
Optical Genome Mapping for Chromosomal Aberrations Detection—False-Negative Results and Contributing Factors

Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjcts
2.2. Procedures
2.2.1. Optical Genome Mapping
2.2.2. Chromosomal Microarray Analysis
2.2.3. Other Classic Cytogenetics and Molecular Genetics Technology
3. Results
3.1. Cohort Description and Aberrations Missed by OGM
3.2. Quality Control Performance
3.3. Sample Type Analysis
3.4. Reference Genome Analysis
3.5. Effective Coverage Analysis
3.6. Type and Location analysis
3.6.1. CNVs
3.6.2. Balanced Translocations
3.6.3. Pericentric Inversion
3.6.4. Isochromosome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eichler, E.E. Genetic Variation, Comparative Genomics, and the Diagnosis of Disease. N. Engl. J. Med. 2019, 381, 64–74. [Google Scholar] [CrossRef]
- Sharp, A.J.; Locke, D.P.; McGrath, S.D.; Cheng, Z.; Bailey, J.A.; Vallente, R.U.; Pertz, L.M.; Clark, R.A.; Schwartz, S.; Segraves, R.; et al. Segmental duplications and copy-number variation in the human genome. Am. J. Hum. Genet. 2005, 77, 78–88. [Google Scholar] [CrossRef]
- Sahajpal, N.S.; Barseghyan, H.; Kolhe, R.; Hastie, A.; Chaubey, A. Optical Genome Mapping as a Next-Generation Cytogenomic Tool for Detection of Structural and Copy Number Variations for Prenatal Genomic Analyses. Genes 2021, 12, 398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, Y.; Xu, Y.; Zhou, R.; Huang, M.; Qiao, F.; Meng, L.; Liu, A.; Zhou, J.; Li, L.; et al. Optical genome mapping for detection of chromosomal aberrations in prenatal diagnosis. Acta Obstet. Gynecol. Scand. 2023, 102, 1053–1062. [Google Scholar] [CrossRef]
- Mantere, T.; Neveling, K.; Pebrel-Richard, C.; Benoist, M.; van der Zande, G.; Kater-Baats, E.; Baatout, I.; van Beek, R.; Yammine, T.; Oorsprong, M.; et al. Optical genome mapping enables constitutional chromosomal aberration detection. Am. J. Hum. Genet. 2021, 108, 1409–1422. [Google Scholar] [CrossRef] [PubMed]
