
Mucopolysaccharidosis Type IIIE: A Real Human Disease or a Diagnostic Pitfall?

 , , ,
, , ,  ,
,  and
and 
Abstract
1. Introduction
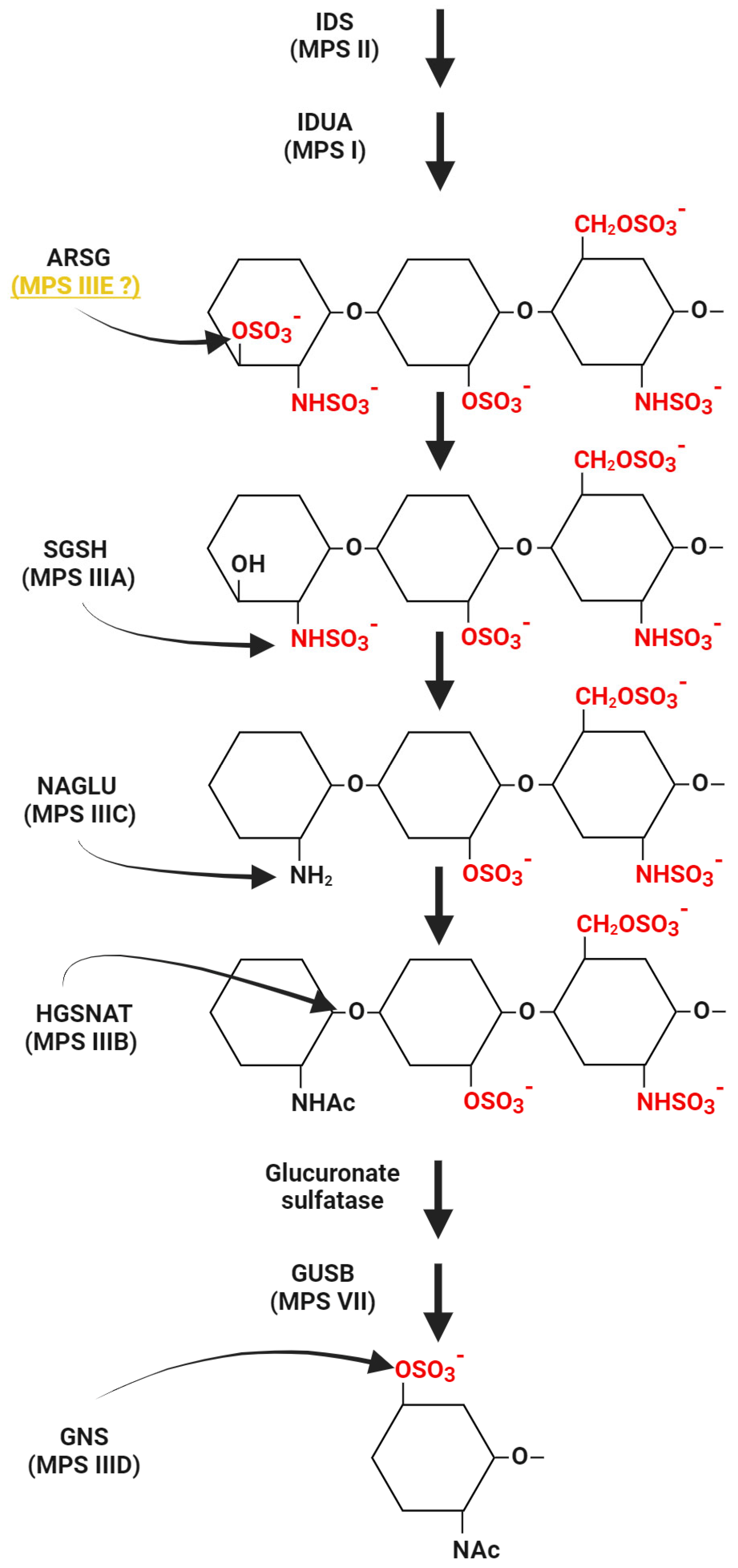
2. Mucopolysaccharidosis
3. MPS IIIE in Humans—Whether It Is or Not?
3.1. Why Yes?
3.1.1. ARSG Activity
3.1.2. Mild/Attenuated Type of MPS
3.2. Why Not?
3.2.1. The Animal Model and Humans
3.2.2. MPS-like Symptoms
3.2.3. One Gene, Different Diseases
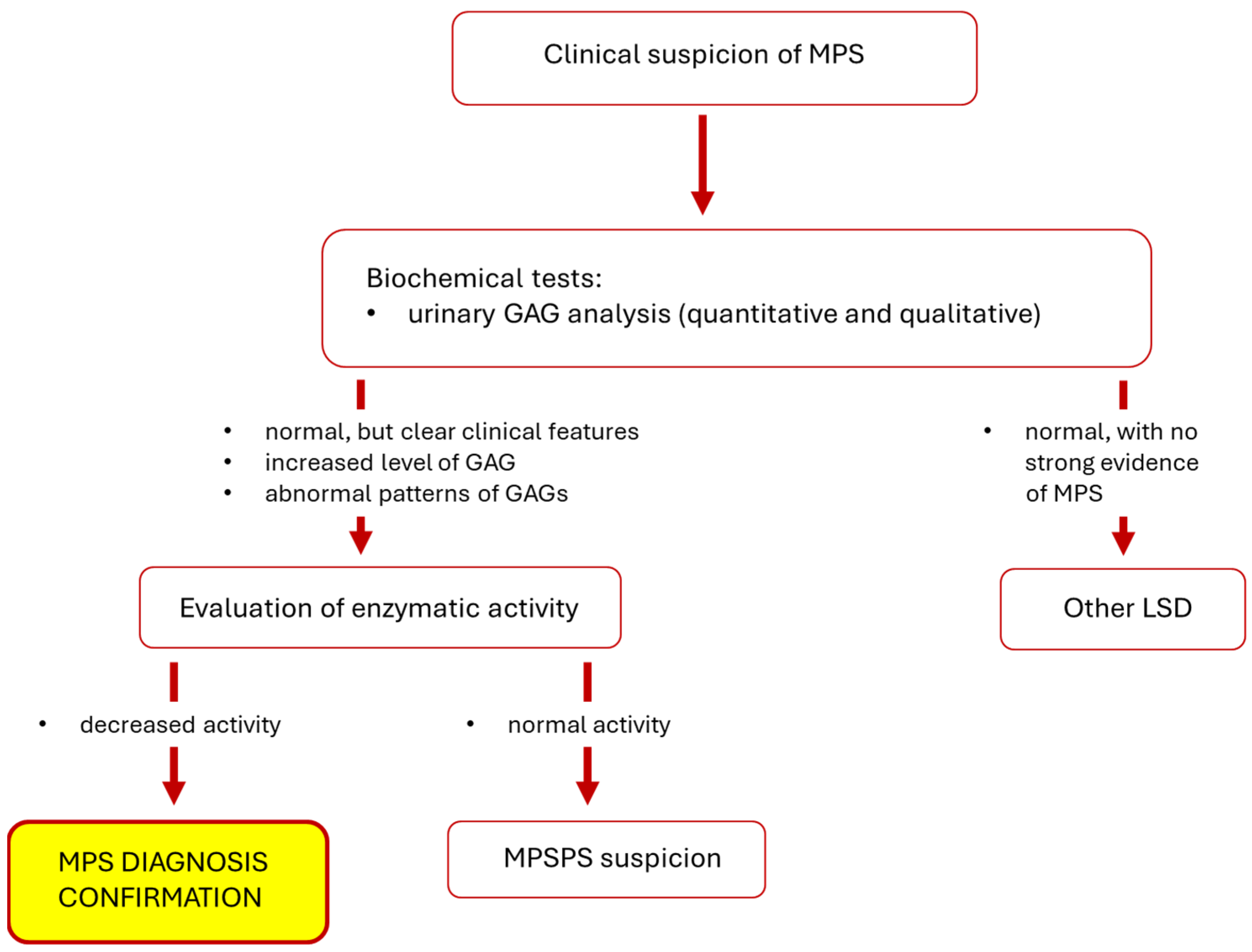
3.2.4. Diagnostics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gaffke, L.; Pierzynowska, K.; Podlacha, M.; Brokowska, J.; Węgrzyn, G. Changes in Cellular Processes Occurring in Mucopolysaccharidoses as Underestimated Pathomechanisms of These Diseases. Cell Biol. Int. 2021, 45, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chi, L. The Alterations and Roles of Glycosaminoglycans in Human Diseases. Polymers 2022, 14, 5014. [Google Scholar] [CrossRef] [PubMed]
- Kubaski, F.; de Oliveira Poswar, F.; Michelin-Tirelli, K.; Burin, M.G.; Rojas-Málaga, D.; Brusius-Facchin, A.C.; Leistner-Segal, S.; Giugliani, R. Diagnosis of Mucopolysaccharidoses. Diagnostics 2020, 10, 172. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewska, K.; Gaffke, L.; Żabińska, M.; Wegrzyn, G.; Pierzynowska, K. Cellular Organelle-Related Transcriptomic Profile Abnormalities in Neuronopathic Types of Mucopolysaccharidosis: A Comparison with Other Neurodegenerative Diseases. Curr. Issues Mol. Biol. 2024, 46, 2678–2700. [Google Scholar] [CrossRef] [PubMed]
- Spahiu, L.; Behluli, E.; Peterlin, B.; Nefic, H.; Hadziselimovic, R.; Liehr, T.; Temaj, G. Mucopolysaccharidosis III: Molecular Basis and Treatment. Pediatr. Endocrinol. Diabetes Metab. 2021, 27, 201–208. [Google Scholar] [CrossRef]
- Rintz, E.; Banacki, M.; Ziemian, M.; Kobus, B.; Wegrzyn, G. Causes of Death in Mucopolysaccharidoses. Mol. Genet. Metab. 2024, 142, 108507. [Google Scholar] [CrossRef] [PubMed]
- Sahin, O.; Thompson, H.P.; Goodman, G.W.; Li, J.; Urayama, A. Mucopolysaccharidoses and the Blood–Brain Barrier. Fluids Barriers CNS 2022, 19, 76. [Google Scholar] [CrossRef]
- Trabszo, C.; Ramms, B.; Chopra, P.; Lüllmann-Rauch, R.; Stroobants, S.; Sproß, J.; Jeschke, A.; Schinke, T.; Boons, G.-J.; Esko, J.D.; et al. Arylsulfatase K Inactivation Causes Mucopolysaccharidosis Due to Deficient Glucuronate Desulfation of Heparan and Chondroitin Sulfate. Biochem. J. 2020, 477, 3433–3451. [Google Scholar] [CrossRef]
- Winner, L.K.; Rogers, M.-L.; Snel, M.F.; Hemsley, K.M. Biomarkers for Predicting Disease Course in Sanfilippo Syndrome: An Urgent Unmet Need in Childhood-Onset Dementia. J. Neurochem. 2023, 166, 481–496. [Google Scholar] [CrossRef]
- Kowalewski, B.; Lamanna, W.C.; Lawrence, R.; Damme, M.; Stroobants, S.; Padva, M.; Kalus, I.; Frese, M.-A.; Lübke, T.; Lüllmann-Rauch, R.; et al. Arylsulfatase G Inactivation Causes Loss of Heparan Sulfate 3-O-Sulfatase Activity and Mucopolysaccharidosis in Mice. Proc. Natl. Acad. Sci. USA 2012, 109, 10310–10315. [Google Scholar] [CrossRef]
