
Molecular Characterization of Advanced-Stage Melanomas in Clinical Practice Using a Laboratory-Developed Next-Generation Sequencing Panel

 , , , , ,
, , , , ,  , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
NGS Analysis
3. Results
3.1. Concomitant Mutations
3.2. Primary and Metastatic Lesions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cancer Genome Atlas, N. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Comito, F.; Aprile, M.; Pagani, R.; Siepe, G.; Sperandi, F.; Gruppioni, E.; Altimari, A.; De Biase, D.; Melotti, B. Clinical characteristics and treatment outcomes of non-V600 E/K BRAF mutant melanoma patients: A single-institution experience. Melanoma Res. 2022, 32, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Owsley, J.; Stein, M.K.; Porter, J.; In, G.K.; Salem, M.; O’Day, S.; Elliott, A.; Poorman, K.; Gibney, G.; VanderWalde, A. Prevalence of class I-III BRAF mutations among 114,662 cancer patients in a large genomic database. Exp. Biol. Med. 2021, 246, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Garnett, M.J.; Rana, S.; Paterson, H.; Barford, D.; Marais, R. Wild-type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol. Cell 2005, 20, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef] [PubMed]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef] [PubMed]
- Hugdahl, E.; Kalvenes, M.B.; Mannelqvist, M.; Ladstein, R.G.; Akslen, L.A. Prognostic impact and concordance of TERT promoter mutation and protein expression in matched primary and metastatic cutaneous melanoma. Br. J. Cancer 2018, 118, 98–105. [Google Scholar] [CrossRef]
- Nagore, E.; Heidenreich, B.; Rachakonda, S.; Garcia-Casado, Z.; Requena, C.; Soriano, V.; Frank, C.; Traves, V.; Quecedo, E.; Sanjuan-Gimenez, J.; et al. TERT promoter mutations in melanoma survival. Int. J. Cancer 2016, 139, 75–84. [Google Scholar] [CrossRef]
