lupus Erythematosus: From Symptoms to Therapeutic Strategies. Diagnostics, 14(11), 1186. https://doi.org/10.3390/diagnostics14111186