- Khateb, S.; Kowalewski, B.; Bedoni, N.; Damme, M.; Pollack, N.; Saada, A.; Obolensky, A.; Ben-Yosef, T.; Gross, M.; Dierks, T.; et al. A Homozygous Founder Missense Variant in Arylsulfatase G Abolishes Its Enzymatic Activity Causing Atypical Usher Syndrome in Humans. Genet. Med. 2018, 20, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Peter, V.G.; Quinodoz, M.; Sadio, S.; Held, S.; Rodrigues, M.; Soares, M.; Sousa, A.B.; Coutinho Santos, L.; Damme, M.; Rivolta, C. New Clinical and Molecular Evidence Linking Mutations in ARSG to Usher Syndrome Type IV. Hum. Mutat. 2021, 42, 261–271. [Google Scholar] [CrossRef]
- Velde, H.M.; Reurink, J.; Held, S.; Li, C.H.Z.; Yzer, S.; Oostrik, J.; Weeda, J.; Haer-Wigman, L.; Yntema, H.G.; Roosing, S.; et al. Usher Syndrome Type IV: Clinically and Molecularly Confirmed by Novel ARSG Variants. Hum. Genet. 2022, 141, 1723–1738. [Google Scholar] [CrossRef] [PubMed]
- Galzerano, D.; Saba, S.; Sergani, A.A.; Vriz, O.; Alghalayini, K.; Ramzan, K.; Elmahi, I.; Cittadini, A.; Salvo, G.D.; Pergola, V. Features and Behavior of Valvular Abnormalities in Adolescent and Adult Patients in Mucopolysaccharidosis: An Echocardiographic Study. Monaldi Arch. Chest Dis. 2021, 91, 1767. [Google Scholar] [CrossRef]
- Zhou, J.; Lin, J.; Leung, W.T.; Wang, L. A Basic Understanding of Mucopolysaccharidosis: Incidence, Clinical Features, Diagnosis, and Management. Intractable Rare Dis. Res. 2020, 9, 1–9. [Google Scholar] [CrossRef]
- Celik, B.; Tomatsu, S.C.; Tomatsu, S.; Khan, S.A. Epidemiology of Mucopolysaccharidoses Update. Diagnostics 2021, 11, 273. [Google Scholar] [CrossRef]
- Ago, Y.; Rintz, E.; Musini, K.S.; Ma, Z.; Tomatsu, S. Molecular Mechanisms in Pathophysiology of Mucopolysaccharidosis and Prospects for Innovative Therapy. Int. J. Mol. Sci. 2024, 25, 1113. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, M.; Arunkumar, N.; Kubaski, F.; Mason, R.W.; Tadao, O.; Tomatsu, S. Clinical Presentation and Diagnosis of Mucopolysaccharidoses. Mol. Genet. Metab. 2018, 125, 4–17. [Google Scholar] [CrossRef]
- Wiśniewska, K.; Wolski, J.; Gaffke, L.; Cyske, Z.; Pierzynowska, K.; Węgrzyn, G. Misdiagnosis in Mucopolysaccharidoses. J. Appl. Genet. 2022, 63, 475–495. [Google Scholar] [CrossRef] [PubMed]
- Arunkumar, N.; Vu, D.C.; Khan, S.; Kobayashi, H.; Ngoc Can, T.B.; Oguni, T.; Watanabe, J.; Tanaka, M.; Yamaguchi, S.; Taketani, T.; et al. Diagnosis of Mucopolysaccharidoses and Mucolipidosis by Assaying Multiplex Enzymes and Glycosaminoglycans. Diagnostics 2021, 11, 1347. [Google Scholar] [CrossRef]
- Mohammed, E.E.A.; Fayez, A.G.; Abdelfattah, N.M.; Fateen, E. Novel Gene-Specific Bayesian Gaussian Mixture Model to Predict the Missense Variants Pathogenicity of Sanfilippo Syndrome. Sci. Rep. 2024, 14, 12148. [Google Scholar] [CrossRef] [PubMed]