- Nagore, E.; Heidenreich, B.; Requena, C.; Garcia-Casado, Z.; Martorell-Calatayud, A.; Pont-Sanjuan, V.; Jimenez-Sanchez, A.I.; Kumar, R. TERT promoter mutations associate with fast-growing melanoma. Pigment. Cell Melanoma Res. 2016, 29, 236–238. [Google Scholar] [CrossRef]
- Shaughnessy, M.; Njauw, C.N.; Artomov, M.; Tsao, H. Classifying Melanoma by TERT Promoter Mutational Status. J. Investig. Dermatol. 2020, 140, 390–394.e1. [Google Scholar] [CrossRef] [PubMed]
- Timis, T.; Bergthorsson, J.T.; Greiff, V.; Cenariu, M.; Cenariu, D. Pathology and Molecular Biology of Melanoma. Curr. Issues Mol. Biol. 2023, 45, 5575–5597. [Google Scholar] [CrossRef] [PubMed]
- Teixido, C.; Castillo, P.; Martinez-Vila, C.; Arance, A.; Alos, L. Molecular Markers and Targets in Melanoma. Cells 2021, 10, 2320. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.; Garraway, L.A.; Fisher, D.E. Malignant melanoma: Genetics and therapeutics in the genomic era. Genes. Dev. 2006, 20, 2149–2182. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Dika, E.; Lambertini, M.; Pellegrini, C.; Veronesi, G.; Melotti, B.; Riefolo, M.; Sperandi, F.; Patrizi, A.; Ricci, C.; Mussi, M.; et al. Cutaneous and Mucosal Melanomas of Uncommon Sites: Where Do We Stand Now? J. Clin. Med. 2021, 10, 478. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017, 548, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Michielin, O.; van Akkooi, A.C.J.; Ascierto, P.A.; Dummer, R.; Keilholz, U.; ESMO Guidelines Committee. Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-updagger. Ann. Oncol. 2019, 30, 1884–1901. [Google Scholar] [CrossRef]
- de Biase, D.; Acquaviva, G.; Visani, M.; Sanza, V.; Argento, C.M.; De Leo, A.; Maloberti, T.; Pession, A.; Tallini, G. Molecular Diagnostic of Solid Tumor Using a Next Generation Sequencing Custom-Designed Multi-Gene Panel. Diagnostics 2020, 10, 250. [Google Scholar] [CrossRef] [PubMed]
- Mosele, F.; Remon, J.; Mateo, J.; Westphalen, C.B.; Barlesi, F.; Lolkema, M.P.; Normanno, N.; Scarpa, A.; Robson, M.; Meric-Bernstam, F.; et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: A report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2020, 31, 1491–1505. [Google Scholar] [CrossRef]