- Sahajpal, N.S.; Mondal, A.K.; Fee, T.; Hilton, B.; Layman, L.; Hastie, A.R.; Chaubey, A.; DuPont, B.R.; Kolhe, R. Clinical Validation and Diagnostic Utility of Optical Genome Mapping in Prenatal Diagnostic Testing. J. Mol. Diagn. 2023, 25, 234–246. [Google Scholar] [CrossRef]
- Dremsek, P.; Schwarz, T.; Weil, B.; Malashka, A.; Laccone, F.; Neesen, J. Optical Genome Mapping in Routine Human Genetic Diagnostics-Its Advantages and Limitations. Genes 2021, 12, 1958. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.A.; Broeckel, U.; Levy, B.; Skinner, S.; Sahajpal, N.S.; Rodriguez, V.; Stence, A.; Awayda, K.; Scharer, G.; Skinner, C.; et al. Multisite Assessment of Optical Genome Mapping for Analysis of Structural Variants in Constitutional Postnatal Cases. J. Mol. Diagn. 2023, 25, 175–188. [Google Scholar] [CrossRef]
- Chan, S.; Lam, E.; Saghbini, M.; Bocklandt, S.; Hastie, A.; Cao, H.; Holmlin, E.; Borodkin, M. Structural Variation Detection and Analysis Using Bionano Optical Mapping. Methods Mol. Biol. 2018, 1833, 193–203. [Google Scholar]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. 2020, 22, 245–257, Erratum in Genet Med. 2021, 23, 2230. [Google Scholar] [CrossRef]
- Li, Y.; Chen, Y.; Zhou, R.; Sang, Z.; Meng, L.; Tan, J.; Qiao, F.; Bao, Q.; Luo, D.; Peng, C.; et al. Systematic analysis of copy-number variations associated with early pregnancy loss. Ultrasound Obstet. Gynecol. 2020, 55, 96–104. [Google Scholar]
- Bates, S.E. Classical cytogenetics: Karyotyping techniques. Methods Mol. Biol. 2011, 767, 177–190. [Google Scholar] [PubMed]
- Zhang, S.; Pei, Z.; Lei, C.; Zhu, S.; Deng, K.; Zhou, J.; Yang, J.; Lu, D.; Sun, X.; Xu, C.; et al. Detection of cryptic balanced chromosomal rearrangements using high-resolution optical genome mapping. J. Med. Genet. 