- Węgrzyn, G.; Jakóbkiewicz-Banecka, J.; Narajczyk, M.; Wiśniewski, A.; Piotrowska, E.; Gabig-Cimińska, M.; Kloska, A.; Słomińska-Wojewódzka, M.; Korzon-Burakowska, A.; Węgrzyn, A. Why Are Behaviors of Children Suffering from Various Neuronopathic Types of Mucopolysaccharidoses Different? Med. Hypotheses 2010, 75, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Ozbay, H.; Ozbek, M.; Sevinçok, D.; Tas, K.; Aksu, H.; Tosun, A. Autism Spectrum Disorder in a Child with Hunter Syndrome. Psychiatry Clin. Psychopharmacol. 2020, 30, 1. [Google Scholar] [CrossRef]
- Kalyoncu, D.; Gümüştekin, R.; Urganci, N. Is Mucopolysaccharidosis a Cause of Sleep and Speech Disorders? Report of Four Cases? JAREM 2020, 10, 204–207. [Google Scholar] [CrossRef]
- Kowalewski, B.; Lübke, T.; Kollmann, K.; Braulke, T.; Reinheckel, T.; Dierks, T.; Damme, M. Molecular Characterization of Arylsulfatase G: Expression, Processing, Glycosylation, Transport, and Activity. J. Biol. Chem. 2014, 289, 27992–28005. [Google Scholar] [CrossRef] [PubMed]
- Kruszewski, K.; Lüllmann-Rauch, R.; Dierks, T.; Bartsch, U.; Damme, M. Degeneration of Photoreceptor Cells in Arylsulfatase G-Deficient Mice. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1120–1131. [Google Scholar] [CrossRef] [PubMed]
- Abitbol, M.; Thibaud, J.-L.; Olby, N.J.; Hitte, C.; Puech, J.-P.; Maurer, M.; Pilot-Storck, F.; Hédan, B.; Dréano, S.; Brahimi, S.; et al. A Canine Arylsulfatase G (ARSG) Mutation Leading to a Sulfatase Deficiency Is Associated with Neuronal Ceroid Lipofuscinosis. Proc. Natl. Acad. Sci. USA 2010, 107, 14775–14780. [Google Scholar] [CrossRef] [PubMed]
- Kido, J.; Nakamura, K.; Era, T. Role of Induced Pluripotent Stem Cells in Lysosomal Storage Diseases. Mol. Cell. Neurosci. 2020, 108, 103540. [Google Scholar] [CrossRef] [PubMed]
- Cocostîrc, V.; Paștiu, A.I.; Pusta, D.L. An Overview of Canine Inherited Neurological Disorders with Known Causal Variants. Animals 2023, 13, 3568. [Google Scholar] [CrossRef]
- Katz, M.L.; Rustad, E.; Robinson, G.O.; Whiting, R.E.H.; Student, J.T.; Coates, J.R.; Narfstrom, K. Canine Neuronal Ceroid Lipofuscinoses: Promising Models for Preclinical Testing of Therapeutic Interventions. Neurobiol. Dis. 2017, 108, 277–287. [Google Scholar] [CrossRef]
- Rintz, E.; Podlacha, M.; Cyske, Z.; Pierzynowska, K.; Węgrzyn, G.; Gaffke, L. Activities of (Poly)Phenolic Antioxidants and Other Natural Autophagy Modulators in the Treatment of Sanfilippo Disease: Remarkable Efficacy of Resveratrol in Cellular and Animal Models. Neurotherapeutics 2023, 20, 254–271. [Google Scholar] [CrossRef]
- Pierzynowska, K.; Deresz, P.; Węgrzyn, G.; Gaffke, L. Dysregulation of Genes Coding for Proteins Involved in Metabolic Processes in Mucopolysaccharidoses, Evidenced by a Transcriptomic Approach. Metab. Brain Dis. 2023, 38, 2133–2144. [Google Scholar] [CrossRef]