- Pegoraro, A.; De Marchi, E.; Ferracin, M.; Orioli, E.; Zanoni, M.; Bassi, C.; Tesei, A.; Capece, M.; Dika, E.; Negrini, M.; et al. P2X7 promotes metastatic spreading and triggers release of miRNA-containing exosomes and microvesicles from melanoma cells. Cell Death Dis. 2021, 12, 1088. [Google Scholar] [CrossRef]
- Mulder, E.; Johansson, I.; Grunhagen, D.J.; Tempel, D.; Rentroia-Pacheco, B.; Dwarkasing, J.T.; Verver, D.; Mooyaart, A.L.; van der Veldt, A.A.M.; Wakkee, M.; et al. Using a Clinicopathologic and Gene Expression (CP-GEP) Model to Identify Stage I–II Melanoma Patients at Risk of Disease Relapse. Cancers 2022, 14, 2854. [Google Scholar] [CrossRef]
- Guo, Y.; Chen, Y.; Zhang, L.; Ma, L.; Jiang, K.; Yao, G.; Zhu, L. TERT Promoter Mutations and Telomerase in Melanoma. J. Oncol. 2022, 2022, 6300329. [Google Scholar] [CrossRef] [PubMed]
- Guterres, A.N.; Villanueva, J. Targeting telomerase for cancer therapy. Oncogene 2020, 39, 5811–5824. [Google Scholar] [CrossRef]
- Bracht, J.W.P.; Karachaliou, N.; Bivona, T.; Lanman, R.B.; Faull, I.; Nagy, R.J.; Drozdowskyj, A.; Berenguer, J.; Fernandez-Bruno, M.; Molina-Vila, M.A.; et al. BRAF Mutations Classes I, II, and III in NSCLC Patients Included in the SLLIP Trial: The Need for a New Pre-Clinical Treatment Rationale. Cancers 2019, 11, 1381. [Google Scholar] [CrossRef]
- Kerkour, T.; Zhou, C.; Hollestein, L.; Mooyaart, A. Genetic Concordance in Primary Cutaneous Melanoma and Matched Metastasis: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2023, 24, 16281. [Google Scholar] [CrossRef]
- Manca, A.; Paliogiannis, P.; Colombino, M.; Casula, M.; Lissia, A.; Botti, G.; Caraco, C.; Ascierto, P.A.; Sini, M.C.; Palomba, G.; et al. Mutational concordance between primary and metastatic melanoma: A next-generation sequencing approach. J. Transl. Med. 2019, 17, 289. [Google Scholar] [CrossRef] [PubMed]