2023, 60, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ma, D.; Liu, G.; Wang, Y.; Liu, A.; Li, L.; Luo, C.; Hu, P.; Xu, Z. Genetic analysis of 62 Chinese families with Duchenne muscular dystrophy and strategies of prenatal diagnosis in a single center. BMC Med. Genet. 2019, 20, 180. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Kim, M.J.; Yang, D.; Song, J.Y.; Im Cho, S.; Park, S.S.; Seong, M.W. Accuracy and Performance Evaluation of Triplet Repeat Primed PCR as an Alternative to Conventional Diagnostic Methods for Fragile X Syndrome. Ann. Lab. Med. 2021, 41, 394–400. [Google Scholar] [CrossRef] [PubMed]
- McGowan-Jordan, J.; Ros, J.H.; Sarah, M. (Eds.) ISCN 2020: An International System for Human Cytogenomic Nomenclature (2020); Karger: Basel, Switzerland, 2020; also in Cytogenet Genome Res. 2020, 160, 341–503. [Google Scholar]
- Hu, P.; Xu, Y.; Zhang, Q.; Zhou, R.; Ji, X.; Wang, Y.; Xu, Z. Prenatal diagnosis of chromosomal abnormalities using optical genome mapping vs. chromosomal microarray. Am. J. Obstet. Gynecol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, Y.; Zhou, J.; Zhou, R.; Liu, A.; Meng, L.; Ji, X.; Hu, P.; Xu, Z. 11q13.3q13.4 deletion plus 9q21.13q21.33 duplication in an affected girl arising from a familial four-way balanced chromosomal translocation. Mol. Genet. Genomic Med. 2023, 11, e2248. [Google Scholar] [CrossRef]
- Goumy, C.; Ouedraogo, Z.G.; Soler, G.; Eymard-Pierre, E.; Laurichesse, H.; Delabaere, A.; Gallot, D.; Bouchet, P.; Perthus, I.; Pebrel-Richard, C.; et al. Optical genome mapping for prenatal diagnosis: A prospective study. Clin. Chim. Acta 2023, 551, 117594. [Google Scholar] [CrossRef] [PubMed]
- Schneider, V.A.; Graves-Lindsay, T.; Howe, K.; Bouk, N.; Chen, H.-C.; Kitts, P.A.; Murphy, T.D.; Pruitt, K.D.; Thibaud-Nissen, F.; Albracht, D.; et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017, 27, 849–864. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Dai, Y.; Yu, H.; Zhao, S.; Samuels, D.C.; Shyr, Y. Improvements and impacts of GRCh38 human reference on high throughput sequencing data analysis. Genomics 2017, 109, 83–90. