

Genetics, Pathophysiology, and Current Challenges in Von Hippel–Lindau Disease Therapeutics

 ,
,  , , ,
, , , 
Abstract
:1. Introduction
2. Von Hippel–Lindau (VHL) Disease: Highlights
2.1. History
2.2. Etiology
2.3. Epidemiology
2.4. Pathophysiology
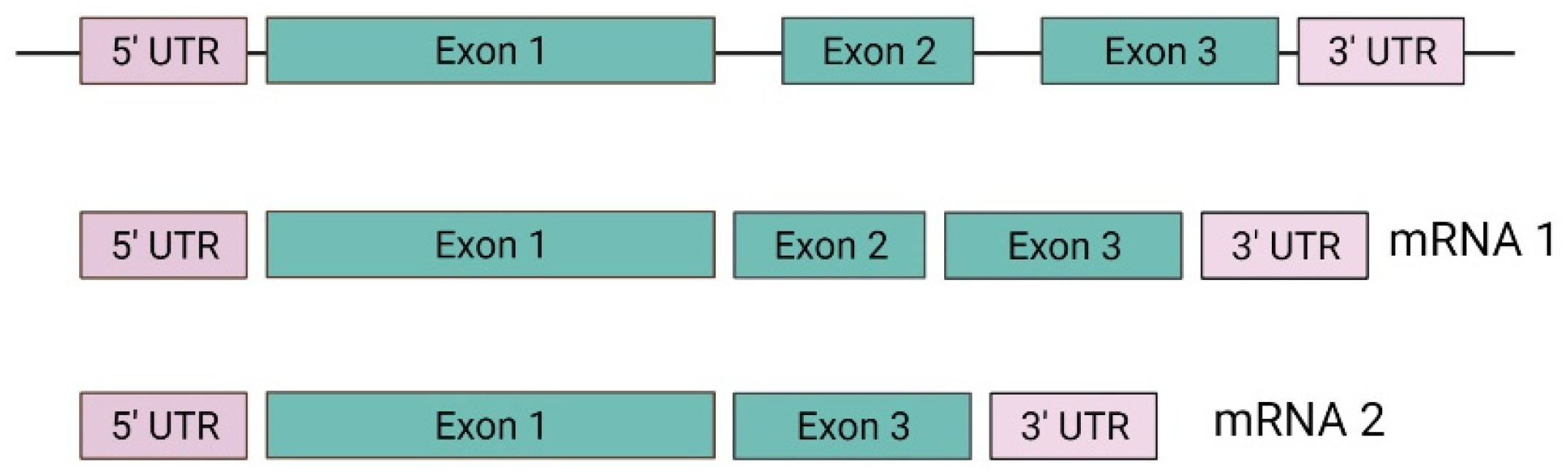
3. Genetics
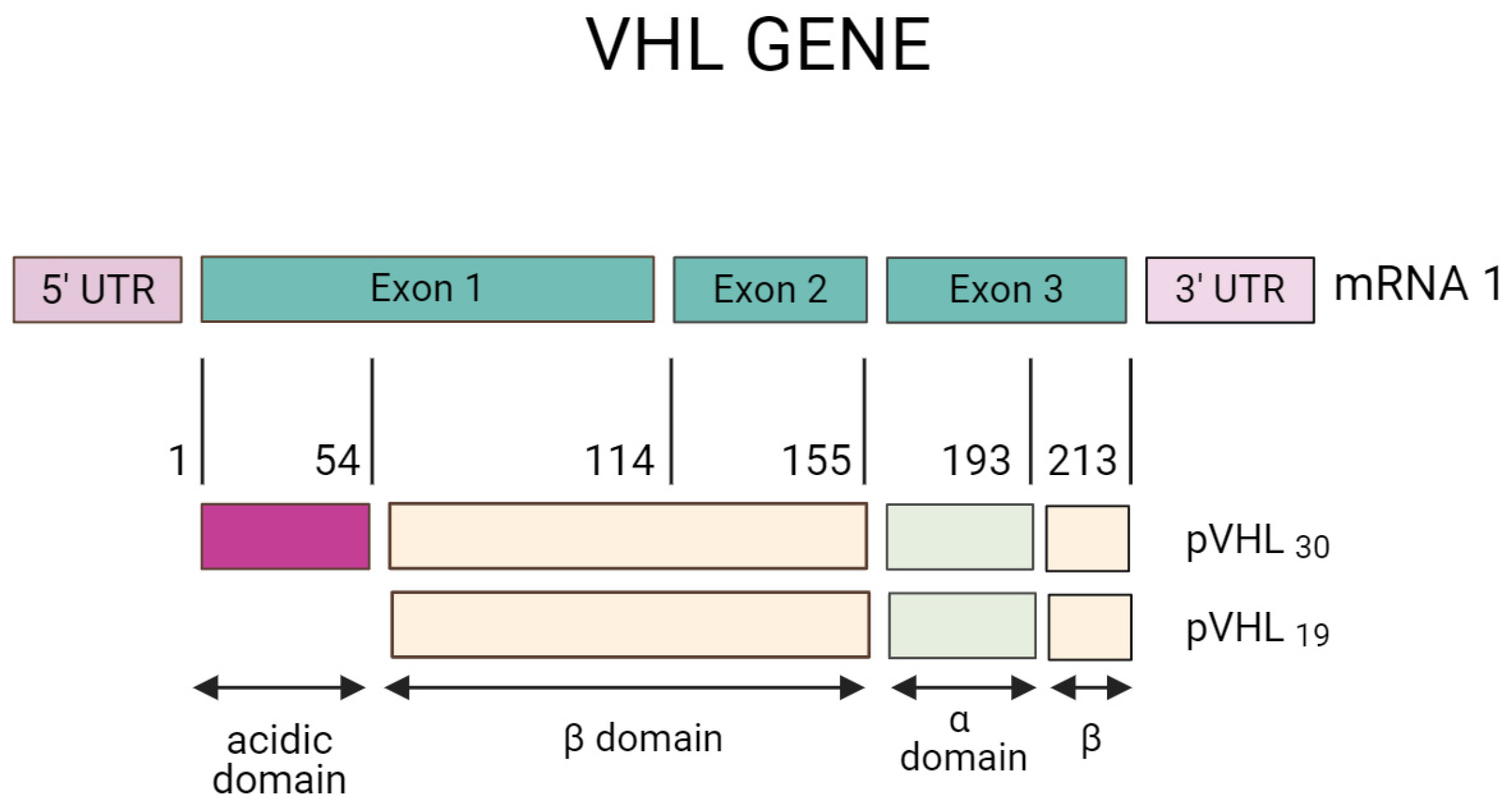
3.1. VHL Gene and Protein
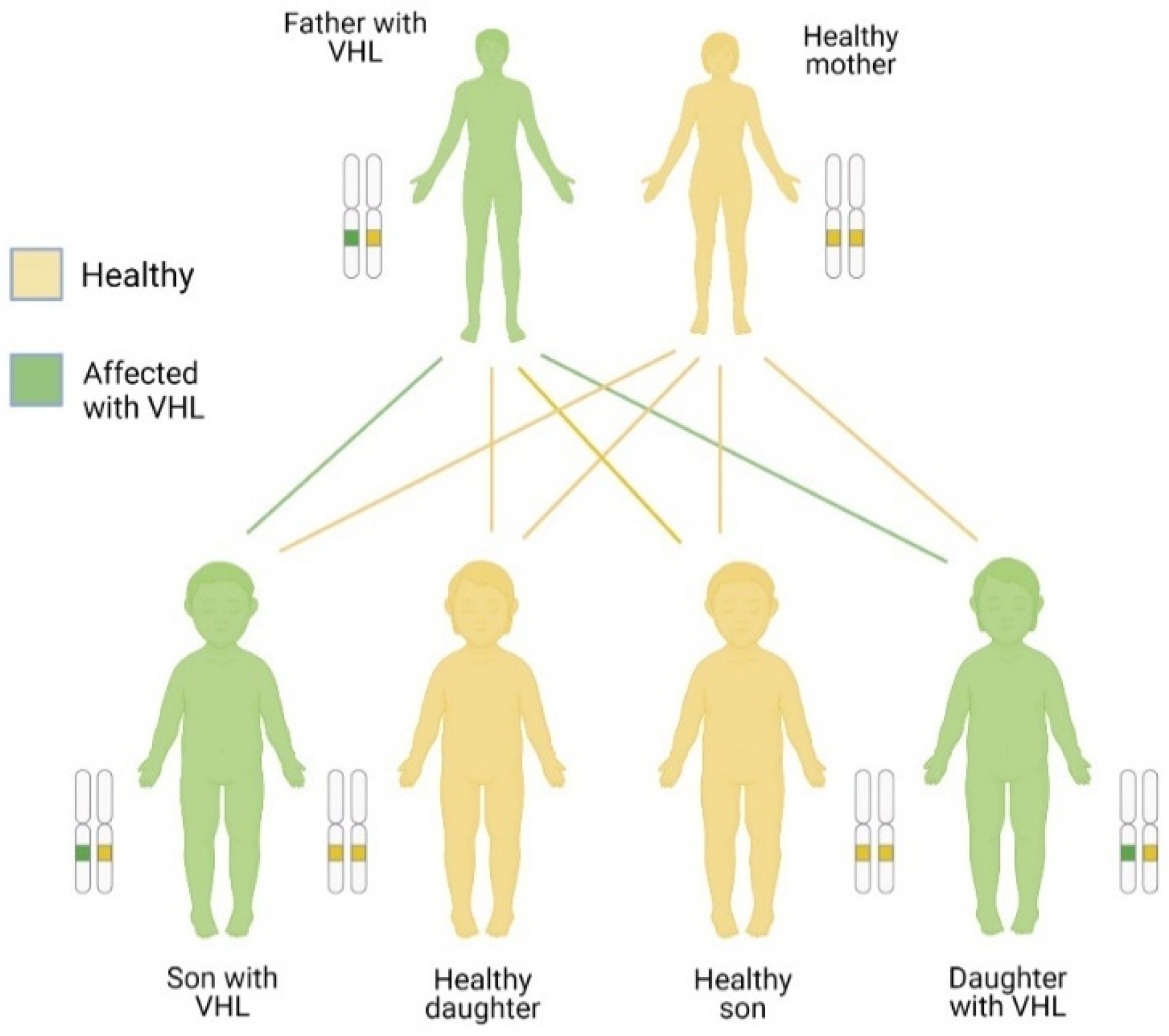
3.2. Inheritance
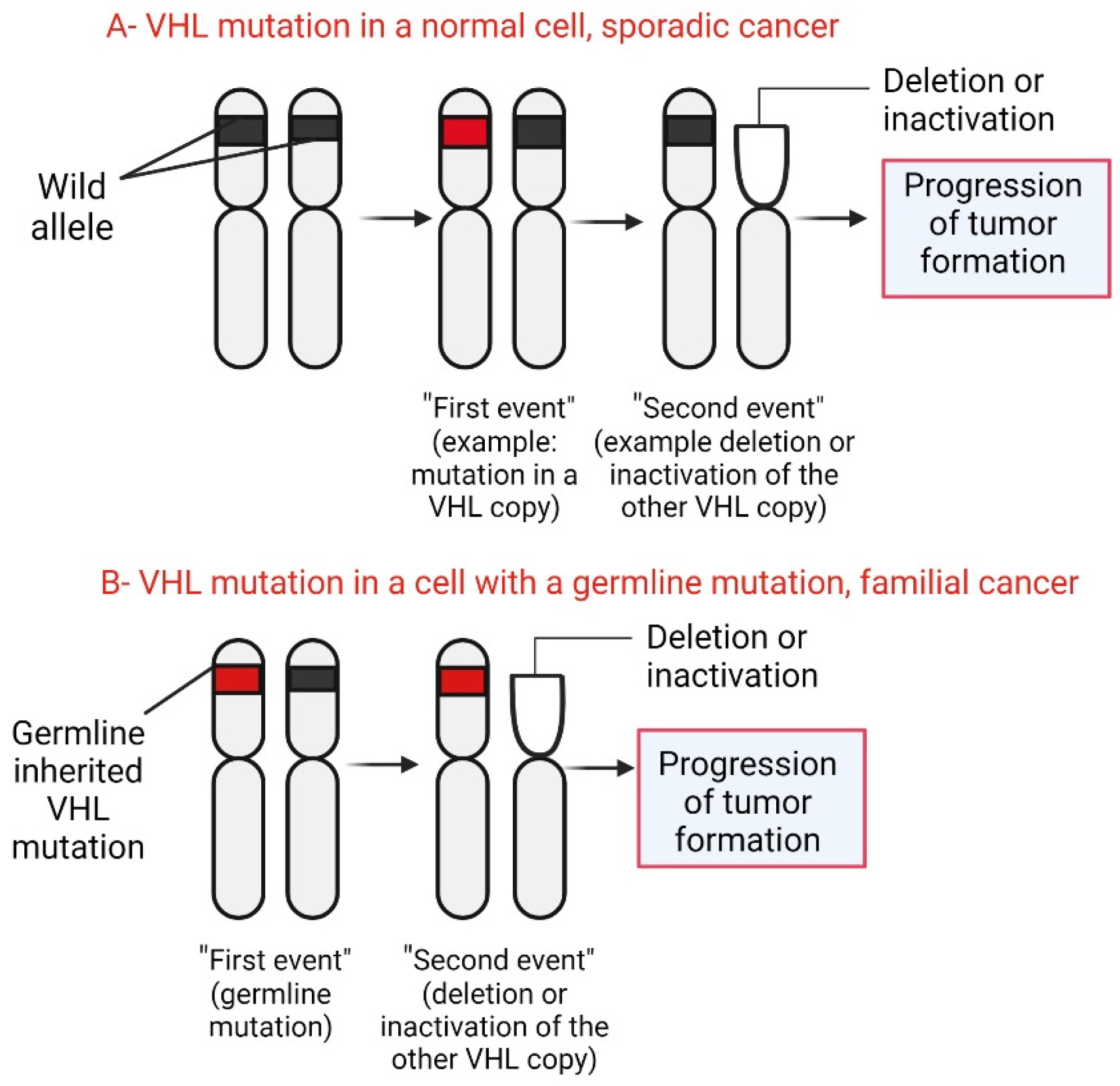
3.3. Mutations
4. Manifestations, Diagnosis, and Treatment of VHL Disease
5. Molecular Basis of VHL Disease
6. Animal Models for the Study of VHL
7. Biomarkers in VHL Disease
8. Effect of Regional Populations on VHL Disease
9. Targeted Therapy in VHL Disease
10. Discussion
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260, 1317–1320. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.; Ma, K.; Zhou, J.; Zhang, J.; Wang, J.; Liu, S.; Zhang, Z.; Cai, L.; Zhang, N.; Gong, K. Frequent Mutations of VHL Gene and the Clinical Phenotypes in the Largest Chinese Cohort with Von Hippel-Lindau Disease. Front. Genet. 2019, 10, 867. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. Molecular basis of the VHL hereditary cancer syndrome. Nat. Rev. Cancer 2002, 2, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Lonser, R.R.; Glenn, G.M.; Walther, M.; Chew, E.Y.; Libutti, S.K.; Linehan, W.M.; Oldfield, E.H. von Hippel-Lindau disease. Lancet 2003, 361, 2059–2067. [Google Scholar] [CrossRef] [PubMed]
- Findeis-Hosey, J.J.; McMahon, K.Q.; Findeis, S.K. Von Hippel-Lindau Disease. J. Pediatr. Genet. 2016, 5, 116–123. [Google Scholar] [CrossRef]
- Castro-Teles, J.; Sousa-Pinto, B.; Rebelo, S.; Pignatelli, D. Pheochromocytomas and paragangliomas in von Hippel-Lindau disease: Not a needle in a haystack. Endocr. Connect. 2021, 10, R293–R304. [Google Scholar] [CrossRef]
- Clark, P.E. The role of VHL in clear-cell renal cell carcinoma and its relation to targeted therapy. Kidney Int. 2009, 76, 939–945. [Google Scholar] [CrossRef]
- Ohh, M.; Taber, C.C.; Ferens, F.G.; Tarade, D. Hypoxia-inducible factor underlies von Hippel-Lindau disease stigmata. eLife 2022, 11, e80774. [Google Scholar] [CrossRef]
- Haase, V.H. The VHL/HIF oxygen-sensing pathway and its relevance to kidney disease. Kidney Int. 2006, 69, 1302–1307. [Google Scholar] [CrossRef]
- Tamura, K.; Kanazashi, Y.; Kawada, C.; Sekine, Y.; Maejima, K.; Ashida, S.; Karashima, T.; Kojima, S.; Parrish, N.F.; Kosugi, S.; et al. Variant spectrum of von Hippel-Lindau disease and its genomic heterogeneity in Japan. Hum. Mol. Genet. 2023, 32, 2046–2054. [Google Scholar] [CrossRef]
- Horton, C.; LaDuca, H.; Deckman, A.; Durda, K.; Jackson, M.; Richardson, M.E.; Tian, Y.; Yussuf, A.; Jasperson, K.; Else, T. Universal Germline Panel Testing for Individuals with Pheochromocytoma and Paraganglioma Produces High Diagnostic Yield. J. Clin. Endocrinol. Metab. 2022, 107, e1917–e1923. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, L.E.; Grzybowski, J.; Grenier, S.; Chao, E.; Downs, G.S.; Farncombe, K.M.; Stockley, T.L.; Mete, O.; Kim, R.H. VHL mosaicism: The added value of multi-tissue analysis. NPJ Genom. Med. 2022, 7, 21. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Varshney, N.; Kebede, A.A.; Owusu-Dapaah, H.; Lather, J.; Kaushik, M.; Bhullar, J.S. A Review of Von Hippel-Lindau Syndrome. J. Kidney Cancer VHL 2017, 4, 20–29. [Google Scholar] [CrossRef]
- Choyke, P.L.; Glenn, G.M.; Walther, M.M.; Patronas, N.J.; Linehan, W.M.; Zbar, B. von Hippel-Lindau disease: Genetic, clinical, and imaging features. Radiology 1995, 194, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Melmon, K.L.; Rosen, S.W. Lindau’s Disease. Review of the Literature and Study of a Large Kindred. Am. J. Med. 1964, 36, 595–617. [Google Scholar] [CrossRef]
- Seizinger, B.R.; Rouleau, G.A.; Ozelius, L.J.; Lane, A.H.; Farmer, G.E.; Lamiell, J.M.; Haines, J.; Yuen, J.W.; Collins, D.; Majoor-Krakauer, D.; et al. Von Hippel-Lindau disease maps to the region of chromosome 3 associated with renal cell carcinoma. Nature 1988, 332, 268–269. [Google Scholar] [CrossRef] [PubMed]
- Gossage, L.; Eisen, T.; Maher, E.R. VHL, the story of a tumour suppressor gene. Nat. Rev. Cancer 2015, 15, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, K.; Makino, Y.; Pereira, T.; Poellinger, L. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000, 19, 4298–4309. [Google Scholar] [CrossRef]
- Richards, F.M.; Schofield, P.N.; Fleming, S.; Maher, E.R. Expression of the von Hippel-Lindau disease tumour suppressor gene during human embryogenesis. Hum. Mol. Genet. 1996, 5, 639–644. [Google Scholar] [CrossRef]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Glasker, S.; Bender, B.U.; Apel, T.W.; van Velthoven, V.; Mulligan, L.M.; Zentner, J.; Neumann, H.P. Reconsideration of biallelic inactivation of the VHL tumour suppressor gene in hemangioblastomas of the central nervous system. J. Neurol. Neurosurg. Psychiatry 2001, 70, 644–648. [Google Scholar] [CrossRef]
- Glasker, S.; Sohn, T.S.; Okamoto, H.; Li, J.; Lonser, R.R.; Oldfield, E.H.; Vortmeyer, A.O.; Zhuang, Z. Second hit deletion size in von Hippel-Lindau disease. Ann. Neurol. 2006, 59, 105–110. [Google Scholar] [CrossRef]
- Maddock, I.R.; Moran, A.; Maher, E.R.; Teare, M.D.; Norman, A.; Payne, S.J.; Whitehouse, R.; Dodd, C.; Lavin, M.; Hartley, N.; et al. A genetic register for von Hippel-Lindau disease. J. Med. Genet. 1996, 33, 120–127. [Google Scholar] [CrossRef]
- Evans, D.G.; Howard, E.; Giblin, C.; Clancy, T.; Spencer, H.; Huson, S.M.; Lalloo, F. Birth incidence and prevalence of tumor-prone syndromes: Estimates from a UK family genetic register service. Am. J. Med. Genet. A 2010, 152A, 327–332. [Google Scholar] [CrossRef]
- Maher, E.R.; Iselius, L.; Yates, J.R.; Littler, M.; Benjamin, C.; Harris, R.; Sampson, J.; Williams, A.; Ferguson-Smith, M.A.; Morton, N. Von Hippel-Lindau disease: A genetic study. J. Med. Genet. 1991, 28, 443–447. [Google Scholar] [CrossRef]
- Neumann, H.P.; Wiestler, O.D. Clustering of features of von Hippel-Lindau syndrome: Evidence for a complex genetic locus. Lancet 1991, 337, 1052–1054. [Google Scholar] [CrossRef] [PubMed]
- Binderup, M.L.; Galanakis, M.; Budtz-Jorgensen, E.; Kosteljanetz, M.; Luise Bisgaard, M. Prevalence, birth incidence, and penetrance of von Hippel-Lindau disease (vHL) in Denmark. Eur. J. Hum. Genet. 2017, 25, 301–307. [Google Scholar] [CrossRef]
- Haase, V.H. The VHL tumor suppressor in development and disease: Functional studies in mice by conditional gene targeting. Semin. Cell Dev. Biol. 2005, 16, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, O.; Levy, A.P.; Jiang, C.; Kaelin, W.G., Jr.; Goldberg, M.A. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc. Natl. Acad. Sci. USA 1996, 93, 10595–10599. [Google Scholar] [CrossRef]
- Krieg, M.; Haas, R.; Brauch, H.; Acker, T.; Flamme, I.; Plate, K.H. Up-regulation of hypoxia-inducible factors HIF-1alpha and HIF-2alpha under normoxic conditions in renal carcinoma cells by von Hippel-Lindau tumor suppressor gene loss of function. Oncogene 2000, 19, 5435–5443. [Google Scholar] [CrossRef]
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 4273–4278. [Google Scholar] [CrossRef]
- Rankin, E.B.; Biju, M.P.; Liu, Q.; Unger, T.L.; Rha, J.; Johnson, R.S.; Simon, M.C.; Keith, B.; Haase, V.H. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J. Clin. Investig. 2007, 117, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Zhao, L.; Gui, Z.; Liu, S.; Liu, C.; Yu, T.; Zhang, L. PI3K/AKT signaling activates HIF1alpha to modulate the biological effects of invasive breast cancer with microcalcification. NPJ Breast Cancer 2023, 9, 93. [Google Scholar] [CrossRef] [PubMed]
