Abstract
Cutaneous melanoma is one of the most lethal tumors among skin cancers, characterized by complex genetic and molecular alterations that result in uncontrolled cell proliferation and metastatic spread. Next-generation sequencing (NGS) enables the simultaneous examination of numerous genes, making this molecular technique essential for melanoma diagnosis, prognostic stratification, and therapy planning. Herein, we present the experience with our laboratory-designed NGS panel for the routine assessment of advanced-stage melanoma. A total of 260 specimens of advanced-stage melanomas were evaluated utilizing a laboratory-developed multi-gene NGS panel, which allowed the investigation of 229 amplicons in 25 oncogene/oncosuppressor genes. The NGS panel proved to be a reliable tool, failing to produce results in only 1.2% of the samples tested. BRAF and TERT were the two more commonly altered genes in 44.0% and 59.9% of samples, respectively. In 59.3% of the mutated cases, at least two concomitant variants were detected. In eight cases, both primary lesion and metastatic disease were analyzed by NGS. In all specimens (8/8, 100%), a perfect concordance in variants harbored by the primary and recurrence lesions was observed. Finally, this study described the validity of a laboratory-developed multi-gene NGS panel built specifically for advanced-stage melanomas in ordinary clinical practice.
1. Introduction
Cutaneous melanoma is one of the most lethal tumors among skin cancers, and its incidence is rising worldwide. It is caused by a complex interplay of genetic and epigenetic alterations that drive its initiation, development, and metastasis. These changes frequently impair essential signaling networks that regulate cell growth, proliferation, differentiation, and survival. The vast majority of melanomas are sporadic, and one of the more common molecular alterations is the mutation of the BRAF gene.
BRAF. One of the most well-known genetic alterations in melanoma is the mutation of the BRAF gene, specifically the p.V600E mutation, which occurs in around 50% of cases. BRAF is a serine/threonine kinase protein involved in the MAP kinase pathway, which controls cell growth and proliferation. The most common variants are BRAF class I mutations (mainly BRAF p.V600E, followed by BRAF p.V600K), which are almost exclusively induced by the oncogenic/pathogenetic key role of UV radiation [1]. However, other non-V600 BRAF variants may be found in advanced-stage melanoma [1,2,3]. These variations in the BRAF gene may cause constitutive protein activation, which results in uncontrolled cell proliferation and tumor formation [3,4].
TERT. Telomerase reverse transcriptase (TERT) mutations are key players in melanoma genesis and progression. TERT activity is typically carefully regulated, but changes can disrupt this control, resulting in unregulated cell proliferation and cancer development. Recurrent mutations in the promoter of the TERT gene were initially detected in melanoma and then in various additional cancer types [5,6,7]. TERT promoter mutations are the most common type of TERT alterations in melanoma, accounting for up to 65% of cutaneous melanoma [1,8]. These mutations increase TERT expression, which helps cancer cells grow and survive. Mutations in the promoter region of TERT are related to reduced disease-free survival, increased tumor recurrence, and an increased rate of metastasis in melanoma [8,9,10,11].
NRAS. Mutations in the NRAS gene are found in about 15–20% of melanomas [1,12]. Hot-spot mutations in the RAS genes are generally at the Q61 codon and less frequently in G12 or G13 [1]. Mutations in RAS genes cause the constitutive activation of the MAPK pathway, resulting in increased cellular proliferation, survival, and resistance to apoptosis [13]. Inhibiting proteins farther down the RAS pathway, such as MEK and ERK, can indirectly prevent the carcinogenic signals triggered by RAS mutations [13].
Other common genetic alterations observed in melanoma are the loss of function of the CDKN2A tumor suppressor gene, TP53 mutations/deletions (thus resulting in the loss of heterozygosis) but with a lower frequency and a lower magnitude effect compared to other solid tumors, and KIT mutations mainly detected in acral and mucosal melanoma [1,12,14,15,16].
