
The Interlinking Metabolic Association between Type 2 Diabetes Mellitus and Cancer: Molecular Mechanisms and Therapeutic Insights



Abstract
:1. Introduction
2. Literature Review and Selection
- Publications that discussed the risk factors for cancer and diabetes.
- Studies that covered metabolic association between various types of cancer and diabetes.
- Studies that covered the potential implication of PI3K/AKT/mTOR signaling in the association between cancer and diabetes.
- Articles published in English within the last two decades, given the rapid advancements.
- Articles not available in full text and studies unrelated to the topic were excluded.
- Studies that focused solely on type 1 diabetes and its association with cancer.
- Studies that focused on the use of inhibitors other than Akt inhibitors and mTOR inhibitors.
- Articles that did not provide substantial information on the association between type 2 diabetes and cancer.
- Publications in languages other than English.
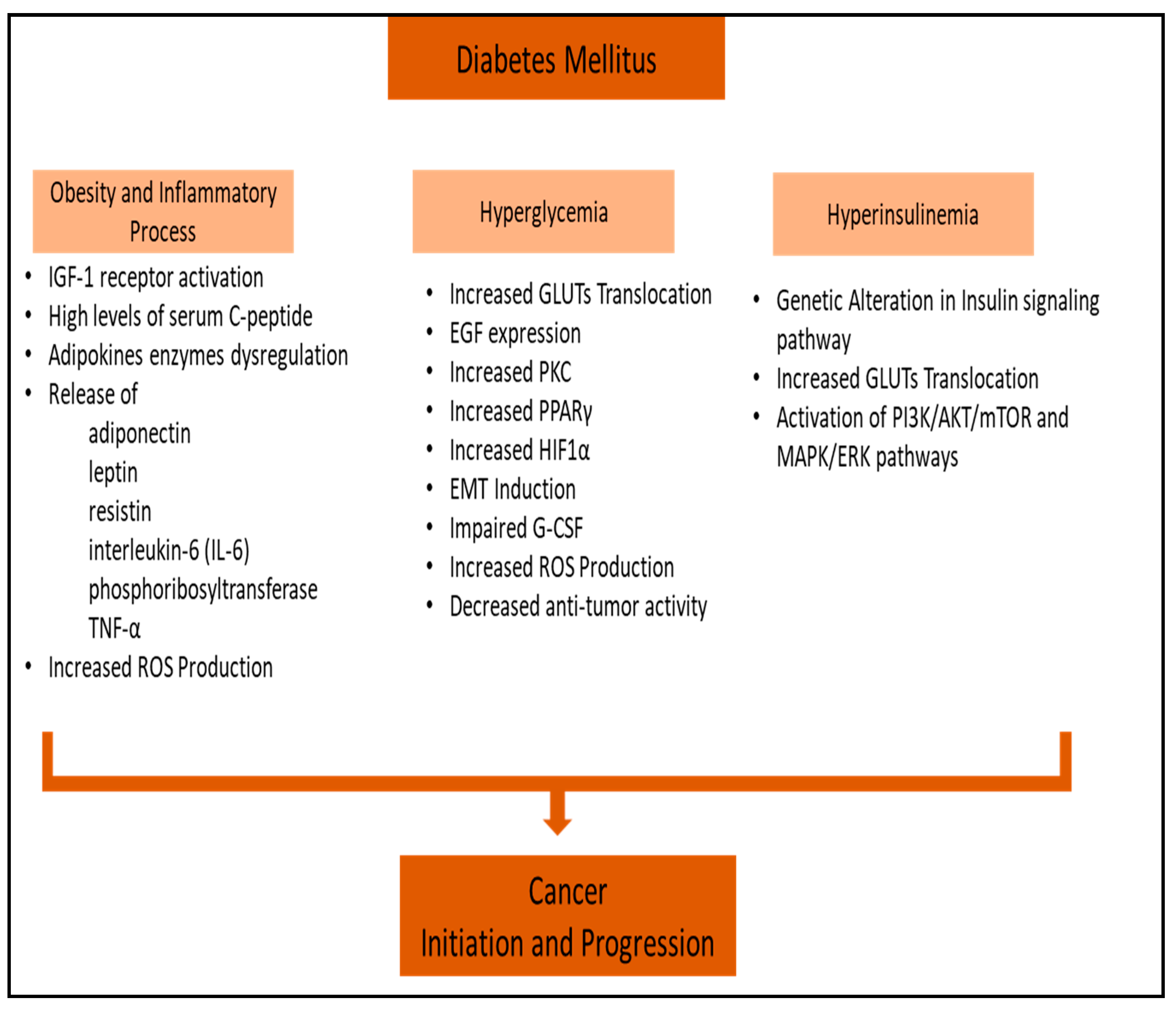
3. Obesity-Associated Diabetes and Inflammatory Processes in Cancer
4. Hyperglycemia and Cancer
4.1. Cancer Cell Proliferation
4.2. Association of Apoptosis and Hyperglycemia in Cancer
4.3. Hyperglycemia and Cancer Metastasis
5. Hyperinsulinemia and Cancer
5.1. PI3K/AKT/mTOR Signaling Pathway in Cancer
5.2. RAS/MAPK/ERK Signaling Pathway in Cancer
6. Insulin-Mediated PI3K Pathway Inhibition in Cancer
6.1. PI3K Inhibitors
6.2. Akt Inhibitors
6.3. mTOR Inhibitors
7. Antidiabetic Drugs and Their Anti-Cancer Effects
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Olatunde, A.; Nigam, M.; Singh, R.K.; Panwar, A.S.; Lasisi, A.; Alhumaydhi, F.A.; Kumar, V.J.; Mishra, A.P.; Sharifi-Rad, J. Cancer and diabetes: The interlinking metabolic pathways and repurposing actions of antidiabetic drugs. Cancer Cell Int. 2021, 21, 499. [Google Scholar] [CrossRef] [PubMed]
- Goyal, R.; Singhal, M.; Jialal, I. Type 2 Diabetes; Statpearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Banday, M.Z.; Sameer, A.S.; Nissar, S. Pathophysiology of diabetes: An overview. Avicenna J. Med. 2020, 10, 174–188. [Google Scholar] [CrossRef]
- Facts & Figures. International Diabetes Federation. 2024. Available online: https://idf.org/about-diabetes/diabetes-facts-figures/ (accessed on 19 February 2024).