- Bae, T.; Hallis, S.P.; Kwak, M.K. Hypoxia, oxidative stress, and the interplay of HIFs and NRF2 signaling in cancer. Exp. Mol. Med. 2024, 56, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Masoud, G.N.; Li, W. HIF-1alpha pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef]
- Chen, C.Y.; Chen, J.; He, L.; Stiles, B.L. PTEN: Tumor Suppressor and Metabolic Regulator. Front. Endocrinol. 2018, 9, 338. [Google Scholar] [CrossRef]
- Singh, B.; Reddy, P.G.; Goberdhan, A.; Walsh, C.; Dao, S.; Ngai, I.; Chou, T.C.; O-Charoenrat, P.; Levine, A.J.; Rao, P.H.; et al. p53 regulates cell survival by inhibiting PIK3CA in squamous cell carcinomas. Genes. Dev. 2002, 16, 984–993. [Google Scholar] [CrossRef]
- Farhan, M.; Wang, H.; Gaur, U.; Little, P.J.; Xu, J.; Zheng, W. FOXO Signaling Pathways as Therapeutic Targets in Cancer. Int. J. Biol. Sci. 2017, 13, 815–827. [Google Scholar] [CrossRef]
- Vortmeyer, A.O.; Falke, E.A.; Glasker, S.; Li, J.; Oldfield, E.H. Nervous system involvement in von Hippel-Lindau disease: Pathology and mechanisms. Acta Neuropathol. 2013, 125, 333–350. [Google Scholar] [CrossRef]
- Albanyan, S.; Giles, R.H.; Gimeno, E.M.; Silver, J.; Murphy, J.; Faghfoury, H.; Morel, C.F.; Machado, J.; Kim, R.H. Characterization of VHL promoter variants in patients suspected of Von Hippel-Lindau disease. Eur. J. Med. Genet. 2019, 62, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.; Le, V.H.; Oyama, T.; Ricketts, C.J.; Ho, T.H.; Cheng, E.H. Chromosome 3p Loss-Orchestrated VHL, HIF, and Epigenetic Deregulation in Clear Cell Renal Cell Carcinoma. J. Clin. Oncol. 2018, 36, 3533–3539. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, J.; Li, L.; Yi, Z.; Lu, R.; Li, C.; Gong, K. Intronic mutation of the VHL gene associated with central nervous system hemangioblastomas in two Chinese families with Von Hippel-Lindau disease: Case report. BMC Med. Genet. 2020, 21, 191. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Zhang, J.; Tan, X.; Huang, Y.; Xu, J.; Silk, N.; Zhang, D.; Liu, Q.; Jiang, J. The VHL/HIF Axis in the Development and Treatment of Pheochromocytoma/Paraganglioma. Front. Endocrinol. 2020, 11, 586857. [Google Scholar] [CrossRef] [PubMed]
- Tarade, D.; He, S.; St-Germain, J.; Petroff, A.; Murphy, A.; Raught, B.; Ohh, M. The long form of pVHL is artifactually modified by serine protease inhibitor AEBSF. Protein Sci. 2020, 29, 1843–1850. [Google Scholar] [CrossRef]
- Buffet, A.; Calsina, B.; Flores, S. Germline mutations in the new E1’ cryptic exon of the VHL gene in patients with tumours of von Hippel-Lindau disease spectrum or with paraganglioma. J. Med. Genet. 2020, 57, 752–759. [Google Scholar] [CrossRef]
- Xie, H.; Ma, K.; Zhang, J.; Hong, B.; Zhou, J.; Li, L.; Zhang, K.; Gong, K.; Cai, L. Novel genetic characterisation and phenotype correlation in von Hippel-Lindau (VHL) disease based on the Elongin C binding site: A large retrospective study. J. Med. Genet. 2020, 57, 744–751. [Google Scholar] [CrossRef]
- Reich, M.; Jaegle, S.; Neumann-Haefelin, E.; Klingler, J.H.; Evers, C.; Daniel, M.; Bucher, F.; Ludwig, F.; Nuessle, S.; Kopp, J.; et al. Genotype-phenotype correlation in von Hippel-Lindau disease. Acta Ophthalmol. 2021, 99, e1492–e1500. [Google Scholar] [CrossRef]
- van der Horst-Schrivers, A.N.A.; Sluiter, W.J.; Kruizinga, R.C.; van Leeuwaarde, R.S.; Giles, R.; Olderode-Berends, M.J.W.; Links, T.P. The incidence of consecutive manifestations in Von Hippel-Lindau disease. Fam. Cancer 2019, 18, 369–376. [Google Scholar] [CrossRef]
- Vocke, C.D.; Ricketts, C.J.; Schmidt, L.S.; Ball, M.W.; Middelton, L.A.; Zbar, B.; Linehan, W.M. Comprehensive characterization of Alu-mediated breakpoints in germline VHL gene deletions and rearrangements in patients from 71 VHL families. Hum. Mutat. 2021, 42, 520–529. [Google Scholar] [CrossRef]
- Cybulski, C.; Krzystolik, K.; Murgia, A.; Gorski, B.; Debniak, T.; Jakubowska, A.; Martella, M.; Kurzawski, G.; Prost, M.; Kojder, I.; et al. Germline mutations in the von Hippel-Lindau (VHL) gene in patients from Poland: Disease presentation in patients with deletions of the entire VHL gene. J. Med. Genet. 2002, 39, E38. [Google Scholar] [CrossRef] [PubMed]
- Tabaro, F.; Minervini, G.; Sundus, F.; Quaglia, F.; Leonardi, E.; Piovesan, D.; Tosatto, S.C. VHLdb: A database of von Hippel-Lindau protein interactors and mutations. Sci. Rep. 2016, 6, 31128. [Google Scholar] [CrossRef]
- Ong, K.R.; Woodward, E.R.; Killick, P.; Lim, C.; Macdonald, F.; Maher, E.R. Genotype-phenotype correlations in von Hippel-Lindau disease. Hum. Mutat. 2007, 28, 143–149. [Google Scholar] [CrossRef]
- Zbar, B.; Kishida, T.; Chen, F.; Schmidt, L.; Maher, E.R.; Richards, F.M.; Crossey, P.A.; Webster, A.R.; Affara, N.A.; Ferguson-Smith, M.A.; et al. Germline mutations in the Von Hippel-Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum. Mutat. 1996, 8, 348–357. [Google Scholar] [CrossRef]
- Chen, F.; Kishida, T.; Yao, M.; Hustad, T.; Glavac, D.; Dean, M.; Gnarra, J.R.; Orcutt, M.L.; Duh, F.M.; Glenn, G.; et al. Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: Correlations with phenotype. Hum. Mutat. 1995, 5, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Kanno, H.; Shuin, T.; Kondo, K.; Ito, S.; Hosaka, M.; Torigoe, S.; Fujii, S.; Tanaka, Y.; Yamamoto, I.; Kim, I.; et al. Molecular genetic diagnosis of von Hippel-Lindau disease: Analysis of five Japanese families. Jpn. J. Cancer Res. 1996, 87, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.S.; Antonello, D.; Martignoni, G.; Racchini, C.; Ventrucci, M.; Scarpa, A. Identification of a novel mutation (c279delC) and a polymorphism (c291C>G) in the von Hippel-Lindau gene in Italian patients. Hum. Mutat. 2000, 15, 582. [Google Scholar] [CrossRef]
- Crossey, P.A.; Richards, F.M.; Foster, K.; Green, J.S.; Prowse, A.; Latif, F.; Lerman, M.I.; Zbar, B.; Affara, N.A.; Ferguson-Smith, M.A.; et al. Identification of intragenic mutations in the von Hippel-Lindau disease tumour suppressor gene and correlation with disease phenotype. Hum. Mol. Genet. 1994, 3, 1303–1308. [Google Scholar] [CrossRef]
- Stolle, C.; Glenn, G.; Zbar, B.; Humphrey, J.S.; Choyke, P.; Walther, M.; Pack, S.; Hurley, K.; Andrey, C.; Klausner, R.; et al. Improved detection of germline mutations in the von Hippel-Lindau disease tumor suppressor gene. Hum. Mutat. 1998, 12, 417–423. [Google Scholar] [CrossRef]
- Rechsteiner, M.P.; von Teichman, A.; Nowicka, A.; Sulser, T.; Schraml, P.; Moch, H. VHL gene mutations and their effects on hypoxia inducible factor HIFalpha: Identification of potential driver and passenger mutations. Cancer Res. 2011, 71, 5500–5511. [Google Scholar] [CrossRef]
- Ruiz-Llorente, S.; Bravo, J.; Cebrian, A.; Cascon, A.; Pollan, M.; Telleria, D.; Leton, R.; Urioste, M.; Rodriguez-Lopez, R.; de Campos, J.M.; et al. Genetic characterization and structural analysis of VHL Spanish families to define genotype-phenotype correlations. Hum. Mutat. 2004, 23, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.R.; Maher, E.R.; Moore, A.T. Clinical characteristics of ocular angiomatosis in von Hippel-Lindau disease and correlation with germline mutation. Arch. Ophthalmol. 1999, 117, 371–378. [Google Scholar] [CrossRef] [PubMed]