Next-generation sequencing (NGS) enables the simultaneous sequencing of numerous genes with a very high depth of coverage. Given the ongoing discovery of molecules as potential targets or molecules that are accountable for treatment resistance mechanisms, using a classic single-gene approach is becoming challenging. The adoption of multi-gene panels is now essential for the molecular investigation of solid tumors, including melanoma. According to the ESMO guidelines, “If the tumour is BRAF wild-type (WT) at the V600 locus (class I BRAF mutant), sequencing the loci of the other known minor BRAF mutations (class II and class III BRAF mutant) to confirm WT status and testing for NRAS and c-kit mutations are recommended [II, C] […]. Alternatively, a clinically validated next-generation sequencing panel covering all key oncogenic drivers is increasingly being carried out” [17,18].
The present study aims to disclose a laboratory-designed multi-gene panel that allows for assessing advanced-stage melanoma in routine clinical practice.
2. Materials and Methods
A total of 260 cases of advanced-stage melanomas were analyzed for routine practice at the Molecular Pathology Laboratory of IRCCS Policilinico di S.Orsola in Bologna, Italy, from January 2021 to December 2023. All samples analyzed were extracted from formalin-fixed and paraffin-embedded (FFPE) histological blocks. Briefly, DNA was extracted from 2 to 3 10 μm thick sections, according to the selection performed by a pathologist on the last Hematoxylin and Eosin (H/E) slide. DNA was quantified using a Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA, USA).
NGS Analysis
The next-generation sequencing (NGS) analysis was performed using a multi-gene panel developed in the Molecular Pathology Laboratory of IRCCS Policlinico di S.Orsola [19]. The panel allows the analysis of the following hot-spot regions of 25 genes for a total of 229 amplicons (15.04 kb, human reference sequence hg19/GRCh37) in the following genes: BRAF (exons 11, 15), CTNNB1 (exon 3), EGFR (exons 12, 18, 19, 20, 21), EIF1AX (exons 1, 2), GNA11 (exons 4, 5), GNAQ (exons 4, 5), GNAS (exons 8, 9), H3F3A (exon 1), HRAS (exons 2, 3), IDH1 (exon 4), IDH2 (exon 4), KIT (exons 8, 9, 11, 13, 17), KRAS (exons 2, 3, 4), MED12 (exons 1, 2), MET (exons 2, 14), MYC (exons 1-3), NRAS (exons 2, 3, 4), PDGFRα (exons 12, 14, 18), PIK3CA (exons 10, 21), PTEN (exon 5), RET (exons 5, 8, 10, 11, 13, 15, 16), RNF43 (exons 2, 8), SMAD4 (exons 6, 9, 10, 11, 12), TERT (promoter region, g.1295141–g.1295471), and TP53 (exons 4, 5, 6, 7, 8, 9).
NGS was performed using the Gene Studio S5 Prime Sequencer (Thermo Fisher Scientific). Briefly, about 30ng of DNA was used per panel for the amplicon library preparation, performed with the AmpliSeq Plus Library Kit 2.0. In specimens where the pathologists highlighted the presence of abundant melanin, 3 μL of betaine 1N was added. Templates were prepared with an Ion Chef Machine and sequenced using an Ion 530 chip. Sequences were analyzed with the Ion Reporter tool (v. 5.18–Thermo Fisher Scientific). The filter chain query was applied as follows: 0.05 ≤ allele frequency ≤ 1.0. Filtered variants were then manually investigated. Only nucleotide variations detected in both strands and at least 5% of the total number of reads analyzed were considered for the mutational calls [19]. The pathogenicity of each mutation was assessed using the Varsome tool (https://varsome.com/, last access: 1 February 2024). Benign/likely benign variants were not considered in the present study. The research was approved by the local institution’s ethics committee. All data used in the present study were completely anonymized and aggregated.