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Dai, P.; Zhu, X.; Pei, Y.; Chen, P.; Li, J.; Gao, Z.; Liang, Y.; Kong, X. Evaluation of optical genome mapping for detecting chromosomal translocation in clinical cytogenetics. Mol. Genet. Genomic Med. 2022, 10, e1936. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jia, Z.; Mao, A.; Xu, B.; Wang, S.; Wang, L.; Liu, S.; Zhang, H.; Zhang, X.; Yu, T.; et al. Analysis of balanced reciprocal translocations in patients with subfertility using single-molecule optical mapping. J. Assist. Reprod. Genet. 2020, 37, 509–516. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Case | Sample Type | Results of SOC Methods | Clinical Significance | Results of OGM (GRCh37 as Reference Genome) | Results of OGM (GRCh38 as Reference Genome) |
|---|---|---|---|---|---|
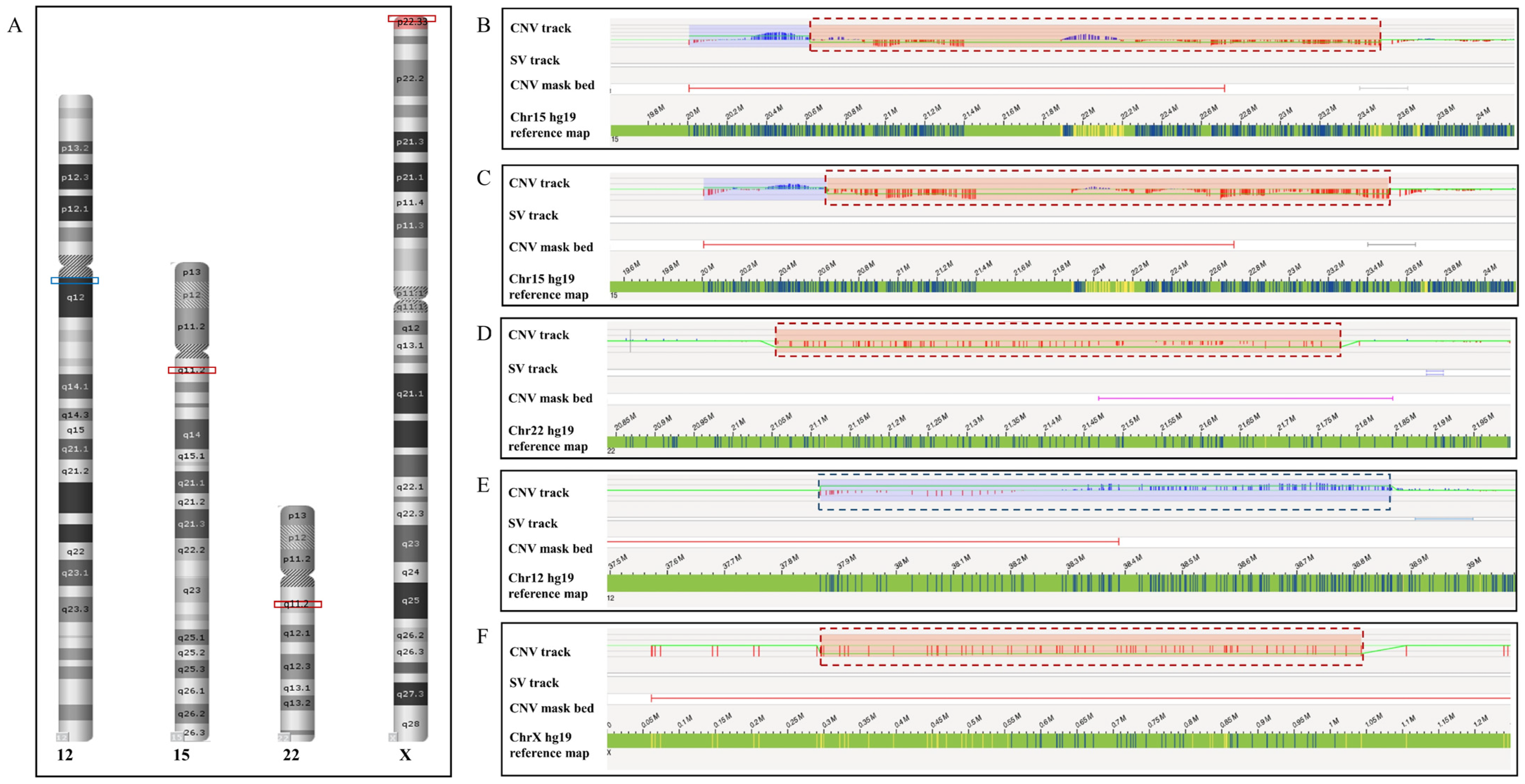
| 1 | Cultured amniotic fluid cells | CMA: arr[hg19]15q11.2(22,770,421–23,191,761) × 1 | VOUS | CNV algorithm: ogm[GRCh37]15q11.1q11.2(20,618,416–23,506,059) × 1 but filtered because confidence was 0 | CNV algorithm: ogm[GRCh38]15q11.2(21,828,570–23,348,707) × 1 but filtered because confidence was 0.3 |
| 2 | Cultured amniotic fluid cells | CMA: arr[hg19]15q11.2(22,770,421–23,281,886) × 1 | VOUS | CNV algorithm: ogm[GRCh37]15q11.1q11.2(20,638,317–23,506,059) × 1 but filtered because confidence was 0 | Detected CNV algorithm: ogm[GRCh38]15q11.2(22,076,863–23,396,245) × 1 |
| 3 | Cultured amniotic fluid cells | CMA: arr[hg19]22q11.21(21,058,888–21,464,764) × 1 | LP | CNV algorithm: ogm[GRCh37]22q11.21(21,054,185–21,779,032) × 1 but filtered because confidence was 0.93 SV algorithm: ogm[GRCh37]22q11.21(21,035,325–21,627,655) × 1 but filtered because frequency was 16.2% in control database | Detected CNV algorithm: ogm[GRCh38]22q11.21(20,699,897–21,424,743) × 1 SV algorithm: ogm[GRCh38]22q11.21(20,681,037–21,273,366) × 1 |