- van Houwelingen, K.P.; van Dijk, B.A.; Hulsbergen-van de Kaa, C.A.; Schouten, L.J.; Gorissen, H.J.; Schalken, J.A.; van den Brandt, P.A.; Oosterwijk, E. Prevalence of von Hippel-Lindau gene mutations in sporadic renal cell carcinoma: Results from The Netherlands cohort study. BMC Cancer 2005, 5, 57. [Google Scholar] [CrossRef]
- Rocha, J.C.; Silva, R.L.; Mendonca, B.B.; Marui, S.; Simpson, A.J.; Camargo, A.A. High frequency of novel germline mutations in the VHL gene in the heterogeneous population of Brazil. J. Med. Genet. 2003, 40, e31. [Google Scholar] [CrossRef] [PubMed]
- Nordstrom-O’Brien, M.; van der Luijt, R.B.; van Rooijen, E.; van den Ouweland, A.M.; Majoor-Krakauer, D.F.; Lolkema, M.P.; van Brussel, A.; Voest, E.E.; Giles, R.H. Genetic analysis of von Hippel-Lindau disease. Hum. Mutat. 2010, 31, 521–537. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Ki, C.S.; Kim, J.W. Improved detection of germline mutations in Korean VHL patients by multiple ligation-dependent probe amplification analysis. J. Korean Med. Sci. 2009, 24, 77–83. [Google Scholar] [CrossRef]
- Wu, P.; Zhang, N.; Wang, X.; Ning, X.; Li, T.; Bu, D.; Gong, K. Family history of von Hippel-Lindau disease was uncommon in Chinese patients: Suggesting the higher frequency of de novo mutations in VHL gene in these patients. J. Hum. Genet. 2012, 57, 238–243. [Google Scholar] [CrossRef]
- Kang, H.C.; Kim, I.J.; Park, J.H.; Shin, Y.; Jang, S.G.; Ahn, S.A.; Park, H.W.; Lim, S.K.; Oh, S.K.; Kim, D.J.; et al. Three novel VHL germline mutations in Korean patients with von Hippel-Lindau disease and pheochromocytomas. Oncol. Rep. 2005, 14, 879–883. [Google Scholar] [CrossRef]
- Clinical Research Group for VHL in Japan. Germline mutations in the von Hippel-Lindau disease (VHL) gene in Japanese VHL. Hum. Mol. Genet. 1995, 4, 2233–2237. [Google Scholar] [CrossRef]
- Neumann, H.P.; Bausch, B.; McWhinney, S.R.; Bender, B.U.; Gimm, O.; Franke, G.; Schipper, J.; Klisch, J.; Altehoefer, C.; Zerres, K.; et al. Germ-line mutations in nonsyndromic pheochromocytoma. N. Engl. J. Med. 2002, 346, 1459–1466. [Google Scholar] [CrossRef]
- Yoshida, M.; Ashida, S.; Kondo, K.; Kobayashi, K.; Kanno, H.; Shinohara, N.; Shitara, N.; Kishida, T.; Kawakami, S.; Baba, M.; et al. Germ-line mutation analysis in patients with von Hippel-Lindau disease in Japan: An extended study of 77 families. Jpn. J. Cancer Res. 2000, 91, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Gnarra, J.R.; Tory, K.; Weng, Y.; Schmidt, L.; Wei, M.H.; Li, H.; Latif, F.; Liu, S.; Chen, F.; Duh, F.M.; et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat. Genet. 1994, 7, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Olschwang, S.; Richard, S.; Boisson, C.; Giraud, S.; Laurent-Puig, P.; Resche, F.; Thomas, G. Germline mutation profile of the VHL gene in von Hippel-Lindau disease and in sporadic hemangioblastoma. Hum. Mutat. 1998, 12, 424–430. [Google Scholar] [CrossRef]
- Glavac, D.; Neumann, H.P.; Wittke, C.; Jaenig, H.; Masek, O.; Streicher, T.; Pausch, F.; Engelhardt, D.; Plate, K.H.; Hofler, H.; et al. Mutations in the VHL tumor suppressor gene and associated lesions in families with von Hippel-Lindau disease from central Europe. Hum. Genet. 1996, 98, 271–280. [Google Scholar] [CrossRef]
- Maher, E.R.; Webster, A.R.; Richards, F.M.; Green, J.S.; Crossey, P.A.; Payne, S.J.; Moore, A.T. Phenotypic expression in von Hippel-Lindau disease: Correlations with germline VHL gene mutations. J. Med. Genet. 1996, 33, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Vortmeyer, A.O.; Lubensky, I.A.; Fogt, F.; Linehan, W.M.; Khettry, U.; Zhuang, Z. Allelic deletion and mutation of the von Hippel-Lindau (VHL) tumor suppressor gene in pancreatic microcystic adenomas. Am. J. Pathol. 1997, 151, 951–956. [Google Scholar]
- Bauters, C.; Vantyghem, M.C.; Leteurtre, E.; Odou, M.F.; Mouton, C.; Porchet, N.; Wemeau, J.L.; Proye, C.; Pigny, P. Hereditary phaeochromocytomas and paragangliomas: A study of five susceptibility genes. J. Med. Genet. 2003, 40, e75. [Google Scholar] [CrossRef]
- Ritter, M.M.; Frilling, A.; Crossey, P.A.; Hoppner, W.; Maher, E.R.; Mulligan, L.; Ponder, B.A.; Engelhardt, D. Isolated familial pheochromocytoma as a variant of von Hippel-Lindau disease. J. Clin. Endocrinol. Metab. 1996, 81, 1035–1037. [Google Scholar] [CrossRef]
- Hes, F.J.; van der Luijt, R.B.; Janssen, A.L.; Zewald, R.A.; de Jong, G.J.; Lenders, J.W.; Links, T.P.; Luyten, G.P.; Sijmons, R.H.; Eussen, H.J.; et al. Frequency of Von Hippel-Lindau germline mutations in classic and non-classic Von Hippel-Lindau disease identified by DNA sequencing, Southern blot analysis and multiplex ligation-dependent probe amplification. Clin. Genet. 2007, 72, 122–129. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, N.; Ning, X.; Li, T.; Wu, P.; Peng, S.; Fan, Y.; Bu, D.; Gong, K. Higher prevalence of novel mutations in VHL gene in Chinese Von Hippel-Lindau disease patients. Urology 2014, 83, 675.e1–675.e6. [Google Scholar] [CrossRef]
- Garcia, A.; Matias-Guiu, X.; Cabezas, R.; Chico, A.; Prat, J.; Baiget, M.; De Leiva, A. Molecular diagnosis of von Hippel-Lindau disease in a kindred with a predominance of familial phaeochromocytoma. Clin. Endocrinol. 1997, 46, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Lee, J.H.; Lee, K.E.; Kim, J.H.; Hong, J.M.; Ra, E.K.; Seo, S.H.; Lee, S.J.; Kim, M.J.; Park, S.S.; et al. Genotype-phenotype analysis of von Hippel-Lindau syndrome in Korean families: HIF-alpha binding site missense mutations elevate age-specific risk for CNS hemangioblastoma. BMC Med. Genet. 2016, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Crossey, P.A.; Eng, C.; Ginalska-Malinowska, M.; Lennard, T.W.; Wheeler, D.C.; Ponder, B.A.; Maher, E.R. Molecular genetic diagnosis of von Hippel-Lindau disease in familial phaeochromocytoma. J. Med. Genet. 1995, 32, 885–886. [Google Scholar] [CrossRef]
- Gross, D.J.; Avishai, N.; Meiner, V.; Filon, D.; Zbar, B.; Abeliovich, D. Familial pheochromocytoma associated with a novel mutation in the von Hippel-Lindau gene. J. Clin. Endocrinol. Metab. 1996, 81, 147–149. [Google Scholar] [CrossRef]
- Bar, M.; Friedman, E.; Jakobovitz, O.; Leibowitz, G.; Lerer, I.; Abeliovich, D.; Gross, D.J. Sporadic phaeochromocytomas are rarely associated with germline mutations in the von Hippel-Lindau and RET genes. Clin. Endocrinol. 1997, 47, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Tisherman, S.E.; Gregg, F.J.; Danowski, T.S. Familial pheochromocytoma. JAMA 1962, 182, 152–156. [Google Scholar] [CrossRef]
- Murgia, A.; Martella, M.; Vinanzi, C.; Polli, R.; Perilongo, G.; Opocher, G. Somatic mosaicism in von Hippel-Lindau Disease. Hum. Mutat. 2000, 15, 114. [Google Scholar] [CrossRef]
- Mete, T.; Berker, D.; Yilmaz, E.; Ozgen, G.; Yalcin, Y.; Tuna, M.; Ciliz, D.; Onen, M.; Aydin, Y.; Guler, S. Clinical presentation of Von Hippel Lindau syndrome type 2B associated with VHL p.A149S mutation in a large Turkish family. Endocrine 2014, 45, 128–135. [Google Scholar] [CrossRef]
- Atuk, N.O.; Stolle, C.; Owen, J.A., Jr.; Carpenter, J.T.; Vance, M.L. Pheochromocytoma in von Hippel-Lindau disease: Clinical presentation and mutation analysis in a large, multigenerational kindred. J. Clin. Endocrinol. Metab. 1998, 83, 117–120. [Google Scholar] [CrossRef]
- Brookes, C.; Prosser, D.O.; Love, J.M.; Gardner, R.J.; Love, D.R. Diagnostic genetics at a distance: Von hippel-lindau disease and a novel mutation. Genet. Res. Int. 2013, 2013, 189196. [Google Scholar] [CrossRef]