3. Results
Of the 260 specimens analyzed by NGS, three (1.2%) were not evaluable due to low quality/quantity DNA. The following evaluations were then performed on a total of 257 specimens with evaluable NGS analysis. In 26 out of 257 specimens (10.1%), no alterations were detected in any of the analyzed genes. In the remnant 231 specimens, overall, 402 pathogenic/likely pathogenic/VUS (P/LP/VUS) variants were identified in 15 genes (Table 1, Figure 1, Supplementary Table S1). No P/LP/VUSs were detected in the remnant genes analyzed in the NGS panel. The BRAF gene and TERT promoter were the more altered markers in the analyzed cohort (Table 1, Figure 1, Supplementary Table S1).

Table 1.
Frequency of altered genes in the analyzed cohort and comparison with data obtained from TCGA.
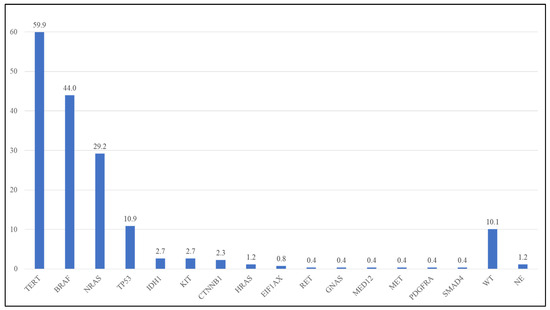
Figure 1.
Frequency of mutations in the analyzed cohort. WT: wild type; NE: Not able to evaluate.
BRAF. BRAF mutations were detected in 113 out of 257 specimens (44.0%). The BRAF p.V600E was the more frequent variant (82/113 BRAF mutated cases—72.6%), followed by p.V600K (21/113, 18.6%), and other rarer BRAF variants (overall 10/113, 8.8%—Table 2). The vast majority (107/113 mutated specimens—94.7%) of the BRAF variants were in exon 15, but in 6 cases (5.3% of BRAF mutated cases), BRAF variants in exon 11 were observed (Table 2, Supplementary Table S1). All variants were pathogenic or likely pathogenic. Three variants (p.V600) were BRAF class I mutations, two were BRAF class II, and 2 were BRAF class III variants (Table 2, Supplementary Table S1).
Table 2.
BRAF variants in the analyzed cohort.
NRAS. The NRAS gene was mutated in 76/260 samples (29.6%). Almost all the NRAS variants (69/75 mutated cases—90.8%) were in exon 3, and the other ones (9.1%) were in exon 2 (Table 3). No variants were detected in NRAS exon 4. All NRAS variants were pathogenic or likely pathogenic. The p.Q61R was the more frequent variant (38/76–50.0% of the NRAS mutated cases), followed by the p.Q61K (24/76–31.6%) (Table 3, Supplementary Table S1).
Table 3.
NRAS variants in the analyzed cohort.
TERT. The TERT promoter was mutated in 154/257 (59.9%) samples, resulting in the gene that was most frequently altered. The c.-146C>T (C250T) was found in 76/154 (49.4%) TERT mutated samples, the c.-124C>T (C228T) in 66 samples (42.9%), c.-138_-139delinsTT in 11 samples (7.1%), and c.-124_-125delinsTT in 1 (0.6%) specimen (Table 4, Supplementary Table S1).
Table 4.
TERT variants in the analyzed cohort.
TP53. TP53 variants were detected in 28/257 cases (10.9%). Mutations are detected in different exons (from 4 to 9) of the TP53 genes and are almost different between them (Supplementary Table S1). One case (3.6%) had a variant in exon 4; six cases had a variant(21.4%) in exon 5; nine(32.1%) in exon 6; seven(25.0%) in exon 7; four(14.3%) in exon 8; and one(3.6%) in exon 9. All but one variant were P/LP mutations (Supplementary Table S1).