- Ryazantsev, S.; Yu, W.-H.; Zhao, H.-Z.; Neufeld, E.F.; Ohmi, K. Lysosomal Accumulation of SCMAS (Subunit c of Mitochondrial ATP Synthase) in Neurons of the Mouse Model of Mucopolysaccharidosis III B. Mol. Genet. Metab. 2007, 90, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Elleder, M.; Sokolová, J.; Hřebíček, M. Follow-up Study of Subunit c of Mitochondrial ATP Synthase (SCMAS) in Batten Disease and in Unrelated Lysosomal Disorders. Acta Neuropathol. 1997, 93, 379–390. [Google Scholar] [CrossRef]
- Del Longo, A.; Piozzi, E.; Schweizer, F. Ocular Features in Mucopolysaccharidosis: Diagnosis and Treatment. Ital. J. Pediatr. 2018, 44, 125. [Google Scholar] [CrossRef]
- Moro, E. Lysosomal Storage Disorders: Molecular Basis and Therapeutic Approaches. Biomolecules 2021, 11, 964. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Pantoom, S.; Petters, J.; Pandey, A.K.; Hermann, A.; Lukas, J. A Molecular Genetics View on Mucopolysaccharidosis Type II. Mutat. Res./Rev. Mutat. Res. 2021, 788, 108392. [Google Scholar] [CrossRef] [PubMed]
- Hampe, C.S.; Yund, B.D.; Orchard, P.J.; Lund, T.C.; Wesley, J.; McIvor, R.S. Differences in MPS I and MPS II Disease Manifestations. Int. J. Mol. Sci. 2021, 22, 7888. [Google Scholar] [CrossRef]
- Wiśniewska, K.; Gaffke, L.; Krzelowska, K.; Węgrzyn, G.; Pierzynowska, K. Differences in Gene Expression Patterns, Revealed by RNA-Seq Analysis, between Various Sanfilippo and Morquio Disease Subtypes. Gene 2022, 812, 146090. [Google Scholar] [CrossRef]
- Nijmeijer, S.C.M.; van den Born, L.I.; Kievit, A.J.A.; Stepien, K.M.; Langendonk, J.; Marchal, J.P.; Roosing, S.; Wijburg, F.A.; Wagenmakers, M.A.E.M. The Attenuated End of the Phenotypic Spectrum in MPS III: From Late-Onset Stable Cognitive Impairment to a Non-Neuronopathic Phenotype. Orphanet. J. Rare Dis. 2019, 14, 249. [Google Scholar] [CrossRef]
- De Falco, A.; Karali, M.; Criscuolo, C.; Testa, F.; Barillari, M.R.; Scarpato, M.; Gaudieri, V.; Cuocolo, A.; Russo, A.; Nigro, V.; et al. Late-Onset Mucopolysaccharidosis Type IIIA Mimicking Usher Syndrome. Am. J. Med. Genet. Part A 2024, 194, e63517. [Google Scholar] [CrossRef]
- Rigoldi, M.; Verrecchia, E.; Manna, R.; Mascia, M.T. Clinical Hints to Diagnosis of Attenuated Forms of Mucopolysaccharidoses. Ital. J. Pediatr. 2018, 44, 132. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.A.; King, B.M.; Thorsen, C.L.; Hassiotis, S.; Beard, H.; Trim, P.J.; Whyte, L.S.; Tamang, S.J.; Duplock, S.K.; Snel, M.F.; et al. A Novel Conditional Sgsh Knockout Mouse Model Recapitulates Phenotypic and Neuropathic Deficits of Sanfilippo Syndrome. J. Inherit. Metab. Dis. 2017, 40, 715–724. [Google Scholar] [CrossRef]