- Ganeshan, D.; Menias, C.O.; Pickhardt, P.J.; Sandrasegaran, K.; Lubner, M.G.; Ramalingam, P.; Bhalla, S. Tumors in von Hippel-Lindau Syndrome: From Head to Toe-Comprehensive State-of-the-Art Review. Radiographics 2018, 38, 849–866. [Google Scholar] [CrossRef] [PubMed]
- Chittiboina, P.; Lonser, R.R. Von Hippel-Lindau disease. Handb. Clin. Neurol. 2015, 132, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Zhang, K.; Ma, K.; Zhou, J.; Gong, Y.; Cai, L.; Gong, K. The Genotype-Phenotype Association of Von Hipple Lindau Disease Based on Mutation Locations: A Retrospective Study of 577 Cases in a Chinese Population. Front. Genet. 2020, 11, 532588. [Google Scholar] [CrossRef]
- Stebbins, C.E.; Kaelin, W.G., Jr.; Pavletich, N.P. Structure of the VHL-ElonginC-ElonginB complex: Implications for VHL tumor suppressor function. Science 1999, 284, 455–461. [Google Scholar] [CrossRef]
- Domene, C.; Illingworth, C.J. Effects of point mutations in pVHL on the binding of HIF-1alpha. Proteins 2012, 80, 733–746. [Google Scholar] [CrossRef]
- Razafinjatovo, C.; Bihr, S.; Mischo, A.; Vogl, U.; Schmidinger, M.; Moch, H.; Schraml, P. Characterization of VHL missense mutations in sporadic clear cell renal cell carcinoma: Hotspots, affected binding domains, functional impact on pVHL and therapeutic relevance. BMC Cancer 2016, 16, 638. [Google Scholar] [CrossRef] [PubMed]
- Pastore, Y.D.; Jelinek, J.; Ang, S.; Guan, Y.; Liu, E.; Jedlickova, K.; Krishnamurti, L.; Prchal, J.T. Mutations in the VHL gene in sporadic apparently congenital polycythemia. Blood 2003, 101, 1591–1595. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.I.; Wang, H.; Foy, R.L.; Ross, J.J.; Cohen, H.T. Tumor suppressor von Hippel-Lindau (VHL) stabilization of Jade-1 protein occurs through plant homeodomains and is VHL mutation dependent. Cancer Res. 2004, 64, 1278–1286. [Google Scholar] [CrossRef]
- Cervio, A.; Villalonga, J.F.; Mormandi, R.; Alcorta, S.C.; Sevlever, G.; Salvat, J. Surgical treatment of cerebellar hemangioblastomas. Surg. Neurol. Int. 2017, 8, 163. [Google Scholar] [CrossRef]
- Klingler, J.H.; Glasker, S.; Bausch, B.; Urbach, H.; Krauss, T.; Jilg, C.A.; Steiert, C.; Puzik, A.; Neumann-Haefelin, E.; Kotsis, F.; et al. Hemangioblastoma and von Hippel-Lindau disease: Genetic background, spectrum of disease, and neurosurgical treatment. Childs Nerv. Syst. 2020, 36, 2537–2552. [Google Scholar] [CrossRef]
- Glasker, S.; Vergauwen, E.; Koch, C.A.; Kutikov, A.; Vortmeyer, A.O. Von Hippel-Lindau Disease: Current Challenges and Future Prospects. Onco Targets Ther. 2020, 13, 5669–5690. [Google Scholar] [CrossRef] [PubMed]
- Karimi, S.; Arabi, A.; Shahraki, T.; Safi, S. Von Hippel-Lindau Disease and the Eye. J. Ophthalmic Vis. Res. 2020, 15, 78–94. [Google Scholar] [CrossRef]
- Singh, A.D.; Shields, C.L.; Shields, J.A. von Hippel-Lindau disease. Surv. Ophthalmol. 2001, 46, 117–142. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Zhao, Y.; Zhang, Z.; Zhang, H. Clinical diagnosis, treatment and screening of the VHL gene in three von Hippel-Lindau disease pedigrees. Exp. Ther. Med. 2020, 20, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Jabi, F. Subclinical Pheochromocytoma and Paraganglioma in an Elderly Patient: A Case Report. JSM Clin. Rep. 2014, 2, 1036. [Google Scholar]
- van Asselt, S.J.; de Vries, E.G.; van Dullemen, H.M.; Brouwers, A.H.; Walenkamp, A.M.; Giles, R.H.; Links, T.P. Pancreatic cyst development: Insights from von Hippel-Lindau disease. Cilia 2013, 2, 3. [Google Scholar] [CrossRef]
- Blansfield, J.A.; Choyke, L.; Morita, S.Y.; Choyke, P.L.; Pingpank, J.F.; Alexander, H.R.; Seidel, G.; Shutack, Y.; Yuldasheva, N.; Eugeni, M.; et al. Clinical, genetic and radiographic analysis of 108 patients with von Hippel-Lindau disease (VHL) manifested by pancreatic neuroendocrine neoplasms (PNETs). Surgery 2007, 142, 814–818; discussion 818.e1–2. [Google Scholar] [CrossRef]
- Maher, E.R.; Neumann, H.P.; Richard, S. von Hippel-Lindau disease: A clinical and scientific review. Eur. J. Hum. Genet. 2011, 19, 617–623. [Google Scholar] [CrossRef]
- Zanoletti, E.; Girasoli, L.; Borsetto, D.; Opocher, G.; Mazzoni, A.; Martini, A. Endolymphatic sac tumour in von Hippel-Lindau disease: Management strategies. Acta Otorhinolaryngol. Ital. 2017, 37, 423–429. [Google Scholar] [CrossRef]
- Butman, J.A.; Kim, H.J.; Baggenstos, M.; Ammerman, J.M.; Dambrosia, J.; Patsalides, A.; Patronas, N.J.; Oldfield, E.H.; Lonser, R.R. Mechanisms of morbid hearing loss associated with tumors of the endolymphatic sac in von Hippel-Lindau disease. JAMA 2007, 298, 41–48. [Google Scholar] [CrossRef]
- Gomella, P.T.; Shin, P.; Srinivasan, R.; Linehan, W.M.; Ball, M.W. Obstructive azoospermia secondary to bilateral epididymal cystadenomas in a patient with von Hippel-Lindau. Urol. Case Rep. 2019, 27, 100922. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, G.Z.; Millo, C.; Sadowski, S.M.; Bagci, U.; Patronas, N.J. Epididymal Cystadenomas in von Hippel-Lindau Disease Showing Increased Activity on 68Ga DOTATATE PET/CT. Clin. Nucl. Med. 2016, 41, 781–782. [Google Scholar] [CrossRef]
- Oliveira, J.D.; Cunha, T.M.; Tereso, A. Tumors of the broad ligament: What and when to suspect such rare location. Radiol. Bras. 2020, 53, 349–355. [Google Scholar] [CrossRef]
- Nogales, F.F.; Goyenaga, P.; Preda, O.; Nicolae, A.; Vieites, B.; Ruiz-Marcellan, M.C.; Pedrosa, A.; Merino, M.J. An analysis of five clear cell papillary cystadenomas of mesosalpinx and broad ligament: Four associated with von Hippel-Lindau disease and one aggressive sporadic type. Histopathology 2012, 60, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Hon, W.C.; Wilson, M.I.; Harlos, K.; Claridge, T.D.; Schofield, C.J.; Pugh, C.W.; Maxwell, P.H.; Ratcliffe, P.J.; Stuart, D.I.; Jones, E.Y. Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature 2002, 417, 975–978. [Google Scholar] [CrossRef] [PubMed]
- McDonough, M.A.; Li, V.; Flashman, E.; Chowdhury, R.; Mohr, C.; Lienard, B.M.; Zondlo, J.; Oldham, N.J.; Clifton, I.J.; Lewis, J.; et al. Cellular oxygen sensing: Crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2). Proc. Natl. Acad. Sci. USA 2006, 103, 9814–9819. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Kim, W.Y.; Lechpammer, M.; Kaelin, W.G., Jr. Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol. 2003, 1, E83. [Google Scholar] [CrossRef]
- Schodel, J.; Ratcliffe, P.J. Mechanisms of hypoxia signalling: New implications for nephrology. Nat. Rev. Nephrol. 2019, 15, 641–659. [Google Scholar] [CrossRef]
- Robb, V.A.; Karbowniczek, M.; Klein-Szanto, A.J.; Henske, E.P. Activation of the mTOR signaling pathway in renal clear cell carcinoma. J. Urol. 2007, 177, 346–352. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef]
- Gingras, A.C.; Raught, B.; Sonenberg, N. Regulation of translation initiation by FRAP/mTOR. Genes. Dev. 2001, 15, 807–826. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes. Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef]
- Kucejova, B.; Pena-Llopis, S.; Yamasaki, T.; Sivanand, S.; Tran, T.A.; Alexander, S.; Wolff, N.C.; Lotan, Y.; Xie, X.J.; Kabbani, W.; et al. Interplay between pVHL and mTORC1 pathways in clear-cell renal cell carcinoma. Mol. Cancer Res. 2011, 9, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Doan, H.; Parsons, A.; Devkumar, S.; Selvarajah, J.; Miralles, F.; Carroll, V.A. HIF-mediated Suppression of DEPTOR Confers Resistance to mTOR Kinase Inhibition in Renal Cancer. iScience 2019, 21, 509–520. [Google Scholar] [CrossRef]