| 4 | Cultured amniotic fluid cells | CMA: arr[hg19]12q11q12(38,012,530–39,027,084) × 3 | VOUS | CNV algorithm: ogm[GRCh37]12q11q12(37,867,370–38,865,742) × 3 but filtered because FCN was 2.797 | Detected CNV algorithm: ogm[GRCh38]12q11q12(37,261,973–38,471,358) × 3 |
| 5 | Cultured amniotic fluid cells | CMA: arr[hg19]Xp22.33(168,551–881,102) × 1 | P | CNV algorithm: ogm[GRCh37]Xp22.33(295,720–1,042,875) × 1 but filtered because confidence was 0 | CNV algorithm: ogm[GRCh38]Xp22.33(544,608–1,439,001) × 1 but filtered because confidence was 0 |
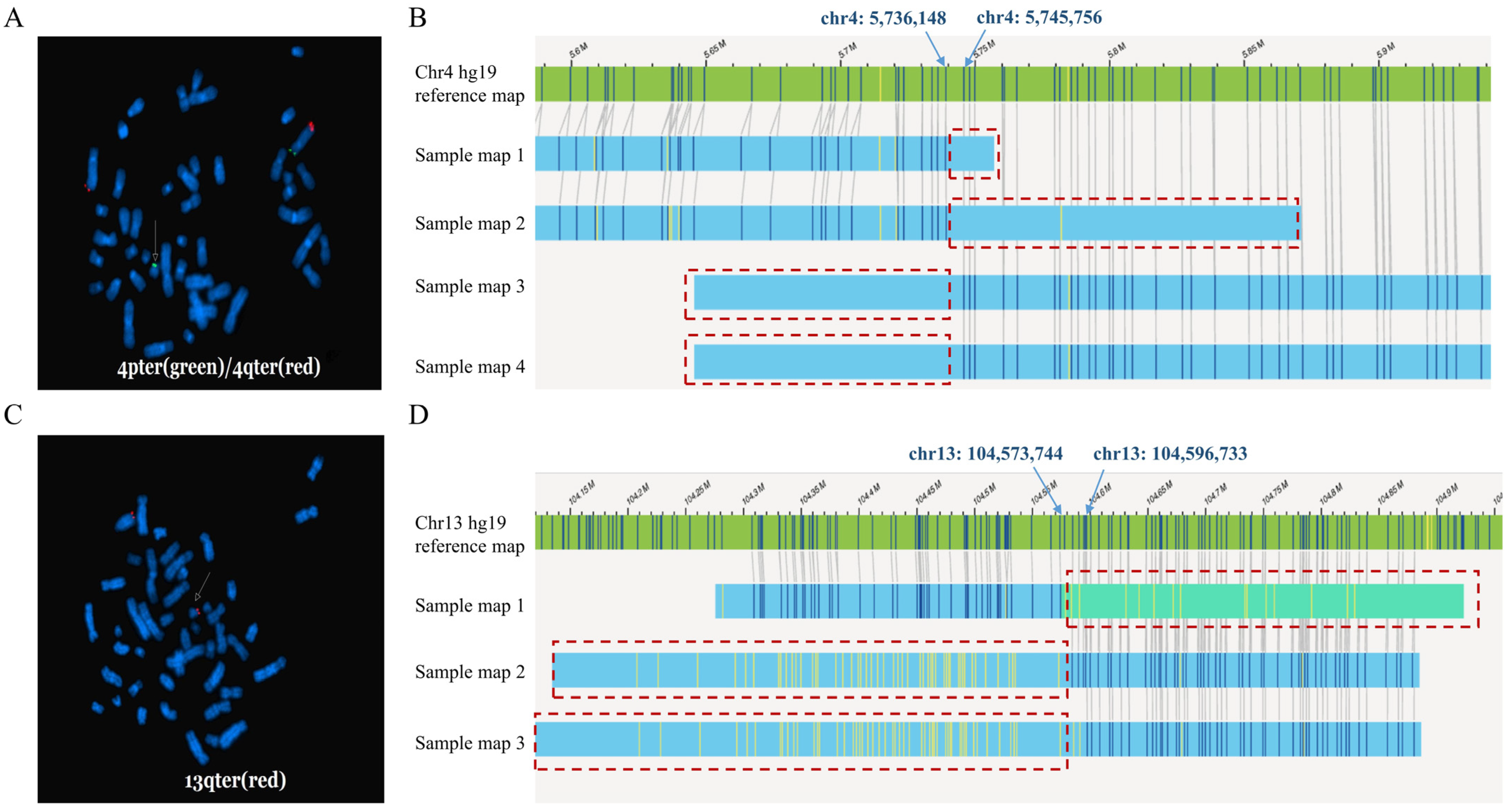
| 6 | Peripheral blood | FISH: ish t(4;?21)(4p−,4q+;21p+) Karyotyping: normal | - | Not detected | Not detected |
| 7 | Peripheral blood | FISH: ish t(13;21)(13q+,21q+) Karyotyping: normal | - | Not detected | Not detected |
| 8 | Cultured amniotic fluid cells | Karyotyping: 46,XN,inv(9)(p12q21) | - | Not detected | Not detected |
| 9 | Peripheral blood | Karyotyping: 46,XN,inv(9)(p12q13) | - | Not detected | Not detected |
| 10 | Cultured amniotic fluid cells | Karyotyping: 45,X [93]/46,X,idic(Y)(q11.22)[7] | P | CNV algorithm: 45,X Not detected idic(Y) mosaicism | CNV algorithm: 45,X Not detected idic(Y) mosaicism |
| Case | Total DNA (≥150 kbp) (Gb) | N50 (≥150 kbp) (kbp) | Map Rate (%) | Average Label Density (N/100 kbp) | Effective Coverage (X) |
|---|---|---|---|---|---|
| 1 | 517.81 | 259.88 | 87.8 | 15.30 | 142.27 |
| 2 | 528.85 | 267.38 | 92.9 | 15.09 | 152.27 |
| 3 | 526.96 | 278.65 | 92.0 | 15.64 | 152.02 |
| 4 | 513.97 | 233.31 | 88.9 | 15.13 | 141.39 |
| 5 | 545.90 | 288.75 | 94.0 | 15.34 | 158.95 |
| 6 | 556.44 | 289.73 | 94.2 | 15.55 | 164.64 |
| 7 | 522.08 | 265.88 | 93.0 | 15.58 | 150.76 |
| 8 | 527.47 | 312.46 | 95.2 | 15.41 | 155.35 |
| 9 | 504.78 | 285.09 | 92.9 | 15.40 | 145.16 |
| 10 | 598.51 | 312.80 | 85.2 | 15.76 | 135.28 |