- Ling, S.; Brown, K.; Miksza, J.K.; Howells, L.; Morrison, A.; Issa, E.; Yates, T.; Khunti, K.; Davies, M.J.; Zaccardi, F. Association of type 2 diabetes with cancer: A meta-analysis with bias analysis for unmeasured confounding in 151 cohorts comprising 32 million people. Diabetes Care 2020, 43, 2313–2322. [Google Scholar] [CrossRef] [PubMed]
- Abudawood, M. Diabetes and cancer: A comprehensive review. J. Res. Med. Sci. 2019, 24, 94. [Google Scholar] [CrossRef] [PubMed]
- Tsilidis, K.K.; Kasimis, J.C.; Lopez, D.S.; Ntzani, E.E.; Ioannidis, J.P. Type 2 diabetes and cancer: Umbrella review of meta-analyses of observational studies. BMJ 2015, 350, g7607. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhang, X.; Ma, Y.; Yuan, C.; Wang, M.; Wu, K.; Tabung, F.K.; Tobias, D.; Hu, F.B.; Giovannucci, E.; et al. Incident type 2 diabetes duration and cancer risk: A prospective study in two us cohorts. J. Natl. Cancer Inst. 2021, 113, 381–389. [Google Scholar] [CrossRef]
- Ediriweera, M.K.; Jayasena, S. The role of reprogrammed glucose metabolism in cancer. Metabolites 2023, 13, 345. [Google Scholar] [CrossRef]
- Zhu, B.; Qu, S. The relationship between diabetes mellitus and cancers and its underlying mechanisms. Front. Endocrinol. 2022, 13, 800995. [Google Scholar] [CrossRef]
- Suh, S.; Kim, K.W. Diabetes and cancer: Cancer should be screened in routine diabetes assessment. Diabetes Metab. J. 2019, 43, 733–743. [Google Scholar] [CrossRef]
- Xu, C.X.; Zhu, H.H.; Zhu, Y.M. Diabetes and cancer: Associations, mechanisms, and implications for medical practice. World J. Diabetes 2014, 5, 372–380. [Google Scholar] [CrossRef]
- Zhang, A.M.Y.; Wellberg, E.A.; Kopp, J.L.; Johnson, J.D. Hyperinsulinemia in obesity, inflammation, and cancer. Diabetes Metab. J. 2021, 45, 622. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Dai, Q.; Su, X.; Fu, J.; Feng, X.; Peng, J. Role of pi3k/akt pathway in cancer: The framework of malignant behavior. Mol. Biol. Rep. 2020, 47, 4587–4629. [Google Scholar] [CrossRef] [PubMed]
- Shahid, R.K.; Ahmed, S.; Le, D.; Yadav, S. Diabetes and cancer: Risk, challenges, management and outcomes. Cancers 2021, 13, 5735. [Google Scholar] [CrossRef]
- Renehan, A.G.; Howell, A. Preventing cancer, cardiovascular disease, and diabetes. Lancet 2005, 365, 1449–1451. [Google Scholar] [CrossRef]
- Lega, I.C.; Lipscombe, L.L. Review: Diabetes, obesity, and cancer-pathophysiology and clinical implications. Endocr. Rev. 2020, 41, 33–52. [Google Scholar] [CrossRef]
- Chandrasekaran, P.; Weiskirchen, R. The role of obesity in type 2 diabetes mellitus-an overview. Int. J. Mol. Sci. 2024, 25, 1882. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Dossus, L.; Kaaks, R. Obesity related hyperinsulinaemia and hyperglycaemia and cancer development. Arch. Physiol. Biochem. 2009, 115, 86–96. [Google Scholar] [CrossRef]
- de Andrade Mesquita, L.; Wayerbacher, L.F.; Schwartsmann, G.; Gerchman, F. Obesity, diabetes, and cancer: Epidemiology, pathophysiology, and potential interventions. Arch. Endocrinol. Metab. 2023, 67, e000647. [Google Scholar] [CrossRef]
- Nam, S.Y.; Lee, E.J.; Kim, K.R.; Cha, B.S.; Song, Y.D.; Lim, S.K.; Lee, H.C.; Huh, K.B. Effect of obesity on total and free insulin-like growth factor (igf)-1, and their relationship to igf-binding protein (bp)-1, igfbp-2, igfbp-3, insulin, and growth hormone. Int. J. Obes. Relat. Metab. Disord. 1997, 21, 355–359. [Google Scholar] [CrossRef]
- Renehan, A.G.; Frystyk, J.; Flyvbjerg, A. Obesity and cancer risk: The role of the insulin-igf axis. Trends Endocrinol. Metab. 2006, 17, 328–336. [Google Scholar] [CrossRef]
- Ma, J.; Li, H.; Giovannucci, E.; Mucci, L.; Qiu, W.; Nguyen, P.L.; Gaziano, J.M.; Pollak, M.; Stampfer, M.J. Prediagnostic body-mass index, plasma c-peptide concentration, and prostate cancer-specific mortality in men with prostate cancer: A long-term survival analysis. Lancet Oncol. 2008, 9, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, E.J.; LeRoith, D. Obesity and diabetes: The increased risk of cancer and cancer-related mortality. Physiol. Rev. 2015, 95, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kim, J.H.; Lee, Y.J. The role of adipokines in tumor progression and its association with obesity. Biomedicines 2024, 12, 97. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; Zwahlen, M.; Egger, M. Adiposity and cancer risk: New mechanistic insights from epidemiology. Nat. Rev. Cancer 2015, 15, 484–498. [Google Scholar] [CrossRef]
- Parida, S.; Siddharth, S.; Sharma, D. Adiponectin, obesity, and cancer: Clash of the bigwigs in health and disease. Int. J. Mol. Sci. 2019, 20, 2519. [Google Scholar] [CrossRef]
- Taliaferro-Smith, L.; Nagalingam, A.; Zhong, D.; Zhou, W.; Saxena, N.K.; Sharma, D. Lkb1 is required for adiponectin-mediated modulation of ampk-s6k axis and inhibition of migration and invasion of breast cancer cells. Oncogene 2009, 28, 2621–2633. [Google Scholar] [CrossRef]
- Saxena, N.K.; Sharma, D. Metastasis suppression by adiponectin: Lkb1 rises up to the challenge. Cell Adhes. Migr. 2010, 4, 358–362. [Google Scholar] [CrossRef]
- Chung, S.J.; Nagaraju, G.P.; Nagalingam, A.; Muniraj, N.; Kuppusamy, P.; Walker, A.; Woo, J.; Gyorffy, B.; Gabrielson, E.; Saxena, N.K.; et al. Adipoq/adiponectin induces cytotoxic autophagy in breast cancer cells through stk11/lkb1-mediated activation of the ampk-ulk1 axis. Autophagy 2017, 13, 1386–1403. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Jimenez, F.; Perez-Perez, A.; de la Cruz-Merino, L.; Sanchez-Margalet, V. Obesity and breast cancer: Role of leptin. Front. Oncol. 2019, 9, 596. [Google Scholar] [CrossRef]
- Mhaidat, N.M.; Alzoubi, K.H.; Kubas, M.A.; Banihani, M.N.; Hamdan, N.; Al-Jaberi, T.M. High levels of leptin and non-high molecular weight-adiponectin in patients with colorectal cancer: Association with chemotherapy and common genetic polymorphisms. Biomed. Rep. 2021, 14, 13. [Google Scholar] [CrossRef]
- Stattin, P.; Lukanova, A.; Biessy, C.; Soderberg, S.; Palmqvist, R.; Kaaks, R.; Olsson, T.; Jellum, E. Obesity and colon cancer: Does leptin provide a link? Int. J. Cancer 2004, 109, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Deng, L.L.; Cui, J.Q.; Shi, L.; Yang, Y.C.; Luo, J.H.; Qin, D.; Wang, L. Association between serum leptin levels and breast cancer risk: An updated systematic review and meta-analysis. Medicine 2018, 97, e11345. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Pan, Q.; Chen, X.; Xu, S.; Luo, X.; Chen, L. The association between obesity related adipokines and risk of breast cancer: A meta-analysis. Oncotarget 2017, 8, 75389–75399. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W.; McPherson, M.; Darlington, L.G. Obesity and cancer: Existing and new hypotheses for a causal connection. EBioMedicine 2018, 30, 14–28. [Google Scholar] [CrossRef]
- Wellen, K.E.; Thompson, C.B. Cellular metabolic stress: Considering how cells respond to nutrient excess. Mol. Cell 2010, 40, 323–332. [Google Scholar] [CrossRef]
- Lee, J.Y.; Sohn, K.H.; Rhee, S.H.; Hwang, D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through toll-like receptor 4. J. Biol. Chem. 2001, 276, 16683–16689. [Google Scholar] [CrossRef]
- Ramos, E.J.; Xu, Y.; Romanova, I.; Middleton, F.; Chen, C.; Quinn, R.; Inui, A.; Das, U.; Meguid, M.M. Is obesity an inflammatory disease? Surgery 2003, 134, 329–335. [Google Scholar] [CrossRef]
- Ramteke, P.; Deb, A.; Shepal, V.; Bhat, M.K. Hyperglycemia associated metabolic and molecular alterations in cancer risk, progression, treatment, and mortality. Cancers 2019, 11, 1402. [Google Scholar] [CrossRef]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martin, C. Pathophysiology of type 2 diabetes mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef]
- Ruze, R.; Liu, T.; Zou, X.; Song, J.; Chen, Y.; Xu, R.; Yin, X.; Xu, Q. Obesity and type 2 diabetes mellitus: Connections in epidemiology, pathogenesis, and treatments. Front. Endocrinol. 2023, 14, 1161521. [Google Scholar] [CrossRef]
- Ryu, T.Y.; Park, J.; Scherer, P.E. Hyperglycemia as a risk factor for cancer progression. Diabetes Metab. J. 2014, 38, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Masur, K.; Vetter, C.; Hinz, A.; Tomas, N.; Henrich, H.; Niggemann, B.; Zanker, K.S. Diabetogenic glucose and insulin concentrations modulate transcriptome and protein levels involved in tumour cell migration, adhesion and proliferation. Br. J. Cancer 2011, 104, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Hahn, T.; Barth, S.; Hofmann, W.; Reich, O.; Lang, I.; Desoye, G. Hyperglycemia regulates the glucose-transport system of clonal choriocarcinoma cells in vitro. A potential molecular mechanism contributing to the adjunct effect of glucose in tumor therapy. Int. J. Cancer 1998, 78, 353–360. [Google Scholar] [CrossRef]