- Ganner, A.; Gehrke, C.; Klein, M.; Thegtmeier, L.; Matulenski, T.; Wingendorf, L.; Wang, L.; Pilz, F.; Greidl, L.; Meid, L.; et al. VHL suppresses RAPTOR and inhibits mTORC1 signaling in clear cell renal cell carcinoma. Sci. Rep. 2021, 11, 14827. [Google Scholar] [CrossRef]
- Rini, B.I.; Flaherty, K. Clinical effect and future considerations for molecularly-targeted therapy in renal cell carcinoma. Urol. Oncol. 2008, 26, 543–549. [Google Scholar] [CrossRef]
- Rini, B.I.; Atkins, M.B. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol. 2009, 10, 992–1000. [Google Scholar] [CrossRef]
- Bellmunt, J.; Pons, F.; Foreshew, A.; Fay, A.P.; Powles, T.; Porta, C.; Bracarda, S.; Lampron, M.E.; Cerbone, L.; Sternberg, C.N.; et al. Sequential targeted therapy after pazopanib therapy in patients with metastatic renal cell cancer: Efficacy and toxicity. Clin. Genitourin. Cancer 2014, 12, 262–269. [Google Scholar] [CrossRef]
- Yao, X.; Tan, J.; Lim, K.J.; Koh, J.; Ooi, W.F.; Li, Z.; Huang, D.; Xing, M.; Chan, Y.S.; Qu, J.Z.; et al. VHL Deficiency Drives Enhancer Activation of Oncogenes in Clear Cell Renal Cell Carcinoma. Cancer Discov. 2017, 7, 1284–1305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wu, T.; Simon, J.; Takada, M.; Saito, R.; Fan, C.; Liu, X.D.; Jonasch, E.; Xie, L.; Chen, X.; et al. VHL substrate transcription factor ZHX2 as an oncogenic driver in clear cell renal cell carcinoma. Science 2018, 361, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Ding, R.; Yan, H.; Zhang, J.; Lin, Z. ZHX2 drives cell growth and migration via activating MEK/ERK signal and induces Sunitinib resistance by regulating the autophagy in clear cell Renal Cell Carcinoma. Cell Death Dis. 2020, 11, 337. [Google Scholar] [CrossRef]
- Liu, X.; Simon, J.M.; Xie, H.; Hu, L.; Wang, J.; Zurlo, G.; Fan, C.; Ptacek, T.S.; Herring, L.; Tan, X.; et al. Genome-wide Screening Identifies SFMBT1 as an Oncogenic Driver in Cancer with VHL Loss. Mol. Cell 2020, 77, 1294–1306.e5. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhu, L.; Xue, S.; Shi, J.; He, C.; Zhang, Q. Novel VHL substrate targets SFMBT1 and ZHX2 may be important prognostic predictors in patients with ccRCC. Oncol. Lett. 2021, 21, 379. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, L.; Elguero, B.; Pacin, D.G.; Senin, S.; Pollak, C.; Garcia Marchinena, P.A.; Jurado, A.M.; Isola, M.; Labanca, M.J.; Palazzo, M.; et al. von Hippel-Lindau mutants in renal cell carcinoma are regulated by increased expression of RSUME. Cell Death Dis. 2019, 10, 266. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Xie, H.; Liu, X.; Potjewyd, F.; James, L.I.; Wilkerson, E.M.; Herring, L.E.; Xie, L.; Chen, X.; Cabrera, J.C.; et al. TBK1 Is a Synthetic Lethal Target in Cancer with VHL Loss. Cancer Discov. 2020, 10, 460–475. [Google Scholar] [CrossRef]
- Kanno, H.; Yoshizumi, T.; Shinonaga, M.; Kubo, A.; Murata, H.; Yao, M. Role of VHL-JAK-STAT signaling pathway in central nervous system hemangioblastoma associated with von Hippel-Lindau disease. J. Neurooncol. 2020, 148, 29–38. [Google Scholar] [CrossRef]
- Perrotta, S.; Nobili, B.; Ferraro, M.; Migliaccio, C.; Borriello, A.; Cucciolla, V.; Martinelli, V.; Rossi, F.; Punzo, F.; Cirillo, P.; et al. Von Hippel-Lindau-dependent polycythemia is endemic on the island of Ischia: Identification of a novel cluster. Blood 2006, 107, 514–519. [Google Scholar] [CrossRef]
- Bento, M.C.; Chang, K.T.; Guan, Y.; Liu, E.; Caldas, G.; Gatti, R.A.; Prchal, J.T. Congenital polycythemia with homozygous and heterozygous mutations of von Hippel-Lindau gene: Five new Caucasian patients. Haematologica 2005, 90, 128–129. [Google Scholar]
- Gnarra, J.R.; Ward, J.M.; Porter, F.D.; Wagner, J.R.; Devor, D.E.; Grinberg, A.; Emmert-Buck, M.R.; Westphal, H.; Klausner, R.D.; Linehan, W.M. Defective placental vasculogenesis causes embryonic lethality in VHL-deficient mice. Proc. Natl. Acad. Sci. USA 1997, 94, 9102–9107. [Google Scholar] [CrossRef] [PubMed]
- van Rooijen, E.; Voest, E.E.; Logister, I.; Bussmann, J.; Korving, J.; van Eeden, F.J.; Giles, R.H.; Schulte-Merker, S. von Hippel-Lindau tumor suppressor mutants faithfully model pathological hypoxia-driven angiogenesis and vascular retinopathies in zebrafish. Dis. Model. Mech. 2010, 3, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Chew, E.Y. Ocular manifestations of von Hippel-Lindau disease: Clinical and genetic investigations. Trans. Am. Ophthalmol. Soc. 2005, 103, 495–511. [Google Scholar] [PubMed]
- Rankin, E.B.; Tomaszewski, J.E.; Haase, V.H. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res. 2006, 66, 2576–2583. [Google Scholar] [CrossRef]
- Ding, M.; Cui, S.; Li, C.; Jothy, S.; Haase, V.; Steer, B.M.; Marsden, P.A.; Pippin, J.; Shankland, S.; Rastaldi, M.P.; et al. Loss of the tumor suppressor Vhlh leads to upregulation of Cxcr4 and rapidly progressive glomerulonephritis in mice. Nat. Med. 2006, 12, 1081–1087. [Google Scholar] [CrossRef]
- Shen, H.C.; Adem, A.; Ylaya, K.; Wilson, A.; He, M.; Lorang, D.; Hewitt, S.M.; Pechhold, K.; Harlan, D.M.; Lubensky, I.A.; et al. Deciphering von Hippel-Lindau (VHL/Vhl)-associated pancreatic manifestations by inactivating Vhl in specific pancreatic cell populations. PLoS ONE 2009, 4, e4897. [Google Scholar] [CrossRef]
- Frew, I.J.; Minola, A.; Georgiev, S.; Hitz, M.; Moch, H.; Richard, S.; Vortmeyer, A.O.; Krek, W. Combined VHLH and PTEN mutation causes genital tract cystadenoma and squamous metaplasia. Mol. Cell Biol. 2008, 28, 4536–4548. [Google Scholar] [CrossRef]
- Hong, S.B.; Furihata, M.; Baba, M.; Zbar, B.; Schmidt, L.S. Vascular defects and liver damage by the acute inactivation of the VHL gene during mouse embryogenesis. Lab. Investig. 2006, 86, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H.; Glickman, J.N.; Socolovsky, M.; Jaenisch, R. Vascular tumors in livers with targeted inactivation of the von Hippel-Lindau tumor suppressor. Proc. Natl. Acad. Sci. USA 2001, 98, 1583–1588. [Google Scholar] [CrossRef]
- Ma, W.; Tessarollo, L.; Hong, S.B.; Baba, M.; Southon, E.; Back, T.C.; Spence, S.; Lobe, C.G.; Sharma, N.; Maher, G.W.; et al. Hepatic vascular tumors, angiectasis in multiple organs, and impaired spermatogenesis in mice with conditional inactivation of the VHL gene. Cancer Res. 2003, 63, 5320–5328. [Google Scholar]
- Park, S.; Chan, C.C. Von Hippel-Lindau disease (VHL): A need for a murine model with retinal hemangioblastoma. Histol. Histopathol. 2012, 27, 975–984. [Google Scholar] [CrossRef]
- Patard, J.J.; Rioux-Leclercq, N.; Masson, D.; Zerrouki, S.; Jouan, F.; Collet, N.; Dubourg, C.; Lobel, B.; Denis, M.; Fergelot, P. Absence of VHL gene alteration and high VEGF expression are associated with tumour aggressiveness and poor survival of renal-cell carcinoma. Br. J. Cancer 2009, 101, 1417–1424. [Google Scholar] [CrossRef]
- Los, M.; Aarsman, C.J.; Terpstra, L.; Wittebol-Post, D.; Lips, C.J.; Blijham, G.H.; Voest, E.E. Elevated ocular levels of vascular endothelial growth factor in patients with von Hippel-Lindau disease. Ann. Oncol. 1997, 8, 1015–1022. [Google Scholar] [CrossRef]
- Salinas-Sanchez, A.S.; Serrano-Oviedo, L.; Nam-Cha, S.Y.; Roche-Losada, O.; Sanchez-Prieto, R.; Gimenez-Bachs, J.M. Prognostic Value of the VHL, HIF-1alpha, and VEGF Signaling Pathway and Associated MAPK (ERK1/2 and ERK5) Pathways in Clear-Cell Renal Cell Carcinoma. A Long-Term Study. Clin. Genitourin. Cancer 2017, 15, e923–e933. [Google Scholar] [CrossRef]
- NIH/FDA. BEST (Biomarkers, EndpointS, and Other Tools) Resource; NIH/FDA: Silver Spring, MD, USA, 2016. Available online: https://www.ncbi.nlm.nih.gov/books/NBK326791/ (accessed on 22 August 2024).