Other variants. Other variants were identified in a total of 34 cases (13.2%) in the following genes: IDH1 (2.7%), CTNNB1 (2.3%), KIT (2.3%), HRAS (1.2%), EIF1AX (0.8%), RET (0.8%), GNAS (0.4%), MED12 (0.4%), MET (0.4%), PDGFRA (0.4%), and SMAD4 (0.4%) (Supplementary Table S1). Interestingly, all the IDH1-detected variants (7 cases—100%) were p.R132C mutations instead of the more common p.R132H IDH1 mutation (Supplementary Table S1).
3.1. Concomitant Mutations
In 137 of the 231 (59.3%) mutated cases, at least two concomitant variants were detected. TERT was the more common gene found to be mutated with other genes, found in 127 of 137 (92.7%) cases with concomitant mutations. Overall, in 82.5% of cases (127 of 154) harboring a TERT promoter variant, at least one other gene was mutated together with TERT (Table 5, Supplementary Table S2). The more frequent matching was between TERT and BRAF, 72 of 127 (56.7%) cases, followed by NRAS (39.4%), TP53 (11.0%), and other genes (IDH1, KIT, CTNNB1—11.0% each) (Table 5, Supplementary Table S2).
Table 5.
Genes more frequently found mutated in combination with other genes.
Of the 113 BRAF-mutated cases, 79 (69.9%) harbored at least one variant in another gene, mainly TERT (72 of 79 cases—91.1%). NRAS was mutated with other genes in 51 out of 76 (67.1%) mutated cases, mainly TERT (50 of 51–98.0%). In three cases, concomitant BRAF/NRAS mutations were observed, but in all these cases, the BRAF variant was a class III mutation, which is known to have low activity compared to the BRAF wild type (Supplementary Table S2).
Of the 28 cases with the TP53 mutation, 22 (78.6%) had at least one other mutation and the combination TP53-TERT was detected in 12 of these 22 (54.5%) samples. Interestingly, all cases harboring IDH1 (n = 7) or CTNNB1 (n = 6) variants had a mutation in at least one other gene. As regards IDH1, 6 out of 7 mutations (85.7%) were concomitant with TERT variants, and all 6 mutated CTNNB1 cases (100%) were also mutated in the TERT promoter.
3.2. Primary and Metastatic Lesions
In eight cases, both primary and metastatic lesions were analyzed by NGS. In all specimens, we detected a perfect concordance in variants harbored by the primary and recurrence lesions (Table 6). Three out of the eight samples harbored a BRAF p.Val600 variant together with a mutation in the TERT promoter region; three samples had an NRAS p.Glu61Arg mutation and a TERT promoter region mutation; one sample had a BRAF and IDH1 mutation; and one case harbored four different variants in BRAF, NRAS, TERT, and TP53 genes (Table 6). Intriguingly, all but one sample had a TERT promoter mutation concomitant with other pathogenic variants. All these specimens were considered only one time in the whole cohort of 260 cases.
Table 6.
Cases in which both primary and metastatic lesions were analyzed.
4. Discussion
To date, the molecular characterization of metastatic melanomas for predictive purposes has primarily relied on the assessment of BRAF mutations for the use of BRAF inhibitors. However, the genetic changes that distinguish these cancers extend beyond the single BRAF p.V600 mutation.
From a technical standpoint, the evaluation of the BRAF mutation alone can be accomplished using “single-marker” approaches, such as real-time or pyrosequencing, which allow the mutation to be studied quickly and affordably. However, if one wants to characterize a larger number of molecular markers in addition to BRAF, such approaches become less cost-effective and are difficult to use in everyday practice due to the multiplicity of tests required for proper characterization. The advent of NGS in molecular diagnostics enabled us to integrate multigene analysis with great analytical sensitivity.
In recent years, a large number of multi-gene panels have become commercially available. However, these panels contain a large number of targets and are typically intended for specific tumors or genes. Creating custom/laboratory-developed multi-gene panels enables the selection of targets based on the demands of the medical community, as supplied by the molecular laboratory. These panels allow for the optimization of the number of specimens that can be evaluated in a single run, reducing expenses.