- Moran, J.M.T.; Le, L.P.; Nardi, V.; Golas, J.; Farahani, A.A.; Signorelli, S.; Onozato, M.L.; Foreman, R.K.; Duncan, L.M.; Lawrence, D.P.; et al. Identification of fusions with potential clinical significance in melanoma. Mod. Pathol. 2022, 35, 1837–1847. [Google Scholar] [CrossRef]
- Huang, R.S.P.; Tse, J.Y.; Harries, L.; Graf, R.P.; Lin, D.I.; Murugesan, K.; Hiemenz, M.C.; Parimi, V.; Janovitz, T.; Decker, B.; et al. Variable Genomic Landscapes of Advanced Melanomas with Heavy Pigmentation. Oncologist 2022, 27, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Pallocca, M.; Molineris, I.; Berrino, E.; Marcozzi, B.; Betti, M.; Levati, L.; D’Atri, S.; Menin, C.; Madonna, G.; Ghiorzo, P.; et al. Comprehensive genomic profiling on metastatic Melanoma: Results from a network screening from 7 Italian Cancer Centres. J. Transl. Med. 2024, 22, 29. [Google Scholar] [CrossRef]
- Pruneri, G.; De Braud, F.; Sapino, A.; Aglietta, M.; Vecchione, A.; Giusti, R.; Marchio, C.; Scarpino, S.; Baggi, A.; Bonetti, G.; et al. Next-Generation Sequencing in Clinical Practice: Is It a Cost-Saving Alternative to a Single-Gene Testing Approach? Pharmacoecon. Open 2021, 5, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Morganti, S.; Tarantino, P.; Ferraro, E.; D’Amico, P.; Viale, G.; Trapani, D.; Duso, B.A.; Curigliano, G. Complexity of genome sequencing and reporting: Next generation sequencing (NGS) technologies and implementation of precision medicine in real life. Crit. Rev. Oncol. Hematol. 2019, 133, 171–182. [Google Scholar] [CrossRef] [PubMed]
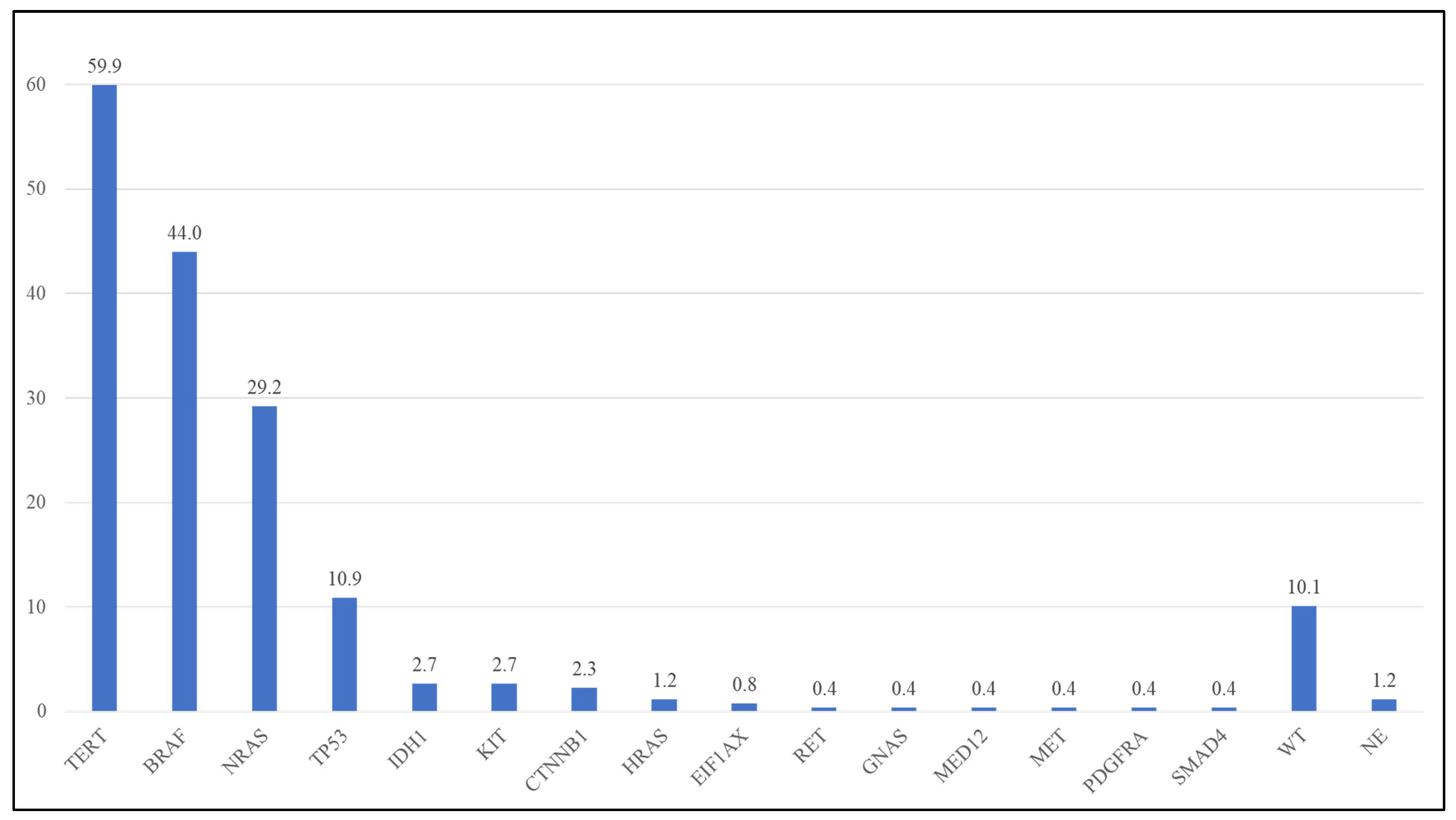

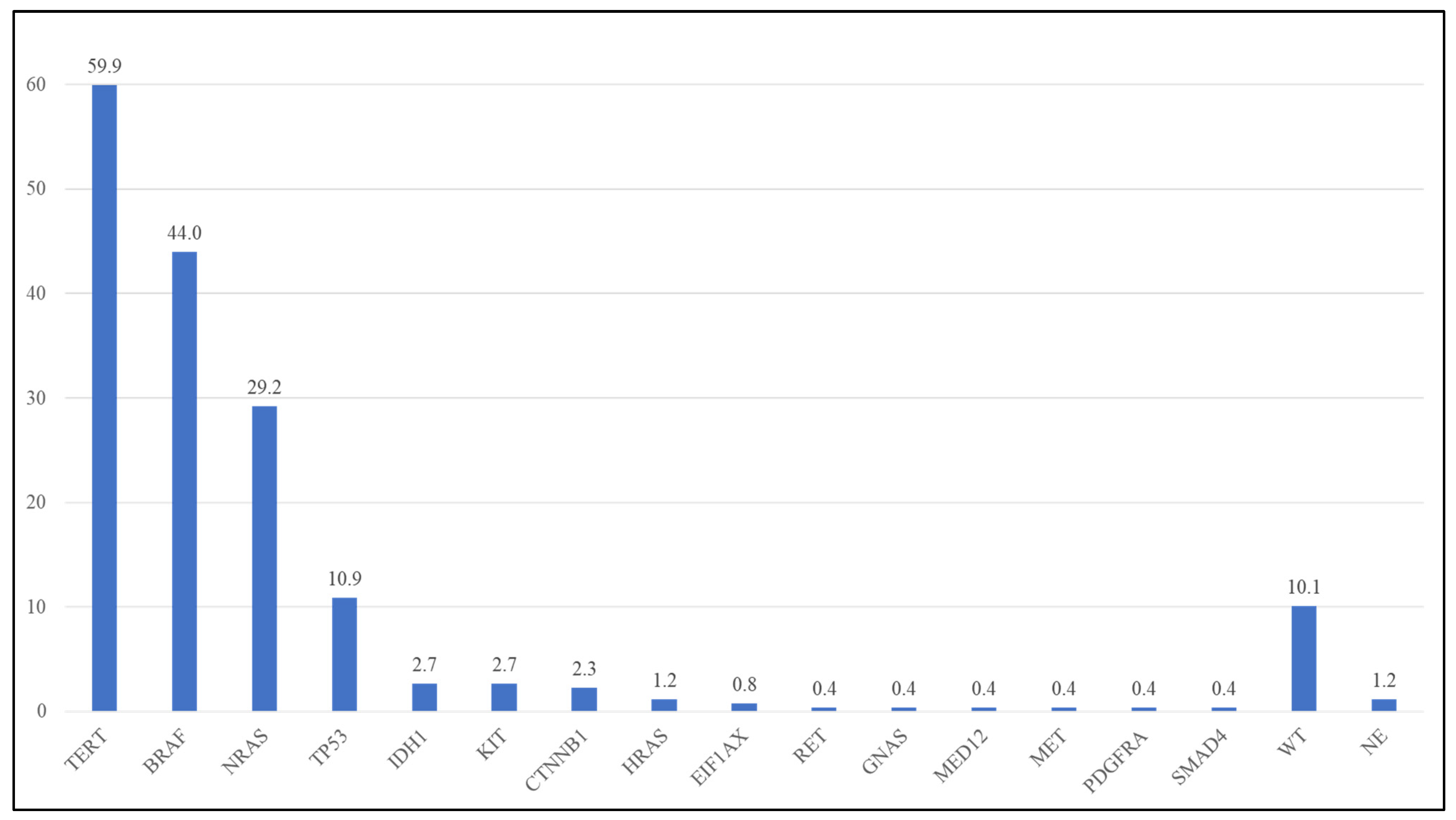{kind=link}
| Gene | Detected Frequency in Present Study (Lab-Developed NGS Panel) (n = 260) | Frequency in TCGA (Whole-Exome Sequencing) [1] * |
|---|---|---|
| TERT | 59.9% | 64.4% ^ |
| BRAF | 44.0% | 51.2% |
| NRAS | 29.2% | 27.3% |
| TP53 | 10.9% | 19.8% |
| KIT | 2.7% | 5.0% |
| IDH1 | 2.7% | 4.1% |
| CTNNB1 | 2.3% | 7.4% |
| HRAS | 1.2% | NA |
| EIF1AX | 0.8% | 1.7% |
| RET | 0.4% | 6.6% |