- Wang, J.Y.; Peng, S.H.; Ning, X.H.; Li, T.; Liu, S.J.; Liu, J.Y.; Hong, B.A.; Qi, N.N.; Peng, X.; Zhou, B.W.; et al. Shorter telomere length increases age-related tumor risks in von Hippel-Lindau disease patients. Cancer Med. 2017, 6, 2131–2141. [Google Scholar] [CrossRef] [PubMed]
- Maroto, P.; Esteban, E.; Parra, E.F.; Mendez-Vidal, M.J.; Domenech, M.; Perez-Valderrama, B.; Calderero, V.; Perez-Gracia, J.L.; Grande, E.; Algaba, F. HIF pathway and c-Myc as biomarkers for response to sunitinib in metastatic clear-cell renal cell carcinoma. OncoTargets Ther. 2017, 10, 4635–4643. [Google Scholar] [CrossRef]
- Qi, Y.; Zhang, Y.; Peng, Z.; Wang, L.; Wang, K.; Feng, D.; He, J.; Zheng, J. SERPINH1 overexpression in clear cell renal cell carcinoma: Association with poor clinical outcome and its potential as a novel prognostic marker. J. Cell. Mol. Med. 2018, 22, 1224–1235. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.; Cai, L.; Wang, J.; Liu, S.; Zhou, J.; Ma, K.; Zhang, J.; Zhou, B.; Peng, X.; Zhang, N.; et al. Differential Expression of PD-L1 between Sporadic and VHL-Associated Hereditary Clear-Cell Renal Cell Carcinoma and Its Correlation with Clinicopathological Features. Clin. Genitourin. Cancer 2019, 17, 97–104.e1. [Google Scholar] [CrossRef] [PubMed]
- Radspieler, M.M.; Schindeldecker, M.; Stenzel, P.; Forsch, S.; Tagscherer, K.E.; Herpel, E.; Hohenfellner, M.; Hatiboglu, G.; Roth, W.; Macher-Goeppinger, S. Lamin-B1 is a senescence-associated biomarker in clear-cell renal cell carcinoma. Oncol. Lett. 2019, 18, 2654–2660. [Google Scholar] [CrossRef]
- Zhang, J.; Yan, A.; Cao, W.; Shi, H.; Cao, K.; Liu, X. Development and validation of a VHL-associated immune prognostic signature for clear cell renal cell carcinoma. Cancer Cell Int. 2020, 20, 584. [Google Scholar] [CrossRef]
- Yang, W.; Zhou, J.; Zhang, K.; Li, L.; Xu, Y.; Ma, K.; Xie, H.; Cai, L.; Gong, Y.; Gong, K. Identification and validation of the clinical roles of the VHL-related LncRNAs in clear cell renal cell carcinoma. J. Cancer 2021, 12, 2702–2714. [Google Scholar] [CrossRef]
- Oosting, S.F.; van Asselt, S.J.; Brouwers, A.H.; Bongaerts, A.H.; Steinberg, J.D.; de Jong, J.R.; Lub-de Hooge, M.N.; van der Horst-Schrivers, A.N.; Walenkamp, A.M.; Hoving, E.W.; et al. 89Zr-Bevacizumab PET Visualizes Disease Manifestations in Patients with von Hippel-Lindau Disease. J. Nucl. Med. 2016, 57, 1244–1250. [Google Scholar] [CrossRef] [PubMed]
- Shell, J.; Tirosh, A.; Millo, C.; Sadowski, S.M.; Assadipour, Y.; Green, P.; Patel, D.; Nilubol, N.; Kebebew, E. The utility of (68)Gallium-DOTATATE PET/CT in the detection of von Hippel-Lindau disease associated tumors. Eur. J. Radiol. 2019, 112, 130–135. [Google Scholar] [CrossRef]
- Xiao, Q.; Lauschke, V.M. The prevalence, genetic complexity and population-specific founder effects of human autosomal recessive disorders. NPJ Genom. Med. 2021, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.I.; Abecasis, G.R.; Cardon, L.R.; Goldstein, D.B.; Little, J.; Ioannidis, J.P.; Hirschhorn, J.N. Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nat. Rev. Genet. 2008, 9, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Mbemi, A.; Khanna, S.; Njiki, S.; Yedjou, C.G.; Tchounwou, P.B. Impact of Gene-Environment Interactions on Cancer Development. Int. J. Environ. Res. Public Health 2020, 17, 8089. [Google Scholar] [CrossRef] [PubMed]
- Marmot, M.; Friel, S.; Bell, R.; Houweling, T.A.; Taylor, S. Commission on Social Determinants of H. Closing the gap in a generation: Health equity through action on the social determinants of health. Lancet 2008, 372, 1661–1669. [Google Scholar] [CrossRef]
- Knickelbein, J.E.; Jacobs-El, N.; Wong, W.T.; Wiley, H.E.; Cukras, C.A.; Meyerle, C.B.; Chew, E.Y. Systemic Sunitinib Malate Treatment for Advanced Juxtapapillary Retinal Hemangioblastomas Associated with von Hippel-Lindau Disease. Ophthalmol. Retina 2017, 1, 181–187. [Google Scholar] [CrossRef]
- Jonasch, E.; McCutcheon, I.E.; Waguespack, S.G.; Wen, S.; Davis, D.W.; Smith, L.A.; Tannir, N.M.; Gombos, D.S.; Fuller, G.N.; Matin, S.F. Pilot trial of sunitinib therapy in patients with von Hippel-Lindau disease. Ann. Oncol. 2011, 22, 2661–2666. [Google Scholar] [CrossRef]
- Hwang, C.K.; Chew, E.Y.; Cukras, C.A.; Keenan, T.D.L.; Wong, W.T.; Linehan, W.M.; Chittiboina, P.; Pacak, K.; Wiley, H.E. Intravitreous treatment of severe ocular von Hippel-Lindau disease using a combination of the VEGF inhibitor, ranibizumab, and PDGF inhibitor, E10030: Results from a phase 1/2 clinical trial. Clin. Exp. Ophthalmol. 2021, 49, 1048–1059. [Google Scholar] [CrossRef]
- Batist, G.; Patenaude, F.; Champagne, P.; Croteau, D.; Levinton, C.; Hariton, C.; Escudier, B.; Dupont, E. Neovastat (AE-941) in refractory renal cell carcinoma patients: Report of a phase II trial with two dose levels. Ann. Oncol. 2002, 13, 1259–1263. [Google Scholar] [CrossRef]
- Escudier, B.; Choueiri, T.K.; Oudard, S.; Szczylik, C.; Negrier, S.; Ravaud, A.; Chevreau, C.; Venner, P.; Champagne, P.; Croteau, D.; et al. Prognostic factors of metastatic renal cell carcinoma after failure of immunotherapy: New paradigm from a large phase III trial with shark cartilage extract AE 941. J. Urol. 2007, 178, 1901–1905. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, B.; Villar Gomez de Las Heras, K.; Aguirre, D.T.; Rodriguez-Padial, L.; Albinana, V.; Recio-Poveda, L.; Cuesta, A.M.; Botella, L.M.; Jimenez-Escribano, R.M. Evaluation of the safety and effectiveness of oral propranolol in patients with von Hippel-Lindau disease and retinal hemangioblastomas: Phase III clinical trial. BMJ Open Ophthalmol. 2019, 4, e000203. [Google Scholar] [CrossRef] [PubMed]
- Panakis, N.; Offiah, C.; Azizan, A.; Roy, A.; Plowman, P.N. Sorafenib therapy and CNS hemangioblastomas in individuals with Von Hippel Lindau syndrome. J. Clin. Oncol. 2008, 26, 2065. [Google Scholar] [CrossRef]
- Jonasch, E.; McCutcheon, I.E.; Gombos, D.S.; Ahrar, K.; Perrier, N.D.; Liu, D.; Robichaux, C.C.; Villarreal, M.F.; Weldon, J.A.; Woodson, A.H.; et al. Pazopanib in patients with von Hippel-Lindau disease: A single-arm, single-centre, phase 2 trial. Lancet Oncol. 2018, 19, 1351–1359. [Google Scholar] [CrossRef]
- Jonasch, E.; Donskov, F.; Iliopoulos, O.; Rathmell, W.K.; Narayan, V.; Maughan, B.L.; Oudard, S.; Else, T.; Maranchie, J.K.; Welsh, S.J.; et al. Phase II study of the oral HIF-2α inhibitor MK-6482 for Von Hippel-Lindau disease–associated renal cell carcinoma. J. Clin. Oncol. 2020, 38, 5003. [Google Scholar] [CrossRef]
- Hasanov, E.; Jonasch, E. MK-6482 as a potential treatment for von Hippel-Lindau disease-associated clear cell renal cell carcinoma. Expert. Opin. Investig. Drugs 2021, 30, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Pilie, P.G.; Matin, S.F.; Woodson, A.H.; Marcott, V.D.; Bird, S.; Slack, R.; Fuller, G.; McCutcheon, I.E.; Jonasch, E. Pilot study of dovitinib in patients with VHL disease. J. Clin. Oncol. 2016, 34, 587. [Google Scholar] [CrossRef]
- Wong, W.T.; Liang, K.J.; Hammel, K.; Coleman, H.R.; Chew, E.Y. Intravitreal ranibizumab therapy for retinal capillary hemangioblastoma related to von Hippel-Lindau disease. Ophthalmology 2008, 115, 1957–1964. [Google Scholar] [CrossRef]
- Stamatakis, L.; Shuch, B.; Singer, E.A.; Nix, J.; Truong, H.; Friend, J.C.; Fowler, S.; Bratslavsky, G.; Metwalli, A.R.; Shih, J.H.; et al. Phase II trial of vandetanib in Von Hippel-Lindau-associated renal cell carcinoma. J. Clin. Oncol. 2013, 31, 4584. [Google Scholar] [CrossRef]
- Ma, K.; Hong, B.; Zhou, J.; Gong, Y.; Wang, J.; Liu, S.; Peng, X.; Zhou, B.; Zhang, J.; Xie, H.; et al. The Efficacy and Safety of Tyrosine Kinase Inhibitors for Von Hippel-Lindau Disease: A Retrospective Study of 32 Patients. Front. Oncol. 2019, 9, 1122. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, C.A. Von Hippel-Lindau syndrome. A pleomorphic condition. Cancer 1999, 86, 2478–2482. [Google Scholar] [CrossRef]
- Nagai, Y.; Ando, H.; Nohara, E.; Yamashita, H.; Takamura, T.; Kobayashi, K. Plasma levels of vascular endothelial growth factor in patients with acromegaly. Horm. Metab. Res. 2000, 32, 326–329. [Google Scholar] [CrossRef]
- Schraml, P.; Struckmann, K.; Hatz, F.; Sonnet, S.; Kully, C.; Gasser, T.; Sauter, G.; Mihatsch, M.J.; Moch, H. VHL mutations and their correlation with tumour cell proliferation, microvessel density, and patient prognosis in clear cell renal cell carcinoma. J. Pathol. 2002, 196, 186–193. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Classification | Associated Mutations | References |
|---|---|---|
| von Hippel–Lindau syndrome Type 1 | p.Asn78Ser | [53] |
| p.Pro81Ser | [54] | |
| p.Trp117Cys | [55] | |
| p.Phe136Ser | [53] | |
| In-frame del | [56] | |
| Stop at 158 | [57] | |
| p.Asn78His | [58] | |
| p.Ser65Trp | [59] | |
| p.Leu89Pro | [60] | |
| p.Ser111Asn | [61] | |
| p.Ser111Arg | [62] | |
| p.Ser80Arg | [63] | |
| p.Pro86Leu | [55] | |
| p.Ser65Leu | [61] | |
| p.Trp88Arg | [64] | |
| In-frame del Phe76 | [54] | |
| p.Gln96Pro | [65] | |
| p.Trp88Ser | [54] | |
| p.Gln73X | [66] | |
| p.Asn90Ile | [67] | |
| p.Leu184Pro | [54] | |
| p.Ile180Val | [60] | |
| p.Cys162Arg | [54] | |
| von Hippel–Lindau syndrome Type 2 | p.Phe119Leu | [68] |
| von Hippel–Lindau syndrome Type 1 and Type 2 | Frameshift | [53,69] |
| p.Leu118Pro | [53,65] | |
| p.Leu178Pro | [54,55] | |
| p.Ser80Asn | [60,70] | |
| p.Gly93Asp | [55,71] | |
| p.Leu158Pro | [54,72] | |
| p.His115Gln | [54,73] | |
| p.Gly114Cys | [53,58] | |
| von Hippel–Lindau syndrome Type 1 and Type 2B | p.Arg113X | [59,71] |
| von Hippel–Lindau syndrome Type 1, Type 2, and Type 2A | p.Tyr98His | [55,74] |