The use of multi-gene NGS panels enables the characterization of various genomic areas while maximizing time and costs. Furthermore, using lab-developed panels allows for the “design” of the panel to be based on clinical needs, incorporating markers such as TERT or TP53 that may not be available in commercial “targeted” panels.
Although the ESMO guidelines for the use of NGS in patients with metastatic cancers do not include melanoma between the advanced neoplasms in which NGS is recommended, it is also true that these guidelines “strongly recommends that clinical research centers perform multigene sequencing as part of their missions to accelerate cancer research and drug development through clinical trials, provide access to innovation to patients and to collect data” [20]. Furthermore, the ESMO guidelines for the characterization of the diagnosis and treatment of melanomas suggest not to be limited to the single analysis of the BRAF V600 locus. In tumors that do not carry this type of mutation, the molecular analysis has to be extended not only to less common BRAF variants but also to other genes, such as NRAS and KIT [18]. The NGS multigene approach allows the simultaneous analysis of these potential hot spots, which is a preferable approach compared to sequential analysis (V600 WT → other BRAF variants not present → analysis of genes other than BRAF); also, in light of the data obtained in this study in which BRAF V600 mutations are commonly found together with TERT promoter variants, it suggests that non-class I BRAF mutations could coexist with variants in other driver genes.
The panel we created for the characterization of melanomas with gene alterations is consistent with what has been described in the literature. TERT was shown to be the most frequently mutant marker, particularly when combined with BRAF. The NGS panel was also demonstrated to be reliable, failing to produce results in only 1.2% of the samples tested.
Although BRAF is one of the most common mutations, our findings show that relying just on BRAF to characterize advanced melanomas is quite limiting. Identifying novel prognostic markers and therapeutic targets is greatly needed, as well as tailored characterization approaches, to detect patients at high risk of disease recurrence [21,22].
In addition to the aforementioned TERT, numerous mutations have been reported in NRAS, TP53, and IDH1, KIT, albeit at a lower frequency. Intriguingly, in the majority of analyzed samples (52.7%), more than one mutation was detected. The more frequent matching was between TERT and BRAF in 72 out of 127 analyzed samples (56.7%). The combination of TERT promoter mutations and BRAF p.V600E is expected to provide a strong genetic basis for tumor aggressiveness [23]. Furthermore, TERT mutations are being studied as possible therapeutic targets due to their role in melanoma progression. Strategies include developing drugs that directly inhibit the TERT function or target the mechanisms that induce TERT alterations [24].
BRAF and NRAS variants were confirmed to be mutually exclusive, except for RAS and BRAF class III variants. These latter variants are known to have low activity compared to the BRAF wild type and cannot directly phosphorylate MEK [3]. In fact, it has been previously described in the literature that BRAF class III variants may co-occur with RAS-activating mutations [17,25].
Interestingly, in all eight cases in which both primary and recurrence lesions were analyzed, a perfect match in the molecular status of the two specimens was observed, as reported in the literature [26,27]. Even if, in our cohort, the primary and metastatic samples showed the same molecular structure, the analysis of other molecular markers could be performed to understand whether variants that are not present in the primary lesion may be acquired in the metastasis.
In those cases that do not harbor any variants in the analyzed genes, other alterations may be present in markers other than those covered by the panel. In fact, although the data demonstrate that this panel was able to identify mutations in almost all advanced melanoma, in 10% of these, no alterations were identified. In these samples, it might be worth investigating other markers, such as using a panel for fusion genes (e.g., ALK fusions) [28] or a Comprehensive Genomic Profiling (CGP) panel [29,30], allowing the identification of mutations in uncommonly altered genes. With our panel, the primary and metastatic samples showed the same molecular structure; however, the extension to other markers could also be performed to understand whether there are any acquired variants in the metastases that are not present in the primary lesion.