- Han, L.; Ma, Q.; Li, J.; Liu, H.; Li, W.; Ma, G.; Xu, Q.; Zhou, S.; Wu, E. High glucose promotes pancreatic cancer cell proliferation via the induction of egf expression and transactivation of egfr. PLoS ONE 2011, 6, e27074. [Google Scholar] [CrossRef]
- Duan, W.; Shen, X.; Lei, J.; Xu, Q.; Yu, Y.; Li, R.; Wu, E.; Ma, Q. Hyperglycemia, a neglected factor during cancer progression. BioMed Res. Int. 2014, 2014, 461917. [Google Scholar] [CrossRef]
- Okumura, M.; Yamamoto, M.; Sakuma, H.; Kojima, T.; Maruyama, T.; Jamali, M.; Cooper, D.R.; Yasuda, K. Leptin and high glucose stimulate cell proliferation in mcf-7 human breast cancer cells: Reciprocal involvement of pkc-alpha and ppar expression. Biochim. Biophys. Acta 2002, 1592, 107–116. [Google Scholar] [CrossRef]
- Ways, D.K.; Kukoly, C.A.; de Vente, J.; Hooker, J.L.; Bryant, W.O.; Posekany, K.J.; Fletcher, D.J.; Cook, P.P.; Parker, P.J. Mcf-7 breast cancer cells transfected with protein kinase c-alpha exhibit altered expression of other protein kinase c isoforms and display a more aggressive neoplastic phenotype. J. Clin. Investig. 1995, 95, 1906–1915. [Google Scholar] [CrossRef]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ros generation are essential for kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, X.; Sang, H.; Zhou, Y.; Shang, C.; Wang, Y.; Zhu, H. Effects of hyperglycemia on the progression of tumor diseases. J. Exp. Clin. Cancer Res. 2019, 38, 327. [Google Scholar] [CrossRef] [PubMed]
- Pothiwala, P.; Jain, S.K.; Yaturu, S. Metabolic syndrome and cancer. Metab. Syndr. Relat. Disord. 2009, 7, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Lopez, R.; Arumugam, A.; Joseph, R.; Monga, K.; Boopalan, T.; Agullo, P.; Gutierrez, C.; Nandy, S.; Subramani, R.; de la Rosa, J.M.; et al. Hyperglycemia enhances the proliferation of non-tumorigenic and malignant mammary epithelial cells through increased leptin/igf1r signaling and activation of akt/mtor. PLoS ONE 2013, 8, e79708. [Google Scholar] [CrossRef]
- Luo, J.; Xiang, Y.; Xu, X.; Fang, D.; Li, D.; Ni, F.; Zhu, X.; Chen, B.; Zhou, M. High glucose-induced ros production stimulates proliferation of pancreatic cancer via inactivating the jnk pathway. Oxid. Med. Cell Longev. 2018, 2018, 6917206. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.A.; Harwood, S.; Varagunam, M.; Raftery, M.J.; Yaqoob, M.M. High glucose-induced oxidative stress causes apoptosis in proximal tubular epithelial cells and is mediated by multiple caspases. FASEB J. 2003, 17, 908–910. [Google Scholar] [CrossRef]
- Ho, F.M.; Lin, W.W.; Chen, B.C.; Chao, C.M.; Yang, C.R.; Lin, L.Y.; Lai, C.C.; Liu, S.H.; Liau, C.S. High glucose-induced apoptosis in human vascular endothelial cells is mediated through nf-kappab and c-jun nh2-terminal kinase pathway and prevented by pi3k/akt/enos pathway. Cell Signal. 2006, 18, 391–399. [Google Scholar] [CrossRef]
- Vaughn, A.E.; Deshmukh, M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat. Cell Biol. 2008, 10, 1477–1483. [Google Scholar] [CrossRef]
- Rudlowski, C.; Moser, M.; Becker, A.J.; Rath, W.; Buttner, R.; Schroder, W.; Schurmann, A. Glut1 mrna and protein expression in ovarian borderline tumors and cancer. Oncology 2004, 66, 404–410. [Google Scholar] [CrossRef]
- Walenta, S.; Mueller-Klieser, W.F. Lactate: Mirror and motor of tumor malignancy. Semin. Radiat. Oncol. 2004, 14, 267–274. [Google Scholar] [CrossRef]
- Kim, J.W.; Dang, C.V. Cancer’s molecular sweet tooth and the warburg effect. Cancer Res. 2006, 66, 8927–8930. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, H. Cellular life span and the warburg effect. Exp. Cell Res. 2008, 314, 1923–1928. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Regulation of mammalian o2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 1999, 15, 551–578. [Google Scholar] [CrossRef]
- Lee, J.W.; Bae, S.H.; Jeong, J.W.; Kim, S.H.; Kim, K.W. Hypoxia-inducible factor (hif-1)alpha: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar] [CrossRef]
- Catrina, S.B.; Okamoto, K.; Pereira, T.; Brismar, K.; Poellinger, L. Hyperglycemia regulates hypoxia-inducible factor-1alpha protein stability and function. Diabetes 2004, 53, 3226–3232. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhou, B.P. New insights of epithelial-mesenchymal transition in cancer metastasis. Acta Biochim. Biophys. Sin. 2008, 40, 643–650. [Google Scholar] [CrossRef]
- Kauppi, J.; Gockel, I.; Rantanen, T.; Hansen, T.; Ristimaki, A.; Lang, H.; Sihvo, E.; Rasanen, J.; Junginger, T.; Salo, J.A. Cause of death during long-term follow-up for superficial esophageal adenocarcinoma. Ann. Surg. Oncol. 2013, 20, 2428–2433. [Google Scholar] [CrossRef]
- Walters, S.; Maringe, C.; Coleman, M.P.; Peake, M.D.; Butler, J.; Young, N.; Bergstrom, S.; Hanna, L.; Jakobsen, E.; Kolbeck, K.; et al. Lung cancer survival and stage at diagnosis in australia, canada, denmark, norway, sweden and the uk: A population-based study, 2004–2007. Thorax 2013, 68, 551–564. [Google Scholar] [CrossRef]
- Walters, S.; Maringe, C.; Butler, J.; Rachet, B.; Barrett-Lee, P.; Bergh, J.; Boyages, J.; Christiansen, P.; Lee, M.; Warnberg, F.; et al. Breast cancer survival and stage at diagnosis in australia, canada, denmark, norway, sweden and the uk, 2000–2007: A population-based study. Br. J. Cancer 2013, 108, 1195–1208. [Google Scholar] [CrossRef]
- Li, W.; Liu, H.; Qian, W.; Cheng, L.; Yan, B.; Han, L.; Xu, Q.; Ma, Q.; Ma, J. Hyperglycemia aggravates microenvironment hypoxia and promotes the metastatic ability of pancreatic cancer. Comput. Struct. Biotechnol. J. 2018, 16, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ma, Q.; Li, J.; Guo, K.; Liu, H.; Han, L.; Ma, G. Hyperglycemia enhances the invasive and migratory activity of pancreatic cancer cells via hydrogen peroxide. Oncol. Rep. 2011, 25, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ma, Q.; Liu, J.; Han, L.; Ma, G.; Liu, H.; Shan, T.; Xie, K.; Wu, E. Hyperglycemia as a mechanism of pancreatic cancer metastasis. FBL 2012, 17, 1761–1774. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Kong, F.; Wu, X.; Ren, Y.; Wu, S.; Wu, K.; Jiang, Z.; Zhang, W. High glucose promotes tumor invasion and increases metastasis-associated protein expression in human lung epithelial cells by upregulating heme oxygenase-1 via reactive oxygen species or the tgf-beta1/pi3k/akt signaling pathway. Cell Physiol. Biochem. 2015, 35, 1008–1022. [Google Scholar] [CrossRef]
- Pickup, M.W.; Owens, P.; Gorska, A.E.; Chytil, A.; Ye, F.; Shi, C.; Weaver, V.M.; Kalluri, R.; Moses, H.L.; Novitskiy, S.V. Development of aggressive pancreatic ductal adenocarcinomas depends on granulocyte colony stimulating factor secretion in carcinoma cells. Cancer Immunol. Res. 2017, 5, 718–729. [Google Scholar] [CrossRef]
- Gallagher, E.J.; LeRoith, D. Hyperinsulinaemia in cancer. Nat. Rev. Cancer 2020, 20, 629–644. [Google Scholar] [CrossRef]
- Gunter, M.J.; Xie, X.; Xue, X.; Kabat, G.C.; Rohan, T.E.; Wassertheil-Smoller, S.; Ho, G.Y.; Wylie-Rosett, J.; Greco, T.; Yu, H.; et al. Breast cancer risk in metabolically healthy but overweight postmenopausal women. Cancer Res. 2015, 75, 270–274. [Google Scholar] [CrossRef]
- Hirose, K.; Hamajima, N.; Takezaki, T.; Miura, S.; Tajima, K. Physical exercise reduces risk of breast cancer in japanese women. Cancer Sci. 2003, 94, 193–199. [Google Scholar] [CrossRef]
- Ma, J.; Giovannucci, E.; Pollak, M.; Leavitt, A.; Tao, Y.; Gaziano, J.M.; Stampfer, M.J. A prospective study of plasma c-peptide and colorectal cancer risk in men. J. Natl. Cancer Inst. 2004, 96, 546–553. [Google Scholar] [CrossRef]
- Hammarsten, J.; Hogstedt, B. Hyperinsulinaemia: A prospective risk factor for lethal clinical prostate cancer. Eur. J. Cancer 2005, 41, 2887–2895. [Google Scholar] [CrossRef]
- Gunter, M.J.; Hoover, D.R.; Yu, H.; Wassertheil-Smoller, S.; Manson, J.E.; Li, J.; Harris, T.G.; Rohan, T.E.; Xue, X.; Ho, G.Y.; et al. A prospective evaluation of insulin and insulin-like growth factor-i as risk factors for endometrial cancer. Cancer Epidemiol. Biomark. Prev. 2008, 17, 921–929. [Google Scholar] [CrossRef]