| von Hippel–Lindau syndrome Type 1, Type 2, and Type 2B | p.Arg161X | [59,75,76] |
| p.Cys162Tyr | [71,77] | |
| p.Val74Gly | [54,74] | |
| p.Cys162Trp | [54,59,73] | |
| von Hippel–Lindau syndrome Type 1, Type 2, and Type 2C | p.Leu188Val | [59,74,78] |
| p.Gly93Ser | [54,70,79] | |
| von Hippel–Lindau syndrome Type 1, Type 2A, and Type 2B | p.Pro86Ser | [65] |
| von Hippel–Lindau syndrome Type 1, Type 2, Type 2A, and Type 2B | p.Arg167Gln | [70,73,74,80] |
| p.Arg167Trp | [59,80,81,82] | |
| von Hippel–Lindau syndrome Type 1, Type 2, Type 2A, Type 2B, and Type 2C | p.Arg167Trp | [54,59,73,83] |
| von Hippel–Lindau syndrome Type 2 and Type 2A | p.Val166Phe | [73,84] |
| p.Arg161Gln | [73,85] | |
| p.Tyr98His | [70,74] | |
| p.Tyr112His | [55,86] | |
| von Hippel–Lindau syndrome Type 2 and Type 2B | p.Arg161Gly | [74,87] |
| von Hippel–Lindau syndrome Type 2, Type 2A, and Type 2B | p.Thr157Ile | [54,71,73] |
| von Hippel–Lindau syndrome Type 2A and Type 2B | p.Ala149Ser | [88,89] |
| Lesion/Frequency in Patients by Age | Symptoms | Diagnosis (MR, Magnetic Resonance; CT, Computed Tomography) | Imagen | Treatment and Management | Refs. |
|---|---|---|---|---|---|
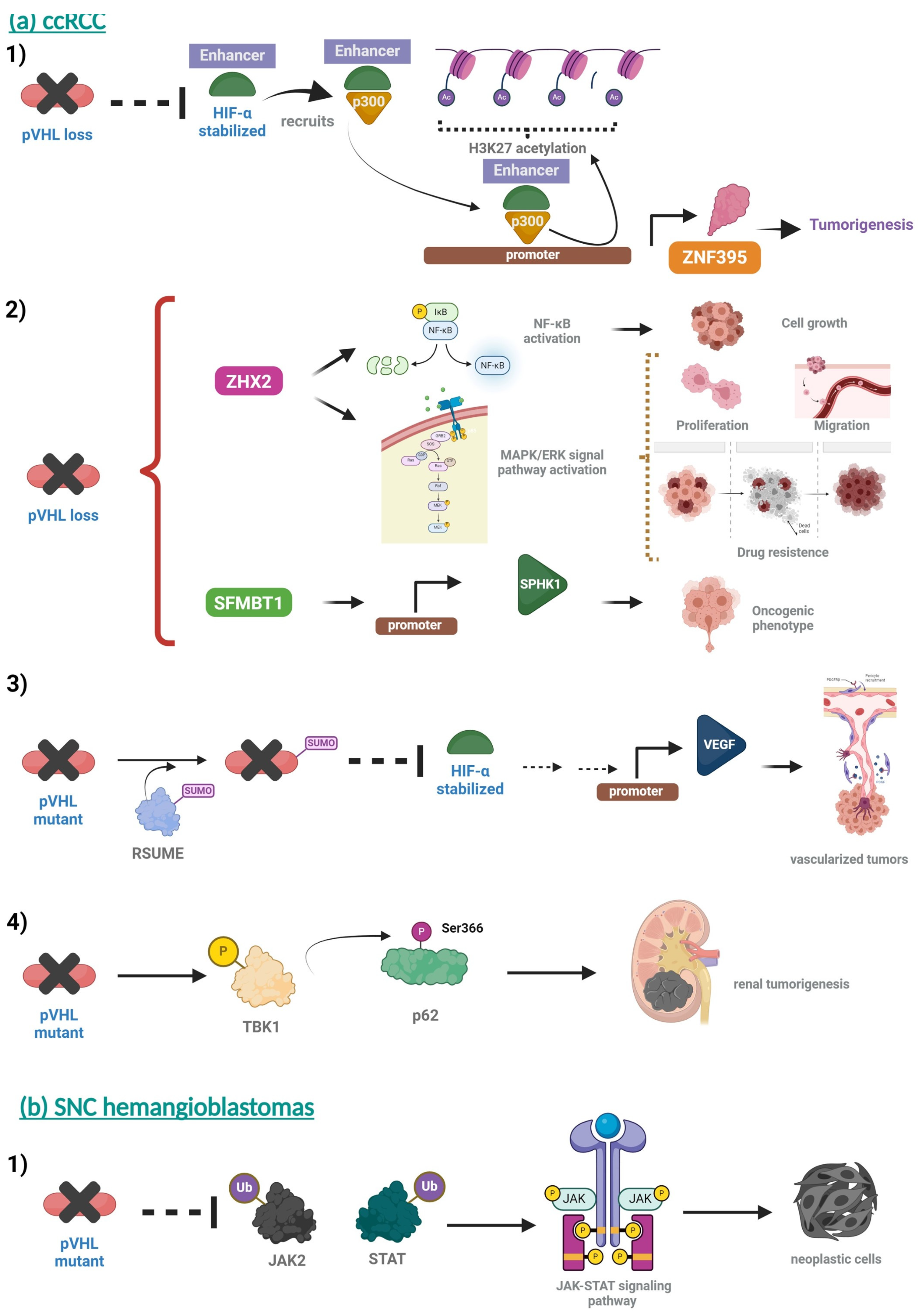
| CNS hemangioblastomas (HBs) cerebellum and spinal cord Early third decade (ages 22–26 years). Hemangioblastomas occur in approximately 60 to 80% of patients with VHL. | Headache, gait imbalance, ataxia, abnormal head position, nausea, vomiting, and papilledema | MR—brain | 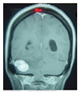 [99] http://creativecommons.org/licenses/by-nc-nd/3.0/ (accessed on 22 August 2024). | Followed by repeated MRI scans in asymptomatic patients. In patients, symptomatic tumors should be surgically removed. | [99,100] |
| Neurological impairment, urinary or bowel abnormalities, singultus, dysphagia, myelopathic disorders, syringomyelia, and polyglobulia. | MR—spinal cord |  [101] http://creativecommons.org/licenses/by-nc-nd/2.0/ (accessed on 22 August 2024). | Depending on the size of the tumor, surgical removal is recommended. | [100,101] | |
| Retinal hemangioblastoma (RH) Median age 21 and 25 years old, with a frequency from 49% to 85%. | Gradual loss of vision | Angiography Ultrasonography | 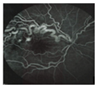 [102] http://creativecommons.org/licenses/by/4.0/ (accessed on 22 August 2024). | Ablative treatment: thermal laser photocoagulation, cryotherapy, radiation, and transpupillary thermotherapy. | [5,102,103] |
| Renal cysts Commonly occurs in the fourth decade of life, but this variant of VHL disease could occurrs as young as 16 years old. 60–70% of the lesions were carcinomas, all with clear cell features (RCCs, Renal Cysts Clear Cells). | Mostly asymptomatic. flank pain or hematuria | Ultrasound, Abdominal MRI, or CT |  [14] http://creativecommons.org/licenses/by/4.0/. | Partial nephrectomy is the option for tumors that have reached 3 cm, reducing the risk of metastasis while maintaining kidney function. VHL RCCs were treated with radical nephrectomies. | [5,14,91,92,101] |
| Pheochromocytoma (PCC) Diagnosticated arround the third decade of life with a frecuency of 33%. | Headache, sweating, palpitation, and hypertension | CT (computational Tomography), MRI (Magnetic Resonance Image) | 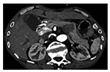 [104] http://creativecommons.org/licenses/by-nc-nd/4.0/. | Initial treatment is with alpha adrenergic blockers such as phenoxybenzamine for the control of hypertension. However, surgical resection remains the definitive treatment. | [5,44,101,104,105] |
| Pancreatic cysts and pancreatic neuroendocrine tumors (NETs) If diagnosis already in fourth decade, 35–75% of patients with VHL disease will develop simple pancreatic cysts. | Jaundice, abdominal pain, pruritis, vomiting and abdominal swelling | MRI, CT | 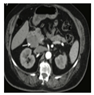 [14] http://creativecommons.org/licenses/by/4.0/. | Pancreatic cysts do not require surgical intervention; PNETs with a potential for metastatic disease are resected with enucleation by Whipple’s procedure or partial pancreatectomy depending on location and size greater than 3 cm. | [14,101,106,107,108] |
| Endolymphatic sac tumors (ELTS) 10 to 15% of patients with VHL disease develop ELSTs. The mean age of onset is 22 years old (range, 12–50 years) and they may be bilateral in 30% of cases. | Facial nerve palsy, vestibulocochlear impairments, tinnitus, vertigo, disequilibrium, and hearing loss | CT |  [109] http://creativecommons.org/licenses/by-nc-nd/3.0/. | Treatment of ELTS requires extensive surgery with adequate bone removal around the area of the macroscopically evident tumor. | [109,110] |
| Epididymal cystadenomas 16–80 years old. Occur in 25–60% of affected men | Mostly asymptomatic | Ultrasonography PET/CT images |  [111] http://creativecommons.org/licenses/by/4.0/. | Surgery is rarely performed in epididymal cystadenomas. Routinely followed by a physical exam and ultrasonography. | [92,111,112] |
| Broad ligament cystadenomas Diagnosticated about the second decade with 10% frecuency. Women unilateral presentation. | Mostly asymptomatic | Transvaginal ultrasound | 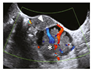 [113] http://creativecommons.org/licenses/by/4.0/. | Being benign lesions, they are usually managed conservatively without surgery. | [113,114] |
| Organ System | Model Organism | Phenotype | Limitations |
|---|---|---|---|
| Central Nervous System (CNS) | Zebrafish | Retinal neovascularization, edema, retinal detachment | No hemangioblastoma development |
| Kidney | Mouse | Renal microcysts, macrocysts, acute nephritis, hematuria, proteinuria, renal failure | Limited to specific kidney regions |
| Pancreas | Mouse | Highly vascularized cysts, microcystic adenomas | Incomplete penetrance, limited to pancreas |
| Reproductive System | Mouse | Clear cell cystadenoma of genital tract | Specific to genital tract epithelium |
| Liver | Mouse | Hepatic hemangiomas, angiectasias, steatosis, inflammatory cell infiltration | Embryonic lethality in some models, limited to liver and other specific organs |
| Biomarker | Sample | Cohort | Results/Remarks | Ref. |
|---|---|---|---|---|
| Telomere length | Blood |
|
| [156] |
| c-Myc/HIF-2α | Formalin-fixed, paraffin-embedded primary tumor samples |
|
| [157] |
| Serpin peptidase inhibitor clade H member 1 (SERPINH1) | Primary ccRCC and adjacent normal kidney tissues |
|
| [158] |
| Programmed death ligand 1 (PD-L1) | Formalin-fixed and paraffin-embedded surgical RCC simples |
|
| [159] |
| Lamin B1 | Sample tissues (primary tumors and normal tissues samples) |
|
| [160] |
| Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4) and PD-1 | Formalin-fixed paraffin embedded (FFPE) specimens collected from radical surgery |
|
| [161] |
| lncRNA FGD5-AS1 | Renal tissue (adjacent normal renal tissue and ccRCC tissue) |
|
| [162] |
| SFMBT1 and ZHX2 | Tumor tissues and adjacent normal tissues |
|
| [135] |
| 89Zr-bevacizumab | Imaging |
|
| [163] |
| 68Gallium-DOTATATE PET/CT | Imaging |
|
| [164] |
| Clinical Trial Identifier and Therapeutic Agent | Clinical Phase | Status | Condition or Disease | Molecular Target | Participants | Outcome | Refs. |
|---|---|---|---|---|---|---|---|
| NCT00673816 Sunitinib | II | Terminated | Advanced Ocular Disease of von Hippel–Lindau Syndrome | Inhibition of multiple receptor tyrosine kinases (RTK), including the vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) | 5 patients receive 9 months of sunitinib malate therapy administered in 6 cycles. Each cycle consisted of a daily oral dose of 50 mg sunitinib malate for 4 weeks followed by a 2-week rest period. (Only one completed treatment). | Change in Best-Correction Visual Acuity (BCVA), Retinal Thickness, and Retinal Angioma Leakage from Baseline to Week 36. The recruitment goal was to enroll five participants; however, the study was terminated after only two participants had been enrolled due to slow recruitment and adverse events. | [169] |
| NCT00330564 Sunitinib | II | Terminated | von Hippel–Lindau syndrome: Renal cell carcinoma and/or Hemangioblastoma | Treatment with SU011248/sunitinib malate (50 mg daily dose for 4 weeks, then 2 weeks off) for 6 months in 15 patients with von Hippel–Lindau (VHL) syndrome who have a measurable lesion undergoing surveillance. | Nine of the fifteen patients completed all four cycles of therapy, and the expected toxic effects were responsible for the necessary dosage reductions and discontinuation of treatment. Renal cell carcinomas responded better to sunitinib therapy than other VHL related lesions using the RECIST measure. The study ended early due to slow accrual. | [170] | |
| NCT02859441 Ranibizumab E10030 | I/II | Completed | von Hippel–Lindau (VHL) Retinal Capillary Hemangiomas (RCH) | E10030, a PDGF-B antagonist, and ranibizumab, a VEGF-A antagonist | This was a single-arm open-label phase 1/2 study, consisting of 3 adults with VHL-associated RH and vision loss. Intravitreous injections of ranibizumab (0.5 mg) and E10030 (1.5 mg) were administered unilaterally and each received 9 injections prior to week 52 and were followed for 104 weeks. | One participant manifested mild episodic ocular hypertension in the study eye. The change in BCVA in the study eye at week 52 for the three participants was –5, –12, and +2 letters. No reduction in RH size was measured at 52 weeks. Variable mild improvements in exudation in two participants at week 16 were not sustained through week 52. Intranavitreous injection with ranibizumab and E10030 demonstrated a reasonable preliminary safety profile, but limited treatment effect. | [171] |