| GNAS | 0.4% | 6.6% |
| MED12 | 0.4% | 5.8% |
| MET | 0.4% | 6.6% |
| PDGFRA | 0.4% | 5.8% |
| SMAD4 | 0.4% | NA |
| BRAF Variant | Exon | Cases (%) n = 113 | ACMG Classification | BRAF Class |
|---|---|---|---|---|
| p.V600E | 15 | 82 (72.6) | P | I |
| p.V600K | 15 | 21 (18.6) | P | I |
| p.V600R | 15 | 3 (2.7) | P | I |
| p.K601E | 15 | 1 (0.9) | LP | II |
| p.G466E | 11 | 2 (1.8) | LP | III |
| p.S467L | 11 | 3 (2.7) | P | III |
| p.G469A | 11 | 1 (0.9) | P | II |
| NRAS Variant | Exon | Cases (%) n = 76 | ACMG Classification |
|---|---|---|---|
| p.Q61R | 3 | 38 (50.0) | P |
| p.Q61K | 3 | 24 (31.6) | P |
| p.Q61L | 3 | 4 (5.3) | P |
| p.Q61H | 3 | 2 (2.7) | P |
| p.G60E | 3 | 1 (1.3) | P |
| p.G12D | 2 | 2 (2.7) | P |
| p.G13R | 2 | 1 (1.3) | P |
| p.G13S | 2 | 1 (1.3) | P |
| p.G13V | 2 | 1 (1.3) | P |
| p.Q22K | 2 | 1 (1.3) | LP |
| TERT Variant | Cases (%) n = 154 | ACMG Classification |
|---|---|---|
| c.-124C>T | 66 (42.9) | P |
| c.-146C>T | 76 (49.4) | LP |
| c.-138_-139delinsTT | 11 (7.1) | LP |
| c.-124_-125delinsTT | 1 (0.6) | LP |
| Gene | Cases out of Total Mutated Samples (%) | Gene More Commonly Mutated with |
|---|---|---|
| BRAF | 79/113 (69.9) | TERT (72/79—91.1%) |
| NRAS | 51/76 (67.1) | TERT (50/51—98.0%) |
| TERT | 127/154 (82.5) | BRAF (72/127—56.7%) |
| TP53 | 24/28 (85.7) | TERT (14/24—58.3%) |
| IDH1 | 7/7 (100) | TERT (6/7—85.7%) |
| CTNNB1 | 6/6 (100) | TERT (6/6—100%) |
| # | Specimen | Variants |
|---|---|---|
| 1 | Primary lesion | BRAF p.Val600Glu TERT c.-124C>T |
| Metastasis | BRAF p.Val600Glu TERT c.-124C>T | |
| 2 | Primary lesion | NRAS p.Gln61Arg TERT c.-124C> |
| Metastasis | NRAS p.Gln61Arg TERT c.-124C>T | |
| 3 | Primary lesion | BRAF p.Val600Lys IDH1 p.Arg132Cys |
| Metastasis | BRAF p.Val600Lys IDH1 p.Arg132Cys | |
| 4 | Primary lesion | BRAF p.Ser467Leu TERT c.-146 C>T NRAS p.Gln61Arg TP53 p.Ile195MetfsTer51 |
| Metastasis | BRAF p.Ser467Leu TERT c.-146 C>T NRAS p.Gln61Arg TP53 p.Ile195MetfsTer51 | |