- Petrova, R.; Patil, A.R.; Trinh, V.; McElroy, K.E.; Bhakta, M.; Tien, J.; Wilson, D.S.; Warren, L.; Stratton, J.R. Disease Pathology Signatures in a Mouse Model of Mucopolysaccharidosis Type IIIB. Sci. Rep. 2023, 13, 16699. [Google Scholar] [CrossRef]
- Marcó, S.; Pujol, A.; Roca, C.; Motas, S.; Ribera, A.; Garcia, M.; Molas, M.; Villacampa, P.; Melia, C.S.; Sánchez, V.; et al. Progressive Neurologic and Somatic Disease in a Novel Mouse Model of Human Mucopolysaccharidosis Type IIIC. Dis. Models Mech. 2016, 9, 999–1013. [Google Scholar] [CrossRef] [PubMed]
- Heon-Roberts, R.; Nguyen, A.L.A.; Pshezhetsky, A.V. Molecular Bases of Neurodegeneration and Cognitive Decline, the Major Burden of Sanfilippo Disease. J. Clin. Med. 2020, 9, 344. [Google Scholar] [CrossRef]
- Bhaumik, M.; Muller, V.J.; Rozaklis, T.; Linda, J.; Dobrenis, K.; Bhattacharyya, R.; Wurzelmann, S.; Finamore, P.; Hopwood, J.J.; Walkley, S.U.; et al. A Mouse Model for Mucopolysaccharidosis Type III A (Sanfilippo Syndrome). Glycobiology 1999, 9, 1389–1396. [Google Scholar] [CrossRef]
- Nolen, R.M.; Hufnagel, R.B.; Friedman, T.B.; Turriff, A.E.; Brewer, C.C.; Zalewski, C.K.; King, K.A.; Wafa, T.; Griffith, A.J.; Brooks, B.; et al. Atypical and Ultra-Rare Usher Syndrome: A Review. Ophthalmic Genet. 2020, 41, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Abad-Morales, V.; Navarro, R.; Burés-Jelstrup, A.; Pomares, E. Identification of a Novel Homozygous ARSG Mutation as the Second Cause of Usher Syndrome Type 4. Am. J. Ophthalmol. Case Rep. 2020, 19, 100736. [Google Scholar] [CrossRef]
- Fowler, N.; El-Rashedy, M.; Chishti, E.; Vander Kooi, C.W.; Maldonado, R. Multimodal Imaging and Genetic Findings in a Case of ARSG-Related Atypical Usher Syndrome. Ophthalmic Genet. 2021, 42, 338–343. [Google Scholar] [CrossRef]
- Igelman, A.D.; Ku, C.; da Palma, M.M.; Georgiou, M.; Schiff, E.R.; Lam, B.L.; Sankila, E.-M.; Ahn, J.; Pyers, L.; Vincent, A.; et al. Expanding the Clinical Phenotype in Patients with Disease Causing Variants Associated with Atypical Usher Syndrome. Ophthalmic Genet. 2021, 42, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Ponzin, D.; Ashworth, J.L.; Fahnehjelm, K.T.; Summers, C.G.; Harmatz, P.R.; Scarpa, M. Diagnosis and Management of Ophthalmologic Features in Patients with Mucopolysaccharidosis. Br. J. Ophthalmol. 2010, 95, 613. [Google Scholar] [CrossRef]
- Wolfberg, J.; Chintalapati, K.; Tomatsu, S.; Nagao, K. Hearing Loss in Mucopolysaccharidoses: Current Knowledge and Future Directions. Diagnostics 2020, 10, 554. [Google Scholar] [CrossRef]
- Kowalewski, B.; Lange, H.; Galle, S.; Dierks, T.; Lübke, T.; Damme, M. Decoding the Consecutive Lysosomal Degradation of 3-O-Sulfate Containing Heparan Sulfate by Arylsulfatase G (ARSG). Biochem. J. 2021, 478, 3221–3237. [Google Scholar] [CrossRef]