| Protocol number CT/AE-941/002 Neovastat (Canadá) | II | Completed | Renal cell carcinoma (RCC) | Inhibition of angiogenesis | 22 patients with a primary diagnosis of refractory CCR. They were treated with Neovastat 240 mL/day (n = 14) compared to patients receiving 60 mL/day (n = 8). | The higher dose of Neovastat administered in this trial is associated with a survival benefit in RCC. Neovastat is well tolerated by advanced cancer patients at doses of 60 and 240 mL/day. | [172] |
| Not definite. Neovastat (Canadá) | III | Completed | Metastatic renal cell carcinoma in whom immunotherapy failed | 300 patients from 48 international centers were randomized to receive 120 mL twice daily of oral Neovastat or placebo Neovastat. The 300 patients who received at least 1 dose of study medication were included in this analysis. | The study of metastatic CRC provides a prognostic model that has a significant impact on risk-adjusted survival. Although external validation in an independent data set is lacking, the results of this trial may lead to a new paradigm for clinical trial design and risk stratification when considering future investigations of patients with metastatic CRC in whom immunotherapy has failed. | [173] | |
| EudraCT Number: 2014-003671-30 Propanolol (Spain) | III | Completed | von Hippel–Lindau disease and retinal hemangioblastomas | VEGF inhibitor | 7 patients were included. All patients received a daily dose of 120 mg propranolol for 1 year. Clinical variables were evaluated at baseline, and at 1, 3, 6, 9, and 12 months. | The number and size of retinal hemangioblastomas remained stable in all patients. The only adverse effect reported was hypotension in one patient. The results suggest that propranolol could be useful for the treatment of retinal hemangioblastomas in patients with VHL, especially when there are retinal exudates. The results of this clinical trial allowed propranolol designation to treat von Hippel–Lindau disease, granted by the European Medicines Agency (EMA). | [174] |
| EudraCT Number: 2007-002132-29 Sorafenib (UK) | II | Terminated | VHL-associated renal cancer | Tyrosine kinase inhibitor | 4 patients with VHL syndrome who had therapy for advanced RCC, received sorafenib orally (400 mg twice a day) for up to six months. | This study concludes that over a 6-month period of sorafenib, at the standard dose used in RCC, there was no response effect in CNS hemangioblastomas in this population of patients. | [175] |
| NCT01436227 Pazopanib | II | Active, not recruiting | von Hippel–Lindau disease genetically confirmed or one disease-related lesion. | Vascular endothelial growth factor receptors (VEGFR) -1, -2, and -3, c-kit and platelet-derived growth factor receptor (PDGF-R) inhibitors | 31 eligible patients were treated with pazopanib 800 mg by mouth daily for 24 weeks. | To ensure timely dissemination of data, the decision was made to close the trial after 31 evaluable patients were accrued. Pazopanib induces a reduction in the burden of the disease in von Hippel–Lindau disease patients. Efficacy data indicate benefit in individuals with renal cell carcinomas and pancreatic lesions, and some potential efficacy signals in hemangioblastomas as well. Pazopanib could be considered in patients with von Hippel–Lindau disease and growing lesions where surgical resection may be required in the relatively near future, or in patients with unresectable lesions where a decrease in tumor size is desired. | [176] |
| NCT03401788 Belzutifan | II | Active, not recruiting | von Hippel–Lindau Disease-associated renal cell Carcinoma | Inhibitor of HIF-2α | 61 patients receive 120 mg of belzutifan orally once a day until progression, intolerable toxicity, or the investigator/patient’s decision to withdraw. | Of the 61 patients, 53 (86.9%) had a decrease in the size of the target lesions. Responses were also observed in CNS, retinal, and pancreatic lesions. MK-6482 showed promising efficacy and tolerability in patients with VHL-associated ccRCC and responses in other VHL-related lesions. | [177,178] |
| NCT03108066 PT2385 | II | Active, not recruiting | von Hippel–Lindau Disease-associated renal cell carcinoma | Inhibitor of HIF-2α | 4 patients were enrolled in each stage of a two-stage design. PT2385 was administered orally at a dose of 800 mg twice daily, with a follow-up of 19 weeks. | All patients had stable disease (SD) as their best response at the latest assessment. PT2385 demonstrated stabilization of disease in VHL-associated clear cell RCC and other tumors, and showed an acceptable safety profile. | See below |
| NCT01266070 Dovitinib | II | Terminated | VHL-related hemangioblastoma | A multityrosine kinase that inhibits FGFR, VEGFR, and PDGFR. | 6 participants received 500 mg/day (5 days in/2 days out of dosing). All participants completed at least two cycles of therapy. | The trial was stopped after six patients due to the activation of the toxicity stopping rule. The lack of response in HBs in this population treated with dovitinib is surprising, and molecular profiling of HB tissue would be extremely useful to help understand the biologic underpinnings of this lack of efficacy. | [179] |
| NCT00089765 Ranibizumab | I | Completed | Angiomas (blood vessel tumors) in patients with von Hippel–Lindau syndrome (VHL) | VEGF-neutralizing agent | 5 patients with retinal capillary hemangioblastomas (RCH) associated with VHL with exudative changes and visual loss. Monthly intravitreal injections of ranibizumab (0.5 mg) were administered over a 6 month course for a total of 7 injections, with additional injections considered until week 52. | The primary outcome was the change in best-corrected visual acuity (BCVA). Secondary outcomes included change in lesion size, change in retinal thickness, and adverse event assessments. Intravitreal ranibizumab, administered as monotherapy every 4 weeks, had minimal beneficial effects on most RCHs related to VHL. Future studies are needed to determine a combination with other therapies for the treatment of ocular tumors associated with VHL. | [180] |
| NCT02108002 Vorinostat | I | Completed | VHL-related hemangioblastoma | Histone deacetylase inhibitor (HDACi) | 7 germline missense VHL patients with symptomatic CNS hemangioblastomas received 400 mg of vorinostat by mouth daily for seven days prior to surgery and subsequently underwent surgical resection. | Vorinostat is well tolerated by patients with symptomatic CNS hemangioblastomas in the context of germline missense VHL disease and shows results in mutated stabilization of the pVHL protein. This suggests that vorinostat may be a promising treatment for patients with a germline mutation. | See below |
| NCT00566995 Vandetanib | II | Completed | VHL-associated renal cell carcinoma | Dual VEGFR2/EGFR inhibitor | 34 participants received a 300 mg/day (starting dose) oral dose of vandetanib for 28 days. | Vandetanib demonstrated antitumor activity. However, the poor tolerability required drug withdrawal in a significant proportion of patients. Newer agents that selectively target VEGF receptors may offer a more tolerable alternative and could optimize clinical benefits in this population. | [181] |
| Medical Ethics Committee of Peking University First Hospital (Beijing, China) TKIs | ND | Completed | von Hippel–Lindau disease | Tyrosine kinase inhibitor (TKI) | 32 patients receiving TKIs were recruited. For sunitinib, a dosage of 50 mg/day was administered orally for 28 days, followed by a 14-day break per cycle for several cycles. For sorafenib, a dose of 800 mg/day divided into two doses was administered orally. For axitinib, a dose of 10 mg/day divided into two doses was administered orally. For pazopanib, a dose of 800 mg/day was administered orally. | A partial response was observed in eleven (31%) of thirty-six renal cell carcinomas, four (27%) of fifteen pancreatic lesions, and one (20%) of five central nervous system (CNS) hemangioblastomas. The mean tumor size decreased significantly for renal cell carcinomas (p = 0.0001), renal cysts (p = 0.027), and pancreatic lesions (p = 0.003) after TKI therapy. Finally, the side effects were acceptable. | [182] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Virgilio, L.; Velazquez-Paniagua, M.; Cuazozon-Ferrer, L.; Silva-Lucero, M.-d.-C.; Gutierrez-Malacara, A.-I.; Padilla-Mendoza, J.-R.; Borbolla-Vázquez, J.; Díaz-Hernández, J.-A.; Jiménez-Orozco, F.-A.; Cardenas-Aguayo, M.-d.-C. Genetics, Pathophysiology, and Current Challenges in Von Hippel–Lindau Disease Therapeutics. Diagnostics 2024, 14, 1909. https://doi.org/10.3390/diagnostics14171909
Gómez-Virgilio L, Velazquez-Paniagua M, Cuazozon-Ferrer L, Silva-Lucero M-d-C, Gutierrez-Malacara A-I, Padilla-Mendoza J-R, Borbolla-Vázquez J, Díaz-Hernández J-A, Jiménez-Orozco F-A, Cardenas-Aguayo M-d-C. Genetics, Pathophysiology, and Current Challenges in Von Hippel–Lindau Disease Therapeutics. Diagnostics. 2024; 14(17):1909. https://doi.org/10.3390/diagnostics14171909
Chicago/Turabian StyleGómez-Virgilio, Laura, Mireya Velazquez-Paniagua, Lucero Cuazozon-Ferrer, Maria-del-Carmen Silva-Lucero, Andres-Ivan Gutierrez-Malacara, Juan-Ramón Padilla-Mendoza, Jessica Borbolla-Vázquez, Job-Alí Díaz-Hernández, Fausto-Alejandro Jiménez-Orozco, and Maria-del-Carmen Cardenas-Aguayo. 2024. "Genetics, Pathophysiology, and Current Challenges in Von Hippel–Lindau Disease Therapeutics" Diagnostics 14, no. 17: 1909. https://doi.org/10.3390/diagnostics14171909