We then provide the validation of a laboratory-developed, custom-designed multi-gene NGS panel. Using this laboratory-developed panel, we were able to analyze multiple types of cancers in the same run. This laboratory-developed NGS panel was designed to cover the diagnostic/prognostic/predictive clinical needs not only for melanomas but also for other tumors, such as CRCs (colorectal carcinomas), thyroid nodules, pancreatic lesions, gliomas, and GISTs (gastrointestinal stromal tumors) [19]. Because this panel is intended for the key gene targets of all the tumors stated above, it may be utilized to analyze several tumor types in a single run, reducing the turn-around time and NGS costs. In comparison to existing/commercial NGS multi-gene panels, the optimized selection of genes and the possibility of analyzing the relevant markers in different tumor types enables a high number of specimens to be analyzed in each run. This versatility is not possible with commercially available multi-gene panels that are dedicated to the in-depth analysis of specific tumors, whereas commercially available comprehensive multi-gene panels include a large number of targets, limiting the number of samples that can be analyzed in the same run. Pooling routine melanoma samples with other tumors enabled us to test 32–40 samples per run, with an average turnaround time of 7.1 working days. The cost of reagents was between EUR 200 and 250 per sample, depending on the number of specimens loaded in a given run, proving that an NGS-based approach may be less costly than a single-gene-based approach [31].
Considering that the analysis of other markers besides BRAF is recommended in melanoma [18], the multi-gene approach using an NGS technique is preferable to sequential testing [31,32]. Therefore, being able to test multiple genes in a single analysis allows for better molecular profiling of melanomas, and through this panel, it is possible to perform this while keeping costs relatively low and reporting times rapid.
In conclusion, this study describes the validation of a custom-designed multi-gene panel capable of analyzing relevant gene targets—25 oncogenes/oncosuppressor genes—in advanced-stage melanomas, which can be successfully used in routine clinical practice for prognostic/predictive clinical purposes.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/diagnostics14080800/s1, Table S1: Details of variants detected in the analyzed cohort using the NGS panel. ACMG: American College of Medical Genetics and Genomics; P: pathogenic; LP: likely pathogenic; VUS: variant of uncertain significance; c.: coding reference sequence; Table S2: Concomitant mutations detected in the analyzed cohort.
Author Contributions
Conceptualization, D.d.B.; methodology, T.M., S.C., V.S., G.C. and C.A.C.; formal analysis, T.M., A.D.L., S.C., V.S., E.G., A.A., G.C. and C.A.C.; data curation, F.C., B.M., P.V.M., E.D., F.V., B.C., G.T. and D.d.B.; writing—original draft preparation, T.M. and D.d.B.; writing—review and editing, G.T. and D.d.B.; supervision, G.T. and D.d.B.; project administration, G.T. and D.d.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The research was approved by the local institution ethics committee (784.2020.Oss.AOUBo, 17 September 2020). All data used in the present study were completely anonymized and aggregated. This study was conducted in accordance with the Declaration of Helsinki.
Informed Consent Statement
All data used in the present study were completely anonymized and aggregated.
Data Availability Statement
All data are included in the article/supplementary material.