- Loftfield, E.; Freedman, N.D.; Lai, G.Y.; Weinstein, S.J.; McGlynn, K.A.; Taylor, P.R.; Mannisto, S.; Albanes, D.; Stolzenberg-Solomon, R.Z. Higher glucose and insulin levels are associated with risk of liver cancer and chronic liver disease mortality among men without a history of diabetes. Cancer Prev. Res. 2016, 9, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Walraven, I.; van’t Riet, E.; Stehouwer, C.D.; Polak, B.C.; Moll, A.C.; Dekker, J.M.; Nijpels, G. Fasting proinsulin levels are significantly associated with 20 year cancer mortality rates. The hoorn study. Diabetologia 2013, 56, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Frasca, F.; Pandini, G.; Sciacca, L.; Pezzino, V.; Squatrito, S.; Belfiore, A.; Vigneri, R. The role of insulin receptors and igf-i receptors in cancer and other diseases. Arch. Physiol. Biochem. 2008, 114, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Baxter, R.C.; Bryson, J.M.; Turtle, J.R. Somatogenic receptors of rat liver: Regulation by insulin. Endocrinology 1980, 107, 1176–1181. [Google Scholar] [CrossRef] [PubMed]
- Le, T.K.C.; Dao, X.D.; Nguyen, D.V.; Luu, D.H.; Bui, T.M.H.; Le, T.H.; Nguyen, H.T.; Le, T.N.; Hosaka, T.; Nguyen, T.T.T. Insulin signaling and its application. Front. Endocrinol. 2023, 14, 1226655. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Gallagher, E.J.; LeRoith, D. The proliferating role of insulin and insulin-like growth factors in cancer. Trends Endocrinol. Metab. 2010, 21, 610–618. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef]
- Savova, M.S.; Mihaylova, L.V.; Tews, D.; Wabitsch, M.; Georgiev, M.I. Targeting pi3k/akt signaling pathway in obesity. Biomed. Pharmacother. 2023, 159, 114244. [Google Scholar] [CrossRef]
- Tsay, A.; Wang, J.C. The role of pik3r1 in metabolic function and insulin sensitivity. Int. J. Mol. Sci. 2023, 24, 12665. [Google Scholar] [CrossRef] [PubMed]
- Brachmann, S.M.; Ueki, K.; Engelman, J.A.; Kahn, R.C.; Cantley, L.C. Phosphoinositide 3-kinase catalytic subunit deletion and regulatory subunit deletion have opposite effects on insulin sensitivity in mice. Mol. Cell. Biol. 2005, 25, 1596–1607. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.F.; Young, P.W.; Yonezawa, K.; Kasuga, M.; Holman, G.D. Inhibition of the translocation of glut1 and glut4 in 3t3-l1 cells by the phosphatidylinositol 3-kinase inhibitor, wortmannin. Biochem. J. 1994, 300 Pt 3, 631–635. [Google Scholar] [CrossRef]
- Asano, T.; Fujishiro, M.; Kushiyama, A.; Nakatsu, Y.; Yoneda, M.; Kamata, H.; Sakoda, H. Role of phosphatidylinositol 3-kinase activation on insulin action and its alteration in diabetic conditions. Biol. Pharm. Bull. 2007, 30, 1610–1616. [Google Scholar] [CrossRef]
- Carracedo, A.; Pandolfi, P.P. The pten-pi3k pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef] [PubMed]
- Stokoe, D.; Stephens, L.R.; Copeland, T.; Gaffney, P.R.; Reese, C.B.; Painter, G.F.; Holmes, A.B.; McCormick, F.; Hawkins, P.T. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase b. Science 1997, 277, 567–570. [Google Scholar] [CrossRef]
- Miinea, C.P.; Sano, H.; Kane, S.; Sano, E.; Fukuda, M.; Peranen, J.; Lane, W.S.; Lienhard, G.E. As160, the akt substrate regulating glut4 translocation, has a functional rab gtpase-activating protein domain. Biochem. J. 2005, 391, 87–93. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. Akt/pkb signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Okoro, D.R.; Rosso, M.; Bargonetti, J. Splicing up mdm2 for cancer proteome diversity. Genes Cancer 2012, 3, 311–319. [Google Scholar] [CrossRef]
- Zafar, A.; Khan, M.J.; Naeem, A. Mdm2- an indispensable player in tumorigenesis. Mol. Biol. Rep. 2023, 50, 6871–6883. [Google Scholar] [CrossRef]
- Cheng, X.; Xia, W.; Yang, J.Y.; Hsu, J.L.; Lang, J.Y.; Chou, C.K.; Du, Y.; Sun, H.L.; Wyszomierski, S.L.; Mills, G.B.; et al. Activation of murine double minute 2 by akt in mammary epithelium delays mammary involution and accelerates mammary tumorigenesis. Cancer Res. 2010, 70, 7684–7689. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Trajkovic-Arsic, M.; Kalideris, E.; Siveke, J.T. The role of insulin and igf system in pancreatic cancer. J. Mol. Endocrinol. 2013, 50, R67–R74. [Google Scholar] [CrossRef]
- Draznin, B. Mitogenic action of insulin: Friend, foe or ‘frenemy’? Diabetologia 2010, 53, 229–233. [Google Scholar] [CrossRef]
- Bedinger, D.H.; Adams, S.H. Metabolic, anabolic, and mitogenic insulin responses: A tissue-specific perspective for insulin receptor activators. Mol. Cell Endocrinol. 2015, 415, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Padma, V.V. An overview of targeted cancer therapy. Biomedicine 2015, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Huemer, F.; Bartsch, R.; Gnant, M. The pi3k/akt/mtor signaling pathway: The roleof pi3k and akt inhibitors in breast cancerf. Curr. Breast Cancer Rep. 2014, 6, 59–70. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific pi3k signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef]
- Alzahrani, A.S. Pi3k/akt/mtor inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef]
- Courtney, K.D.; Corcoran, R.B.; Engelman, J.A. The pi3k pathway as drug target in human cancer. J. Clin. Oncol. 2010, 28, 1075–1083. [Google Scholar] [CrossRef]
- Pons-Tostivint, E.; Thibault, B.; Guillermet-Guibert, J. Targeting pi3k signaling in combination cancer therapy. Trends Cancer 2017, 3, 454–469. [Google Scholar] [CrossRef]
- Akinleye, A.; Avvaru, P.; Furqan, M.; Song, Y.; Liu, D. Phosphatidylinositol 3-kinase (pi3k) inhibitors as cancer therapeutics. J. Hematol. Oncol. 2013, 6, 88. [Google Scholar] [CrossRef] [PubMed]
- Pirali, T.; Ciraolo, E.; Aprile, S.; Massarotti, A.; Berndt, A.; Griglio, A.; Serafini, M.; Mercalli, V.; Landoni, C.; Campa, C.C.; et al. Identification of a potent phosphoinositide 3-kinase pan inhibitor displaying a strategic carboxylic acid group and development of its prodrugs. ChemMedChem 2017, 12, 1542–1554. [Google Scholar] [CrossRef]
- Ando, Y.; Iwasa, S.; Takahashi, S.; Saka, H.; Kakizume, T.; Natsume, K.; Suenaga, N.; Quadt, C.; Yamada, Y. Phase i study of alpelisib (byl719), an alpha-specific pi3k inhibitor, in japanese patients with advanced solid tumors. Cancer Sci. 2019, 110, 1021–1031. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. Pi3k and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef]
- Fritsch, C.; Huang, A.; Chatenay-Rivauday, C.; Schnell, C.; Reddy, A.; Liu, M.; Kauffmann, A.; Guthy, D.; Erdmann, D.; De Pover, A.; et al. Characterization of the novel and specific pi3kalpha inhibitor nvp-byl719 and development of the patient stratification strategy for clinical trials. Mol. Cancer Ther. 2014, 13, 1117–1129. [Google Scholar] [CrossRef]
- Juric, D.; Rodon, J.; Tabernero, J.; Janku, F.; Burris, H.A.; Schellens, J.H.M.; Middleton, M.R.; Berlin, J.; Schuler, M.; Gil-Martin, M.; et al. Phosphatidylinositol 3-kinase alpha-selective inhibition with alpelisib (byl719) in pik3ca-altered solid tumors: Results from the first-in-human study. J. Clin. Oncol. 2018, 36, 1291–1299. [Google Scholar] [CrossRef]
- Lampson, B.L.; Brown, J.R. Pi3kdelta-selective and pi3kalpha/delta-combinatorial inhibitors in clinical development for b-cell non-hodgkin lymphoma. Expert. Opin. Investig. Drugs 2017, 26, 1267–1279. [Google Scholar] [CrossRef]
- Gayathri Nagaraj, C.X.M.; Luo, J.; Ellis, M.J. A phase i study of bkm120, a novel oral selective phosphatidylinositol-3-kinase (pi3k) inhibitor, in combination with fulvestrant in postmenopausal women with estrogen receptor positive metastatic breast cancer. ASCO Annual Meeting I. J. Clin. Oncol. 2012, 30 (Suppl. S15), TPS664. [Google Scholar] [CrossRef]
- Gopal, A.K.; Kahl, B.S.; de Vos, S.; Wagner-Johnston, N.D.; Schuster, S.J.; Jurczak, W.J.; Flinn, I.W.; Flowers, C.R.; Martin, P.; Viardot, A.; et al. Pi3kdelta inhibition by idelalisib in patients with relapsed indolent lymphoma. N. Engl. J. Med. 2014, 370, 1008–1018. [Google Scholar] [CrossRef]
- Herman, S.E.; Gordon, A.L.; Wagner, A.J.; Heerema, N.A.; Zhao, W.; Flynn, J.M.; Jones, J.; Andritsos, L.; Puri, K.D.; Lannutti, B.J.; et al. Phosphatidylinositol 3-kinase-delta inhibitor cal-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood 2010, 116, 2078–2088. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting pi3k in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed]
- FDA News. FDA Approves New Treatment for Adults with Relapsed Follicular Lymphoma. 2017. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-adults-relapsed-follicular-lymphoma (accessed on 23 July 2024).