| 5 | Primary lesion | NRAS p.Gln61Arg TERT c.-146C>T |
| Metastasis | NRAS p.Gln61Arg TERT c.-146C>T | |
| 6 | Primary lesion | NRAS p.Gln61Arg TERT c.-146C>T |
| Metastasis | NRAS p.Gln61Arg TERT c.-146C>T | |
| 7 | Primary lesion | BRAF p.Val600Glu TERT c.-146C>T |
| Metastasis | BRAF p.Val600Glu TERT c.-146C>T | |
| 8 | Primary lesion | BRAF p.Val600Lys TERT c.-124C>T |
| Metastasis | BRAF p.Val600Lys TERT c.-124C>T |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maloberti, T.; De Leo, A.; Coluccelli, S.; Sanza, V.; Gruppioni, E.; Altimari, A.; Comito, F.; Melotti, B.; Marchese, P.V.; Dika, E.; et al. Molecular Characterization of Advanced-Stage Melanomas in Clinical Practice Using a Laboratory-Developed Next-Generation Sequencing Panel. Diagnostics 2024, 14, 800. https://doi.org/10.3390/diagnostics14080800
Maloberti T, De Leo A, Coluccelli S, Sanza V, Gruppioni E, Altimari A, Comito F, Melotti B, Marchese PV, Dika E, et al. Molecular Characterization of Advanced-Stage Melanomas in Clinical Practice Using a Laboratory-Developed Next-Generation Sequencing Panel. Diagnostics. 2024; 14(8):800. https://doi.org/10.3390/diagnostics14080800
Chicago/Turabian StyleMaloberti, Thais, Antonio De Leo, Sara Coluccelli, Viviana Sanza, Elisa Gruppioni, Annalisa Altimari, Francesca Comito, Barbara Melotti, Paola Valeria Marchese, Emi Dika, and et al. 2024. "Molecular Characterization of Advanced-Stage Melanomas in Clinical Practice Using a Laboratory-Developed Next-Generation Sequencing Panel" Diagnostics 14, no. 8: 800. https://doi.org/10.3390/diagnostics14080800
APA StyleMaloberti, T., De Leo, A., Coluccelli, S., Sanza, V., Gruppioni, E., Altimari, A., Comito, F., Melotti, B., Marchese, P. V., Dika, E., Venturi, F., Corti, B., Ciccimarra, G., Ciceu, C. A., Tallini, G., & de Biase, D. (2024). Molecular Characterization of Advanced-Stage Melanomas in Clinical Practice Using a Laboratory-Developed Next-Generation Sequencing Panel. Diagnostics, 14(8), 800. https://doi.org/10.3390/diagnostics14080800