Acknowledgments
The work reported in this publication is supported by the Italian Ministry of Health, RC-2024-2790027.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Cancer Genome Atlas, N. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Comito, F.; Aprile, M.; Pagani, R.; Siepe, G.; Sperandi, F.; Gruppioni, E.; Altimari, A.; De Biase, D.; Melotti, B. Clinical characteristics and treatment outcomes of non-V600 E/K BRAF mutant melanoma patients: A single-institution experience. Melanoma Res. 2022, 32, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Owsley, J.; Stein, M.K.; Porter, J.; In, G.K.; Salem, M.; O’Day, S.; Elliott, A.; Poorman, K.; Gibney, G.; VanderWalde, A. Prevalence of class I-III BRAF mutations among 114,662 cancer patients in a large genomic database. Exp. Biol. Med. 2021, 246, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Garnett, M.J.; Rana, S.; Paterson, H.; Barford, D.; Marais, R. Wild-type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol. Cell 2005, 20, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef] [PubMed]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef] [PubMed]
- Hugdahl, E.; Kalvenes, M.B.; Mannelqvist, M.; Ladstein, R.G.; Akslen, L.A. Prognostic impact and concordance of TERT promoter mutation and protein expression in matched primary and metastatic cutaneous melanoma. Br. J. Cancer 2018, 118, 98–105. [Google Scholar] [CrossRef]
- Nagore, E.; Heidenreich, B.; Rachakonda, S.; Garcia-Casado, Z.; Requena, C.; Soriano, V.; Frank, C.; Traves, V.; Quecedo, E.; Sanjuan-Gimenez, J.; et al. TERT promoter mutations in melanoma survival. Int. J. Cancer 2016, 139, 75–84. [Google Scholar] [CrossRef]
- Nagore, E.; Heidenreich, B.; Requena, C.; Garcia-Casado, Z.; Martorell-Calatayud, A.; Pont-Sanjuan, V.; Jimenez-Sanchez, A.I.; Kumar, R. TERT promoter mutations associate with fast-growing melanoma. Pigment. Cell Melanoma Res. 2016, 29, 236–238. [Google Scholar] [CrossRef]
- Shaughnessy, M.; Njauw, C.N.; Artomov, M.; Tsao, H. Classifying Melanoma by TERT Promoter Mutational Status. J. Investig. Dermatol. 2020, 140, 390–394.e1. [Google Scholar] [CrossRef] [PubMed]
- Timis, T.; Bergthorsson, J.T.; Greiff, V.; Cenariu, M.; Cenariu, D. Pathology and Molecular Biology of Melanoma. Curr. Issues Mol. Biol. 2023, 45, 5575–5597. [Google Scholar] [CrossRef] [PubMed]
- Teixido, C.; Castillo, P.; Martinez-Vila, C.; Arance, A.; Alos, L. Molecular Markers and Targets in Melanoma. Cells 2021, 10, 2320. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.; Garraway, L.A.; Fisher, D.E. Malignant melanoma: Genetics and therapeutics in the genomic era. Genes. Dev. 2006, 20, 2149–2182. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Dika, E.; Lambertini, M.; Pellegrini, C.; Veronesi, G.; Melotti, B.; Riefolo, M.; Sperandi, F.; Patrizi, A.; Ricci, C.; Mussi, M.; et al. Cutaneous and Mucosal Melanomas of Uncommon Sites: Where Do We Stand Now? J. Clin. Med. 2021, 10, 478. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017, 548, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Michielin, O.; van Akkooi, A.C.J.; Ascierto, P.A.; Dummer, R.; Keilholz, U.; ESMO Guidelines Committee. Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-updagger. Ann. Oncol. 2019, 30, 1884–1901. [Google Scholar] [CrossRef]
- de Biase, D.; Acquaviva, G.; Visani, M.; Sanza, V.; Argento, C.M.; De Leo, A.; Maloberti, T.; Pession, A.; Tallini, G. Molecular Diagnostic of Solid Tumor Using a Next Generation Sequencing Custom-Designed Multi-Gene Panel. Diagnostics 2020, 10, 250. [Google Scholar] [CrossRef] [PubMed]