- FDA News. Center for Drug Evaluation and Research. Duvelisib (Copiktra, Verastem, Inc.) for Adult Patients with Relapsed. U.S. Food and Drug Administration. 2018. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/duvelisib-copiktra-verastem-inc-adult-patients-relapsed-or-refractory-chronic-lymphocytic-leukemia (accessed on 23 July 2024).
- Juric, D.; Castel, P.; Griffith, M.; Griffith, O.L.; Won, H.H.; Ellis, H.; Ebbesen, S.H.; Ainscough, B.J.; Ramu, A.; Iyer, G.; et al. Convergent loss of pten leads to clinical resistance to a pi(3)kalpha inhibitor. Nature 2015, 518, 240–244. [Google Scholar] [CrossRef]
- Haddadi, N.; Lin, Y.; Travis, G.; Simpson, A.M.; Nassif, N.T.; McGowan, E.M. Pten/ptenp1: ‘Regulating the regulator of rtk-dependent pi3k/akt signalling’, new targets for cancer therapy. Mol. Cancer 2018, 17, 37. [Google Scholar] [CrossRef] [PubMed]
- Aziz, S.A.; Jilaveanu, L.B.; Zito, C.; Camp, R.L.; Rimm, D.L.; Conrad, P.; Kluger, H.M. Vertical targeting of the phosphatidylinositol-3 kinase pathway as a strategy for treating melanoma. Clin. Cancer Res. 2010, 16, 6029–6039. [Google Scholar] [CrossRef]
- Calero, R.; Morchon, E.; Martinez-Argudo, I.; Serrano, R. Synergistic anti-tumor effect of 17aag with the pi3k/mtor inhibitor nvp-bez235 on human melanoma. Cancer Lett. 2017, 406, 1–11. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, Q.; He, X.; Romigh, T.; Altemus, J.; Eng, C. Activation of ar sensitizes breast carcinomas to nvp-bez235’s therapeutic effect mediated by pten and klln upregulation. Mol. Cancer Ther. 2014, 13, 517–527. [Google Scholar] [CrossRef]
- Martinelli, E.; Troiani, T.; D’Aiuto, E.; Morgillo, F.; Vitagliano, D.; Capasso, A.; Costantino, S.; Ciuffreda, L.P.; Merolla, F.; Vecchione, L.; et al. Antitumor activity of pimasertib, a selective mek 1/2 inhibitor, in combination with pi3k/mtor inhibitors or with multi-targeted kinase inhibitors in pimasertib-resistant human lung and colorectal cancer cells. Int. J. Cancer 2013, 133, 2089–2101. [Google Scholar] [CrossRef]
- Yang, X.; Niu, B.; Wang, L.; Chen, M.; Kang, X.; Wang, L.; Ji, Y.; Zhong, J. Autophagy inhibition enhances colorectal cancer apoptosis induced by dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor nvp-bez235. Oncol. Lett. 2016, 12, 102–106. [Google Scholar] [CrossRef]
- Cho, D.C.; Cohen, M.B.; Panka, D.J.; Collins, M.; Ghebremichael, M.; Atkins, M.B.; Signoretti, S.; Mier, J.W. The efficacy of the novel dual pi3-kinase/mtor inhibitor nvp-bez235 compared with rapamycin in renal cell carcinoma. Clin. Cancer Res. 2010, 16, 3628–3638. [Google Scholar] [CrossRef]
- Cheng, H.; Li, C.; Bailey, S.; Baxi, S.M.; Goulet, L.; Guo, L.; Hoffman, J.; Jiang, Y.; Johnson, T.O.; Johnson, T.W.; et al. Discovery of the highly potent pi3k/mtor dual inhibitor pf-04979064 through structure-based drug design. ACS Med. Chem. Lett. 2013, 4, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Mehta, P.P.; Yin, M.J.; Sun, S.; Zou, A.; Chen, J.; Rafidi, K.; Feng, Z.; Nickel, J.; Engebretsen, J.; et al. Pf-04691502, a potent and selective oral inhibitor of pi3k and mtor kinases with antitumor activity. Mol. Cancer Ther. 2011, 10, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Mallon, R.; Feldberg, L.R.; Lucas, J.; Chaudhary, I.; Dehnhardt, C.; Santos, E.D.; Chen, Z.; Santos, O.D.; Ayral-Kaloustian, S.; Venkatesan, A.; et al. Antitumor efficacy of pki-587, a highly potent dual pi3k/mtor kinase inhibitor. Clin. Cancer Res. 2011, 17, 3193–3203. [Google Scholar] [CrossRef]
- Colombo, I.; Genta, S.; Martorana, F.; Guidi, M.; Frattini, M.; Samartzis, E.P.; Brandt, S.; Gaggetta, S.; Moser, L.; Pascale, M.; et al. Phase i dose-escalation study of the dual pi3k-mtorc1/2 inhibitor gedatolisib in combination with paclitaxel and carboplatin in patients with advanced solid tumors. Clin. Cancer Res. 2021, 27, 5012–5019. [Google Scholar] [CrossRef]
- Herzog, A.; Bian, Y.; Broek, R.V.; Hall, B.; Coupar, J.; Cheng, H.; Sowers, A.L.; Cook, J.D.; Mitchell, J.B.; Chen, Z.; et al. Pi3k/mtor inhibitor pf-04691502 antitumor activity is enhanced with induction of wild-type tp53 in human xenograft and murine knockout models of head and neck cancer. Clin. Cancer Res. 2013, 19, 3808–3819. [Google Scholar] [CrossRef]
- Soares, H.P.; Ming, M.; Mellon, M.; Young, S.H.; Han, L.; Sinnet-Smith, J.; Rozengurt, E. Dual pi3k/mtor inhibitors induce rapid overactivation of the mek/erk pathway in human pancreatic cancer cells through suppression of mtorc2. Mol. Cancer Ther. 2015, 14, 1014–1023. [Google Scholar] [CrossRef]
- Freitag, H.; Christen, F.; Lewens, F.; Grass, I.; Briest, F.; Iwaszkiewicz, S.; Siegmund, B.; Grabowski, P. Inhibition of mtor’s catalytic site by pki-587 is a promising therapeutic option for gastroenteropancreatic neuroendocrine tumor disease. Neuroendocrinology 2017, 105, 90–104. [Google Scholar] [CrossRef] [PubMed]
- Shariff, A.I.; Syed, S.; Shelby, R.A.; Force, J.; Clarke, J.M.; D’Alessio, D.; Corsino, L. Novel cancer therapies and their association with diabetes. J. Mol. Endocrinol. 2019, 62, R187–R199. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R.; et al. Suppression of insulin feedback enhances the efficacy of pi3k inhibitors. Nature 2018, 560, 499–503. [Google Scholar] [CrossRef]
- Ewa Świderska, J.S.; Wróblewski, A.; Szemraj, J.; Drzewoski, J.; Śliwińska, A. Role of pi3k/akt pathway in insulin-mediated glucose uptake. In Blood Glucose Levels; Books on Demand: Norderstedt, Germany, 2018. [Google Scholar]
- Goncalves, M.D.; Hopkins, B.D.; Cantley, L.C. Phosphatidylinositol 3-kinase, growth disorders, and cancer. N. Engl. J. Med. 2018, 379, 2052–2062. [Google Scholar] [CrossRef]
- Nair, K.S.; Cheson, B. The role of idelalisib in the treatment of relapsed and refractory chronic lymphocytic leukemia. Ther. Adv. Hematol. 2016, 7, 69–84. [Google Scholar] [CrossRef]
- Staal, S.P. Molecular cloning of the akt oncogene and its human homologues akt1 and akt2: Amplification of akt1 in a primary human gastric adenocarcinoma. Proc. Natl. Acad. Sci. USA 1987, 84, 5034–5037. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting pi3k/akt signal transduction for cancer therapy. Signal. Transduct. Target Ther. 2021, 6, 425. [Google Scholar] [CrossRef] [PubMed]
- Nunnery, S.E.; Mayer, I.A. Management of toxicity to isoform alpha-specific pi3k inhibitors. Ann. Oncol. 2019, 30 (Suppl. S10), x21–x26. [Google Scholar] [CrossRef]