- Verheyen, S.; Blatterer, J.; Speicher, M.R.; Bhavani, G.S.; Boons, G.-J.; Ilse, M.-B.; Andrae, D.; Sproß, J.; Vaz, F.M.; Kircher, S.G.; et al. Novel Subtype of Mucopolysaccharidosis Caused by Arylsulfatase K (ARSK) Deficiency. J. Med. Genet. 2022, 59, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Rustad, C.F.; Prescott, T.E.; Merckoll, E.; Kristensen, E.; Salvador, C.L.; Nordgarden, H.; Tveten, K. Phenotypic Expansion of ARSK-Related Mucopolysaccharidosis. Am. J. Med. Genet. Part A 2022, 188, 3369–3373. [Google Scholar] [CrossRef]
- Sun, M.; Kaminsky, C.K.; Deppe, P.; Ilse, M.-B.; Vaz, F.M.; Plecko, B.; Lübke, T.; Randolph, L.M. A Novel Homozygous Missense Variant in ARSK Causes MPS X, a New Subtype of Mucopolysaccharidosis. Genes Dis. 2024, 11, 101025. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Maksimova, N.; Otomo, T.; Kato, H.; Imai, A.; Asano, Y.; Kobayashi, K.; Nojima, S.; Nakaya, A.; Hamada, Y.; et al. Mutation in VPS33A Affects Metabolism of Glycosaminoglycans: A New Type of Mucopolysaccharidosis with Severe Systemic Symptoms. Hum. Mol. Genet. 2017, 26, 173–183. [Google Scholar] [CrossRef]
- Vasilev, F.; Sukhomyasova, A.; Otomo, T. Mucopolysaccharidosis-Plus Syndrome. Int. J. Mol. Sci. 2020, 21, 421. [Google Scholar] [CrossRef]
- Faraguna, M.C.; Musto, F.; Crescitelli, V.; Iascone, M.; Spaccini, L.; Tonduti, D.; Fedeli, T.; Kullmann, G.; Canonico, F.; Cattoni, A.; et al. Mucopolysaccharidosis-Plus Syndrome, a Rapidly Progressive Disease: Favorable Impact of a Very Prolonged Steroid Treatment on the Clinical Course in a Child. Genes 2022, 13, 442. [Google Scholar] [CrossRef]
- Caciotti, A.; Donati, M.A.; Procopio, E.; Filocamo, M.; Kleijer, W.; Wuyts, W.; Blaumeiser, B.; d’Azzo, A.; Simi, L.; Orlando, C.; et al. GM1 Gangliosidosis: Molecular Analysis of Nine Patients and Development of an RT-PCR Assay for GLB1 Gene Expression Profiling. Hum. Mutat. 2007, 28, 204. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Jin, D.-K. GLB1-Related Disorders: GM1 Gangliosidosis and Morquio B Disease. J. Genet. Med. 2021, 18, 16–23. [Google Scholar] [CrossRef]
- Kingma, S.D.K.; Ceulemans, B.; Kenis, S.; Jonckheere, A.I. Are GMI Gangliosidosis and Morquio Type B Two Different Disorders or Part of One Phenotypic Spectrum? JIMD Rep. 2021, 59, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Haer-Wigman, L.; Newman, H.; Leibu, R.; Bax, N.M.; Baris, H.N.; Rizel, L.; Banin, E.; Massarweh, A.; Roosing, S.; Lefeber, D.J.; et al. Non-Syndromic Retinitis Pigmentosa Due to Mutations in the Mucopolysaccharidosis Type IIIC Gene, Heparan-Alpha-Glucosaminide N-Acetyltransferase (HGSNAT). Hum. Mol. Genet. 2015, 24, 3742–3751. [Google Scholar] [CrossRef] [PubMed]
- Holanda, I.P.; Rim, P.H.H.; Rare Genomes Project Consortium; Guaragna, M.S.; Gil-da-Silva-Lopes, V.L.; Steiner, C.E. Syndromic Retinitis Pigmentosa: A 15-Patient Study. Genes 2024, 15, 516. [Google Scholar] [CrossRef]
- Tomatsu, S.; Pitz, S.; Hampel, U. Ophthalmological Findings in Mucopolysaccharidoses. J. Clin. Med. 2019, 8, 1467. [Google Scholar] [CrossRef]