| Clinically Reported Variations | Aberrations Detected by SOC Methods | Taking GRCh37 as Reference Genome | Taking GRCh38 as Reference Genome | p Value † | ||
|---|---|---|---|---|---|---|
| Aberrations Missed by OGM | False-Negative Rate of OGM | Aberrations Missed by OGM | False-Negative Rate of OGM | |||
| Triploidy | 1 | 0 | 0.0% | 0 | 0.0% | - |
| Aneuploidy | 31 | 0 | 0.0% | 0 | 0.0% | - |
| Copy number variation | 102 | 5 | 4.9% | 2 | 2.0% | 0.442 ‡ |
| Balanced translocation | 10 | 2 | 20.0% | 2 | 20.0% | 1.000 § |
| Microscopic | 5 | 0 | 0.0% | 0 | 0.0% | - |
| Submicroscopic | 5 | 2 | 40.0% | 2 | 40/0% | 1.000 § |
| Unbalanced translocation | 3 | 0 | 0.0% | 0 | 0.0% | - |
| Microscopic | 1 | 0 | 0.0% | 0 | 0.0% | - |
| Submicroscopic | 2 | 0 | 0.0% | 0 | 0.0% | - |
| Pericentric inversion | 2 | 2 | 100.0% | 2 | 100.0% | 1.000 § |
| Isochromosome | 3 | 1 | 33.3% | 1 | 33.3% | 1.000 § |
| ROH | 1 | 0 | 0.0% | 0 | 0.0% | - |
| FMR1 full mutation | 1 | 0 | 0.0% | 0 | 0.0% | - |
| Total | 154 | 10 | 6.5% | 7 | 4.5% | 0.454 ¶ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Zhang, Q.; Wang, Y.; Zhou, R.; Ji, X.; Meng, L.; Luo, C.; Liu, A.; Jiao, J.; Chen, H.; et al. Optical Genome Mapping for Chromosomal Aberrations Detection—False-Negative Results and Contributing Factors. Diagnostics 2024, 14, 165. https://doi.org/10.3390/diagnostics14020165
Xu Y, Zhang Q, Wang Y, Zhou R, Ji X, Meng L, Luo C, Liu A, Jiao J, Chen H, et al. Optical Genome Mapping for Chromosomal Aberrations Detection—False-Negative Results and Contributing Factors. Diagnostics. 2024; 14(2):165. https://doi.org/10.3390/diagnostics14020165
Chicago/Turabian StyleXu, Yiyun, Qinxin Zhang, Yan Wang, Ran Zhou, Xiuqing Ji, Lulu Meng, Chunyu Luo, An Liu, Jiao Jiao, Hao Chen, and et al. 2024. "Optical Genome Mapping for Chromosomal Aberrations Detection—False-Negative Results and Contributing Factors" Diagnostics 14, no. 2: 165. https://doi.org/10.3390/diagnostics14020165