- West, K.A.; Castillo, S.S.; Dennis, P.A. Activation of the pi3k/akt pathway and chemotherapeutic resistance. Drug Resist. Update 2002, 5, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.M.; Kunnimalaiyaan, S.; Gamblin, T.C.; Kunnimalaiyaan, M. Mk2206 inhibits hepatocellular carcinoma cellular proliferation via induction of apoptosis and cell cycle arrest. J. Surg. Res. 2014, 191, 280–285. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V. Development of pi3k/akt/mtor pathway inhibitors and their application in personalized therapy for non-small-cell lung cancer. J. Thorac. Oncol. 2012, 7, 1315–1326. [Google Scholar] [CrossRef]
- Hirai, H.; Sootome, H.; Nakatsuru, Y.; Miyama, K.; Taguchi, S.; Tsujioka, K.; Ueno, Y.; Hatch, H.; Majumder, P.K.; Pan, B.S.; et al. Mk-2206, an allosteric akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 2010, 9, 1956–1967. [Google Scholar] [CrossRef]
- Konopleva, M.Y.; Walter, R.B.; Faderl, S.H.; Jabbour, E.J.; Zeng, Z.; Borthakur, G.; Huang, X.; Kadia, T.M.; Ruvolo, P.P.; Feliu, J.B.; et al. Preclinical and early clinical evaluation of the oral akt inhibitor, mk-2206, for the treatment of acute myelogenous leukemia. Clin. Cancer Res. 2014, 20, 2226–2235. [Google Scholar] [CrossRef]
- Yap, T.A.; Yan, L.; Patnaik, A.; Fearen, I.; Olmos, D.; Papadopoulos, K.; Baird, R.D.; Delgado, L.; Taylor, A.; Lupinacci, L.; et al. First-in-man clinical trial of the oral pan-akt inhibitor mk-2206 in patients with advanced solid tumors. J. Clin. Oncol. 2011, 29, 4688–4695. [Google Scholar] [CrossRef]
- Davies, B.R.; Greenwood, H.; Dudley, P.; Crafter, C.; Yu, D.H.; Zhang, J.; Li, J.; Gao, B.; Ji, Q.; Maynard, J.; et al. Preclinical pharmacology of azd5363, an inhibitor of akt: Pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol. Cancer Ther. 2012, 11, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zheng, Y.; Faheem, A.; Sun, T.; Li, C.; Li, Z.; Zhao, D.; Wu, C.; Liu, J. A novel akt inhibitor, azd5363, inhibits phosphorylation of akt downstream molecules, and activates phosphorylation of mtor and smg-1 dependent on the liver cancer cell type. Oncol. Lett. 2016, 11, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Davies, B.R.; Han, S.; Zhou, M.; Bai, Y.; Zhang, J.; Xu, Y.; Tang, L.; Wang, H.; Liu, Y.J.; et al. The akt inhibitor azd5363 is selectively active in pi3kca mutant gastric cancer, and sensitizes a patient-derived gastric cancer xenograft model with pten loss to taxotere. J. Transl. Med. 2013, 11, 241. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.C.; Liu, Y.; Jacobs, R.; Rider, M.H. A novel pkb/akt inhibitor, mk-2206, effectively inhibits insulin-stimulated glucose metabolism and protein synthesis in isolated rat skeletal muscle. Biochem. J. 2012, 447, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. Mtor signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Popova, N.V.; Jucker, M. The role of mtor signaling as a therapeutic target in cancer. Int. J. Mol. Sci. 2021, 22, 1743. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, F.; Zou, S.; Qu, L. Rapamycin: A bacteria-derived immunosuppressant that has anti-atherosclerotic effects and its clinical application. Front. Pharmacol. 2018, 9, 1520. [Google Scholar] [CrossRef]
- Edwards, S.R.; Wandless, T.J. The rapamycin-binding domain of the protein kinase mammalian target of rapamycin is a destabilizing domain. J. Biol. Chem. 2007, 282, 13395–13401. [Google Scholar] [CrossRef]
- Stanfel, M.N.; Shamieh, L.S.; Kaeberlein, M.; Kennedy, B.K. The tor pathway comes of age. Biochim. Biophys. Acta 2009, 1790, 1067–1074. [Google Scholar] [CrossRef]
- de Braud, F.; Machiels, J.H.; Boggiani, D.; Rottey, S.W.H.; Duca, M.; Laruelle, M.; Salvagni, S.; Damian, S.; Lapeire, L.D.F.; Tiseo, M.; et al. A phase 1 study of mtorc1/2 inhibitor bi 860585 as a single agent or with exemestane or paclitaxel in patients with advanced solid tumors. Cancers 2020, 12, 1425. [Google Scholar] [CrossRef]
- Capelan, M.; Kumar, P.; Tolcher, A.; Zivi, A.W.; Desai, M.; Papadopoulos, K.P.; Senaldi, P.; Patnaik, A.; Banerji, U.; Rasco, D.D. Abstract C173: A first-in-human phase I study of DS-3078A, an oral torc1/2 inhibitor, in patients with advanced solid tumors: Preliminary results. Mol. Cancer Ther. 2013, 12 (Suppl. S11), C173. [Google Scholar] [CrossRef]
- Yang, H.; Zhao, J.; Zhao, M.; Zhao, L.; Zhou, L.N.; Duan, Y.; Li, G. Gdc-0349 inhibits non-small cell lung cancer cell growth. Cell Death Dis. 2020, 11, 951. [Google Scholar] [CrossRef] [PubMed]
- Voss, M.H.; Gordon, M.S.; Mita, M.; Rini, B.; Makker, V.; Macarulla, T.; Smith, D.C.; Cervantes, A.; Puzanov, I.; Pili, R.; et al. Phase 1 study of mtorc1/2 inhibitor sapanisertib (tak-228) in advanced solid tumours, with an expansion phase in renal, endometrial or bladder cancer. Br. J. Cancer 2020, 123, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Wolin, E.; Mita, A.; Mahipal, A.; Meyer, T.; Bendell, J.; Nemunaitis, J.; Munster, P.N.; Paz-Ares, L.; Filvaroff, E.H.; Li, S.; et al. A phase 2 study of an oral mtorc1/mtorc2 kinase inhibitor (cc-223) for non-pancreatic neuroendocrine tumors with or without carcinoid symptoms. PLoS ONE 2019, 14, e0221994. [Google Scholar] [CrossRef] [PubMed]
- Chresta, C.M.; Davies, B.R.; Hickson, I.; Harding, T.; Cosulich, S.; Critchlow, S.E.; Vincent, J.P.; Ellston, R.; Jones, D.; Sini, P.; et al. Azd8055 is a potent, selective, and orally bioavailable atp-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010, 70, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Murugan, A.K.; Liu, R.; Xing, M. Identification and characterization of two novel oncogenic mtor mutations. Oncogene 2019, 38, 5211–5226. [Google Scholar] [CrossRef]
- Kwiatkowski, D.J.; Choueiri, T.K.; Fay, A.P.; Rini, B.I.; Thorner, A.R.; de Velasco, G.; Tyburczy, M.E.; Hamieh, L.; Albiges, L.; Agarwal, N.; et al. Mutations in tsc1, tsc2, and mtor are associated with response to rapalogs in patients with metastatic renal cell carcinoma. Clin. Cancer Res. 2016, 22, 2445–2452. [Google Scholar] [CrossRef]
- Wagle, N.; Grabiner, B.C.; Van Allen, E.M.; Amin-Mansour, A.; Taylor-Weiner, A.; Rosenberg, M.; Gray, N.; Barletta, J.A.; Guo, Y.; Swanson, S.J.; et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N. Engl. J. Med. 2014, 371, 1426–1433. [Google Scholar] [CrossRef]
- Murugan, A.K.; Humudh, E.A.; Qasem, E.; Al-Hindi, H.; Almohanna, M.; Hassan, Z.K.; Alzahrani, A.S. Absence of somatic mutations of the mtor gene in differentiated thyroid cancer. Meta Gene 2015, 6, 69–71. [Google Scholar] [CrossRef]
- Hassan, B.; Akcakanat, A.; Sangai, T.; Evans, K.W.; Adkins, F.; Eterovic, A.K.; Zhao, H.; Chen, K.; Chen, H.; Do, K.A.; et al. Catalytic mtor inhibitors can overcome intrinsic and acquired resistance to allosteric mtor inhibitors. Oncotarget 2014, 5, 8544–8557. [Google Scholar] [CrossRef]
- Bahar, M.E.; Kim, H.J.; Kim, D.R. Targeting the ras/raf/mapk pathway for cancer therapy: From mechanism to clinical studies. Signal. Transduct. Target Ther. 2023, 8, 455. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, Z.; Luo, T.; Shi, H. Targeting the pi3k/akt/mtor and raf/mek/erk pathways for cancer therapy. Mol. Biomed. 2022, 3, 47. [Google Scholar] [CrossRef]