- Schiff, E.R.; Daich Varela, M.; Robson, A.G.; Pierpoint, K.; Ba-Abbad, R.; Nutan, S.; Zein, W.M.; Ullah, E.; Huryn, L.A.; Tuupanen, S.; et al. A Genetic and Clinical Study of Individuals with Nonsyndromic Retinopathy Consequent upon Sequence Variants in HGSNAT, the Gene Associated with Sanfilippo C Mucopolysaccharidosis. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Kammenga, J.E. The Background Puzzle: How Identical Mutations in the Same Gene Lead to Different Disease Symptoms. FEBS J. 2017, 284, 3362–3373. [Google Scholar] [CrossRef]
- Kohlschütter, A.; Schulz, A.; Bartsch, U.; Storch, S. Current and Emerging Treatment Strategies for Neuronal Ceroid Lipofuscinoses. CNS Drugs 2019, 33, 315–325. [Google Scholar] [CrossRef]
- Bauwens, M.; Storch, S.; Weisschuh, N.; Ceuterick-de Groote, C.; De Rycke, R.; Guillemyn, B.; De Jaegere, S.; Coppieters, F.; Van Coster, R.; Leroy, B.P.; et al. Functional Characterization of Novel MFSD8 Pathogenic Variants Anticipates Neurological Involvement in Juvenile Isolated Maculopathy. Clin. Genet. 2020, 97, 426–436. [Google Scholar] [CrossRef]
- Khan, K.N.; El-Asrag, M.E.; Ku, C.A.; Holder, G.E.; McKibbin, M.; Arno, G.; Poulter, J.A.; Carss, K.; Bommireddy, T.; Bagheri, S.; et al. Specific Alleles of CLN7/MFSD8, a Protein That Localizes to Photoreceptor Synaptic Terminals, Cause a Spectrum of Nonsyndromic Retinal Dystrophy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2906–2914. [Google Scholar] [CrossRef]
- Ku, C.A.; Hull, S.; Arno, G.; Vincent, A.; Carss, K.; Kayton, R.; Weeks, D.; Anderson, G.W.; Geraets, R.; Parker, C.; et al. Detailed Clinical Phenotype and Molecular Genetic Findings in CLN3-Associated Isolated Retinal Degeneration. JAMA Ophthalmol. 2017, 135, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Magliyah, M.S.; Geuer, S.; Alsalamah, A.K.; Lenzner, S.; Drasdo, M.; Schatz, P. Association of the Recurrent Rare Variant c.415T>C p.Phe139Leu in CLN5 With a Recessively Inherited Macular Dystrophy. JAMA Ophthalmol. 2021, 139, 1–5. [Google Scholar] [CrossRef]
- Lehman, T.J.A.; Miller, N.; Norquist, B.; Underhill, L.; Keutzer, J. Diagnosis of the Mucopolysaccharidoses. Rheumatology 2011, 50, v41–v48. [Google Scholar] [CrossRef] [PubMed]
- Chih-Kuang, C.; Shuan-Pei, L.; Shyue-Jye, L.; Tuen-Jen, W. MPS Screening Methods, the Berry Spot and Acid Turbidity Tests, Cause a High Incidence of False-Negative Results in Sanfilippo and Morquio Syndromes. J. Clin. Lab. Anal. 2002, 16, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Langereis, E.J.; van den Berg, I.E.T.; Halley, D.J.J.; Poorthuis, B.J.H.M.; Vaz, F.M.; Wokke, J.H.J.; Linthorst, G.E. Considering Fabry, but Diagnosing MPS I: Difficulties in the Diagnostic Process. JIMD Rep. 2012, 9, 117–120. [Google Scholar] [CrossRef]