- Mosele, F.; Remon, J.; Mateo, J.; Westphalen, C.B.; Barlesi, F.; Lolkema, M.P.; Normanno, N.; Scarpa, A.; Robson, M.; Meric-Bernstam, F.; et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: A report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2020, 31, 1491–1505. [Google Scholar] [CrossRef]
- Pegoraro, A.; De Marchi, E.; Ferracin, M.; Orioli, E.; Zanoni, M.; Bassi, C.; Tesei, A.; Capece, M.; Dika, E.; Negrini, M.; et al. P2X7 promotes metastatic spreading and triggers release of miRNA-containing exosomes and microvesicles from melanoma cells. Cell Death Dis. 2021, 12, 1088. [Google Scholar] [CrossRef]
- Mulder, E.; Johansson, I.; Grunhagen, D.J.; Tempel, D.; Rentroia-Pacheco, B.; Dwarkasing, J.T.; Verver, D.; Mooyaart, A.L.; van der Veldt, A.A.M.; Wakkee, M.; et al. Using a Clinicopathologic and Gene Expression (CP-GEP) Model to Identify Stage I–II Melanoma Patients at Risk of Disease Relapse. Cancers 2022, 14, 2854. [Google Scholar] [CrossRef]
- Guo, Y.; Chen, Y.; Zhang, L.; Ma, L.; Jiang, K.; Yao, G.; Zhu, L. TERT Promoter Mutations and Telomerase in Melanoma. J. Oncol. 2022, 2022, 6300329. [Google Scholar] [CrossRef] [PubMed]
- Guterres, A.N.; Villanueva, J. Targeting telomerase for cancer therapy. Oncogene 2020, 39, 5811–5824. [Google Scholar] [CrossRef]
- Bracht, J.W.P.; Karachaliou, N.; Bivona, T.; Lanman, R.B.; Faull, I.; Nagy, R.J.; Drozdowskyj, A.; Berenguer, J.; Fernandez-Bruno, M.; Molina-Vila, M.A.; et al. BRAF Mutations Classes I, II, and III in NSCLC Patients Included in the SLLIP Trial: The Need for a New Pre-Clinical Treatment Rationale. Cancers 2019, 11, 1381. [Google Scholar] [CrossRef]
- Kerkour, T.; Zhou, C.; Hollestein, L.; Mooyaart, A. Genetic Concordance in Primary Cutaneous Melanoma and Matched Metastasis: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2023, 24, 16281. [Google Scholar] [CrossRef]
- Manca, A.; Paliogiannis, P.; Colombino, M.; Casula, M.; Lissia, A.; Botti, G.; Caraco, C.; Ascierto, P.A.; Sini, M.C.; Palomba, G.; et al. Mutational concordance between primary and metastatic melanoma: A next-generation sequencing approach. J. Transl. Med. 2019, 17, 289. [Google Scholar] [CrossRef] [PubMed]
- Moran, J.M.T.; Le, L.P.; Nardi, V.; Golas, J.; Farahani, A.A.; Signorelli, S.; Onozato, M.L.; Foreman, R.K.; Duncan, L.M.; Lawrence, D.P.; et al. Identification of fusions with potential clinical significance in melanoma. Mod. Pathol. 2022, 35, 1837–1847. [Google Scholar] [CrossRef]
- Huang, R.S.P.; Tse, J.Y.; Harries, L.; Graf, R.P.; Lin, D.I.; Murugesan, K.; Hiemenz, M.C.; Parimi, V.; Janovitz, T.; Decker, B.; et al. Variable Genomic Landscapes of Advanced Melanomas with Heavy Pigmentation. Oncologist 2022, 27, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Pallocca, M.; Molineris, I.; Berrino, E.; Marcozzi, B.; Betti, M.; Levati, L.; D’Atri, S.; Menin, C.; Madonna, G.; Ghiorzo, P.; et al. Comprehensive genomic profiling on metastatic Melanoma: Results from a network screening from 7 Italian Cancer Centres. J. Transl. Med. 2024, 22, 29. [Google Scholar] [CrossRef]
- Pruneri, G.; De Braud, F.; Sapino, A.; Aglietta, M.; Vecchione, A.; Giusti, R.; Marchio, C.; Scarpino, S.; Baggi, A.; Bonetti, G.; et al. Next-Generation Sequencing in Clinical Practice: Is It a Cost-Saving Alternative to a Single-Gene Testing Approach? Pharmacoecon. Open 2021, 5, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Morganti, S.; Tarantino, P.; Ferraro, E.; D’Amico, P.; Viale, G.; Trapani, D.; Duso, B.A.; Curigliano, G. Complexity of genome sequencing and reporting: Next generation sequencing (NGS) technologies and implementation of precision medicine in real life. Crit. Rev. Oncol. Hematol. 2019, 133, 171–182. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).