- Verges, B.; Cariou, B. Mtor inhibitors and diabetes. Diabetes Res. Clin. Pract. 2015, 110, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Fraenkel, M.; Ketzinel-Gilad, M.; Ariav, Y.; Pappo, O.; Karaca, M.; Castel, J.; Berthault, M.F.; Magnan, C.; Cerasi, E.; Kaiser, N.; et al. Mtor inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes 2008, 57, 945–957. [Google Scholar] [CrossRef]
- Yim, C.; Mansell, K.; Hussein, N.; Arnason, T. Current cancer therapies and their influence on glucose control. World J. Diabetes 2021, 12, 1010–1025. [Google Scholar] [CrossRef]
- Kleinert, M.; Sylow, L.; Fazakerley, D.J.; Krycer, J.R.; Thomas, K.C.; Oxboll, A.J.; Jordy, A.B.; Jensen, T.E.; Yang, G.; Schjerling, P.; et al. Acute mtor inhibition induces insulin resistance and alters substrate utilization in vivo. Mol. Metab. 2014, 3, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Rachdi, L.; Balcazar, N.; Osorio-Duque, F.; Elghazi, L.; Weiss, A.; Gould, A.; Chang-Chen, K.J.; Gambello, M.J.; Bernal-Mizrachi, E. Disruption of tsc2 in pancreatic beta cells induces beta cell mass expansion and improved glucose tolerance in a torc1-dependent manner. Proc. Natl. Acad. Sci. USA 2008, 105, 9250–9255. [Google Scholar] [CrossRef]
- Asahara, S.I.; Inoue, H.; Watanabe, H.; Kido, Y. Roles of mtor in the regulation of pancreatic beta-cell mass and insulin secretion. Biomolecules 2022, 12, 614. [Google Scholar] [CrossRef]
- Dabrowski, M. Diabetes, antidiabetic medications and cancer risk in type 2 diabetes: Focus on sglt-2 inhibitors. Int. J. Mol. Sci. 2021, 22, 1680. [Google Scholar] [CrossRef]
- Soranna, D.; Scotti, L.; Zambon, A.; Bosetti, C.; Grassi, G.; Catapano, A.; La Vecchia, C.; Mancia, G.; Corrao, G. Cancer risk associated with use of metformin and sulfonylurea in type 2 diabetes: A meta-analysis. Oncologist 2012, 17, 813–822. [Google Scholar] [CrossRef]
- Noto, H.; Goto, A.; Tsujimoto, T.; Noda, M. Cancer risk in diabetic patients treated with metformin: A systematic review and meta-analysis. PLoS ONE 2012, 7, e33411. [Google Scholar] [CrossRef] [PubMed]
- Currie, C.J.; Poole, C.D.; Gale, E.A. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia 2009, 52, 1766–1777. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef]
- Nathan, D.M.; Buse, J.B.; Davidson, M.B.; Ferrannini, E.; Holman, R.R.; Sherwin, R.; Zinman, B. Medical management of hyperglycaemia in type 2 diabetes mellitus: A consensus algorithm for the initiation and adjustment of therapy: A consensus statement from the american diabetes association and the european association for the study of diabetes. Diabetologia 2009, 52, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.J. Metformin: Historical overview. Diabetologia 2017, 60, 1566–1576. [Google Scholar] [CrossRef]
- Quinn, B.J.; Kitagawa, H.; Memmott, R.M.; Gills, J.J.; Dennis, P.A. Repositioning metformin for cancer prevention and treatment. Trends Endocrinol. Metab. 2013, 24, 469–480. [Google Scholar] [CrossRef]
- Alimova, I.N.; Liu, B.; Fan, Z.; Edgerton, S.M.; Dillon, T.; Lind, S.E.; Thor, A.D. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle 2009, 8, 909–915. [Google Scholar] [CrossRef]
- Buzzai, M.; Jones, R.G.; Amaravadi, R.K.; Lum, J.J.; DeBerardinis, R.J.; Zhao, F.; Viollet, B.; Thompson, C.B. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007, 67, 6745–6752. [Google Scholar] [CrossRef] [PubMed]
- Noto, H. Systematic review of cancer risk in diabetes mellitus. Nihon Rinsho 2012, 70 (Suppl. S5), 542–545. [Google Scholar]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an amp kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef]
- Heckman-Stoddard, B.M.; DeCensi, A.; Sahasrabuddhe, V.V.; Ford, L.G. Repurposing metformin for the prevention of cancer and cancer recurrence. Diabetologia 2017, 60, 1639–1647. [Google Scholar] [CrossRef] [PubMed]
- Morgillo, F.; Sasso, F.C.; Della Corte, C.M.; Festino, L.; Manzo, A.; Martinelli, E.; Troiani, T.; Capuano, A.; Ciardiello, F. Metformin in lung cancer: Rationale for a combination therapy. Expert Opin. Investig. Drugs 2013, 22, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of amp-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Cerezo, M.; Tichet, M.; Abbe, P.; Ohanna, M.; Lehraiki, A.; Rouaud, F.; Allegra, M.; Giacchero, D.; Bahadoran, P.; Bertolotto, C.; et al. Metformin blocks melanoma invasion and metastasis development in ampk/p53-dependent manner. Mol. Cancer Ther. 2013, 12, 1605–1615. [Google Scholar] [CrossRef]
- Lv, Z.; Guo, Y. Metformin and its benefits for various diseases. Front. Endocrinol. 2020, 11, 191. [Google Scholar] [CrossRef]
- Abdul, M.; Hoosein, N. Expression and activity of potassium ion channels in human prostate cancer. Cancer Lett. 2002, 186, 99–105. [Google Scholar] [CrossRef]
- Algire, C.; Moiseeva, O.; Deschenes-Simard, X.; Amrein, L.; Petruccelli, L.; Birman, E.; Viollet, B.; Ferbeyre, G.; Pollak, M.N. Metformin reduces endogenous reactive oxygen species and associated DNA damage. Cancer Prev. Res. 2012, 5, 536–543. [Google Scholar] [CrossRef]
- Soccio, R.E.; Chen, E.R.; Lazar, M.A. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab. 2014, 20, 573–591. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, E.; Wahl, R. Chemotherapy and chemoprevention by thiazolidinediones. BioMed Res. Int. 2015, 2015, 845340. [Google Scholar] [CrossRef]
- Moumita Nath, S.N.; Choudhury, Y. The impact of thiazolidinediones on the risk for prostate cancer in patients with type 2 diabetes mellitus: A review and meta-analysis. Meta Gene 2021, 27, 100840. [Google Scholar] [CrossRef]
- Xiao, Y.; Yuan, T.; Yao, W.; Liao, K. 3t3-l1 adipocyte apoptosis induced by thiazolidinediones is peroxisome proliferator-activated receptor-gamma-dependent and mediated by the caspase-3-dependent apoptotic pathway. FEBS J. 2010, 277, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Wang, W.; Wang, H.; Zhu, Y.; Lei, C. Ppargamma activation serves as therapeutic strategy against bladder cancer via inhibiting pi3k-akt signaling pathway. BMC Cancer 2019, 19, 204. [Google Scholar] [CrossRef] [PubMed]
- Ciaramella, V.; Sasso, F.C.; Di Liello, R.; Corte, C.M.D.; Barra, G.; Viscardi, G.; Esposito, G.; Sparano, F.; Troiani, T.; Martinelli, E.; et al. Activity and molecular targets of pioglitazone via blockade of proliferation, invasiveness and bioenergetics in human nsclc. J. Exp. Clin. Cancer Res. 2019, 38, 178. [Google Scholar] [CrossRef] [PubMed]