- Klingelhöfer, D.; Braun, M.; Seeger-Zybok, R.K.; Quarcoo, D.; Brüggmann, D.; Groneberg, D.A. Global Research on Fabry’s Disease: Demands for a Rare Disease. Mol. Genet. Genom. Med. 2020, 8, e1163. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| MPS Type | Defective Gene | Deficient Enzyme | Stored GAG b | Neurological Symptoms c |
|---|---|---|---|---|
| MPS I | IDUA | α-L-iduronidase | HS, DS | Impaired cognitive function, language, and speech abilities, behavioral abnormalities (excessive silencing), sleeping problems, and/or epileptic seizures |
| MPS II | IDS | Iduronate-2-sulfatase | HS, DS | Developmental delay, mental retardation, and behavior problems (aggression, over-excitability) |
| MPS IIIA | SGSH | Heparan-N-sulfatase | HS | Developmental delay, cognitive impairment, behavioral disorders (impulsivity, aggression, anxiety disorders, autistic behavior), sleeping problems |
| MPS IIIB | NAGLU | α-N-acetylglucosaminidase | ||
| MPS IIIC | HGSNAT | Heparan α-Glucosaminide N-acetyltransferase | ||
| MPS IIID | GNS | N-Acetylglucosamine-6-sulfatase | ||
| MPS IVA | GLANS | N-Acetylglucosamine- 6-sulfate sulfatase | C6S, KS | Absence or mild neurological disorders as a consequence of secondary disturbances |
| MPS IVB | GLB1 | β-Galactosidase | KS | |
| MPS VI | ARSB | N-acetylglucosamine-4-sulfatase (arylsulfatase B) | DS, C4S | None |
| MPS VII | GUSB | β-Glucuronidase | HS, DS, C4S, C6S | Impaired cognitive, language, and speech abilities, behavioral abnormalities, sleep problems, and/or epileptic seizures |
| MPS IX | HYAL1 | Hyaluronidase | Hyaluronan | None |
| MPS X | ARSK | Arylsulfatase K | DS | None |
| Typical MPS | Mild/Attenuated MPS | ||
|---|---|---|---|
Somatic
| Neurological
| Somatic
| Neurological
|
| Why YES? | Why NOT? |
|---|---|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiśniewska, K.; Wolski, J.; Żabińska, M.; Szulc, A.; Gaffke, L.; Pierzynowska, K.; Węgrzyn, G. Mucopolysaccharidosis Type IIIE: A Real Human Disease or a Diagnostic Pitfall? Diagnostics 2024, 14, 1734. https://doi.org/10.3390/diagnostics14161734
Wiśniewska K, Wolski J, Żabińska M, Szulc A, Gaffke L, Pierzynowska K, Węgrzyn G. Mucopolysaccharidosis Type IIIE: A Real Human Disease or a Diagnostic Pitfall? Diagnostics. 2024; 14(16):1734. https://doi.org/10.3390/diagnostics14161734
Chicago/Turabian StyleWiśniewska, Karolina, Jakub Wolski, Magdalena Żabińska, Aneta Szulc, Lidia Gaffke, Karolina Pierzynowska, and Grzegorz Węgrzyn. 2024. "Mucopolysaccharidosis Type IIIE: A Real Human Disease or a Diagnostic Pitfall?" Diagnostics 14, no. 16: 1734. https://doi.org/10.3390/diagnostics14161734
APA StyleWiśniewska, K., Wolski, J., Żabińska, M., Szulc, A., Gaffke, L., Pierzynowska, K., & Węgrzyn, G. (2024). Mucopolysaccharidosis Type IIIE: A Real Human Disease or a Diagnostic Pitfall? Diagnostics, 14(16), 1734. https://doi.org/10.3390/diagnostics14161734