- Sola, D.; Rossi, L.; Schianca, G.P.; Maffioli, P.; Bigliocca, M.; Mella, R.; Corliano, F.; Fra, G.P.; Bartoli, E.; Derosa, G. Sulfonylureas and their use in clinical practice. Arch. Med. Sci. 2015, 11, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Pasello, G.; Urso, L.; Conte, P.; Favaretto, A. Effects of sulfonylureas on tumor growth: A review of the literature. Oncologist 2013, 18, 1118–1125. [Google Scholar] [CrossRef]
- Payen, L.; Delugin, L.; Courtois, A.; Trinquart, Y.; Guillouzo, A.; Fardel, O. The sulphonylurea glibenclamide inhibits multidrug resistance protein (mrp1) activity in human lung cancer cells. Br. J. Pharmacol. 2001, 132, 778–784. [Google Scholar] [CrossRef]
- Mizuno, C.S.; Chittiboyina, A.G.; Kurtz, T.W.; Pershadsingh, H.A.; Avery, M.A. Type 2 diabetes and oral antihyperglycemic drugs. Curr. Med. Chem. 2008, 15, 61–74. [Google Scholar] [CrossRef]
- Ashcroft, F.M. Atp-sensitive potassium channelopathies: Focus on insulin secretion. J. Clin. Investig. 2005, 115, 2047–2058. [Google Scholar] [CrossRef]
- Nunez, M.; Medina, V.; Cricco, G.; Croci, M.; Cocca, C.; Rivera, E.; Bergoc, R.; Martin, G. Glibenclamide inhibits cell growth by inducing g0/g1 arrest in the human breast cancer cell line mda-mb-231. BMC Pharmacol. Toxicol. 2013, 14, 6. [Google Scholar] [CrossRef]
- Wondergem, R.; Cregan, M.; Strickler, L.; Miller, R.; Suttles, J. Membrane potassium channels and human bladder tumor cells: Ii. Growth properties. J. Membr. Biol. 1998, 161, 257–262. [Google Scholar] [CrossRef]
- Malhi, H.; Irani, A.N.; Rajvanshi, P.; Suadicani, S.O.; Spray, D.C.; McDonald, T.V.; Gupta, S. Katp channels regulate mitogenically induced proliferation in primary rat hepatocytes and human liver cell lines. Implications for liver growth control and potential therapeutic targeting. J. Biol. Chem. 2000, 275, 26050–26057. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.; Hunke, S. Atp-binding-cassette (abc) transport systems: Functional and structural aspects of the atp-hydrolyzing subunits/domains. FEMS Microbiol. Rev. 1998, 22, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.K.; Wang, Y.J.; Gupta, P.; Chen, Z.S. Multidrug resistance proteins (mrps) and cancer therapy. AAPS J. 2015, 17, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Lautier, D.; Canitrot, Y.; Deeley, R.G.; Cole, S.P. Multidrug resistance mediated by the multidrug resistance protein (mrp) gene. Biochem. Pharmacol. 1996, 52, 967–977. [Google Scholar] [CrossRef]
- Xu, K.; Sun, G.; Li, M.; Chen, H.; Zhang, Z.; Qian, X.; Li, P.; Xu, L.; Huang, W.; Wang, X. Glibenclamide targets sulfonylurea receptor 1 to inhibit p70s6k activity and upregulate klf4 expression to suppress non-small cell lung carcinoma. Mol. Cancer Ther. 2019, 18, 2085–2096. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Li, J.; Ding, J.; Wang, Z.; Duan, L.; Hu, G. Glibenclamide exerts an antitumor activity through reactive oxygen species-c-jun nh2-terminal kinase pathway in human gastric cancer cell line mgc-803. Biochem. Pharmacol. 2008, 76, 1705–1715. [Google Scholar] [CrossRef]
- Lin, Y.; Zhang, Y.; Wang, S.; Cao, L.; Zhao, R.; Ma, X.; Yang, Q.; Zhang, L.; Yang, Q. Pharmacological targets of sglt2 inhibition on prostate cancer mediated by circulating metabolites: A drug-target mendelian randomization study. Front. Pharmacol. 2024, 15, 1443045. [Google Scholar] [CrossRef]
- Wang, L.; Wang, W.; Kaelber, D.C.; Xu, R.; Berger, N.A. Glp-1 receptor agonists and colorectal cancer risk in drug-naive patients with type 2 diabetes, with and without overweight/obesity. JAMA Oncol. 2024, 10, 256–258. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Target | Inhibitor | Disease Condition | Sample Size | Clinical Phase | Clinical Trial ID * |
|---|---|---|---|---|---|
| PI3K | Idelalisib (CAL-101) | Non-Hodgkin’s Lymphoma | 125 | II | NCT01282424 |
| PI3K | Pictilisib (GDC-0941) | Non-Hodgkin’s Lymphoma | 60 | I | NCT00876122 |
| PI3K | BYL719 (Alpelisib) | Advanced Solid Malignancies | 221 | I | NCT01219699 |
| PI3K | Copanlisib (BAY80-6946) | Advanced Cancer | 57 | I | NCT00962611 |
| PI3K | Duvelisib (IPI-145) | Advanced Hematologic Malignancies | 210 | I | NCT01476657 |
| PI3K | Buparlisib (BKM120) | Relapsed or Refractory Non-Hodgkin Lymphoma | 7 | I | NCT01719250 |
| PI3K | Pilaralisib (XL147) | Endometrial Carcinoma | 67 | II | NCT01013324 |
| Dual PI3K/mTOR | BEZ235 | Advanced Solid Tumors | 33 | I | NCT01343498 |
| Dual PI3K/mTOR | PF-04691502 PF-05212384 | Recurrent Endometrial Cancer | 67 | II | NCT01420081 |
| Dual PI3K/mTOR | GDC-0980 | Refractory Solid Tumors or Non-Hodgkin’s Lymphoma | 121 | I | NCT00854152 |
| mTOR | AZD8055 | Gliomas | 22 | I | NCT01316809 |
| mTOR | Temsirolimus | Breast Neoplasms | 108 | II | NCT00062751 |
| mTOR | Sirolimus | Advanced Cancers | 40 | I | NCT00707135 |
| mTOR | BI860585 | Advanced and/or Metastatic Solid Tumors | 90 | I | NCT01938846 |
| mTOR | DS-3078a | Advanced Solid Tumors or Lymphomas | 32 | I | NCT01588678 |
| mTOR | GDC-0349 | Advanced or Metastatic Solid Tumors or Non-Hodgkin’s Lymphoma | 10 | I | NCT01356173 |
| mTOR | Sapanisertib (MLN0128) | Advanced Malignancies | 198 | I | NCT01058707 |
| mTOR | CC-223 | Advanced Solid Tumors, Non-Hodgkin Lymphoma, or Multiple Myeloma | 226 | I/II | NCT01177397 |
| mTOR | Everolimus | Advanced Solid Tumors | 30 | II | NCT02201212 |
| mTOR | Ridaforolimus | Refractory or Advanced Malignancies | 147 | I | NCT00112372 |
| mTOR | Deforolimus (AP23573) | Relapsed or Refractory Hematologic Malignancies | 57 | II | NCT00086125 |
| AKT | GSK690693 | Solid Tumors and Lymphoma | 70 | I | NCT00493818 |
| AKT | MK-2206 | Metastatic Neuroendocrine Tumors | 11 | II | NCT01169649 |
| AKT | AZD5363 | Advanced Solid Tumors | 12 | I | NCT03310541 |
| AKT | ARQ092 | Advanced Solid Tumors and Recurrent Malignant Lymphoma | 120 | I | NCT01473095 |
| AKT | BAY1125976 | Neoplasm | 79 | I | NCT01915576 |
| AKT | Perifosine | Recurrent/Progressive Malignant Gliomas | 32 | II | NCT00590954 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asiri, A.; Al Qarni, A.; Bakillah, A. The Interlinking Metabolic Association between Type 2 Diabetes Mellitus and Cancer: Molecular Mechanisms and Therapeutic Insights. Diagnostics 2024, 14, 2132. https://doi.org/10.3390/diagnostics14192132
Asiri A, Al Qarni A, Bakillah A. The Interlinking Metabolic Association between Type 2 Diabetes Mellitus and Cancer: Molecular Mechanisms and Therapeutic Insights. Diagnostics. 2024; 14(19):2132. https://doi.org/10.3390/diagnostics14192132
Chicago/Turabian StyleAsiri, Abutaleb, Ali Al Qarni, and Ahmed Bakillah. 2024. "The Interlinking Metabolic Association between Type 2 Diabetes Mellitus and Cancer: Molecular Mechanisms and Therapeutic Insights" Diagnostics 14, no. 19: 2132. https://doi.org/10.3390/diagnostics14192132