
Cardiac Manifestations in Fabry Disease: A Case Report on Two Siblings


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
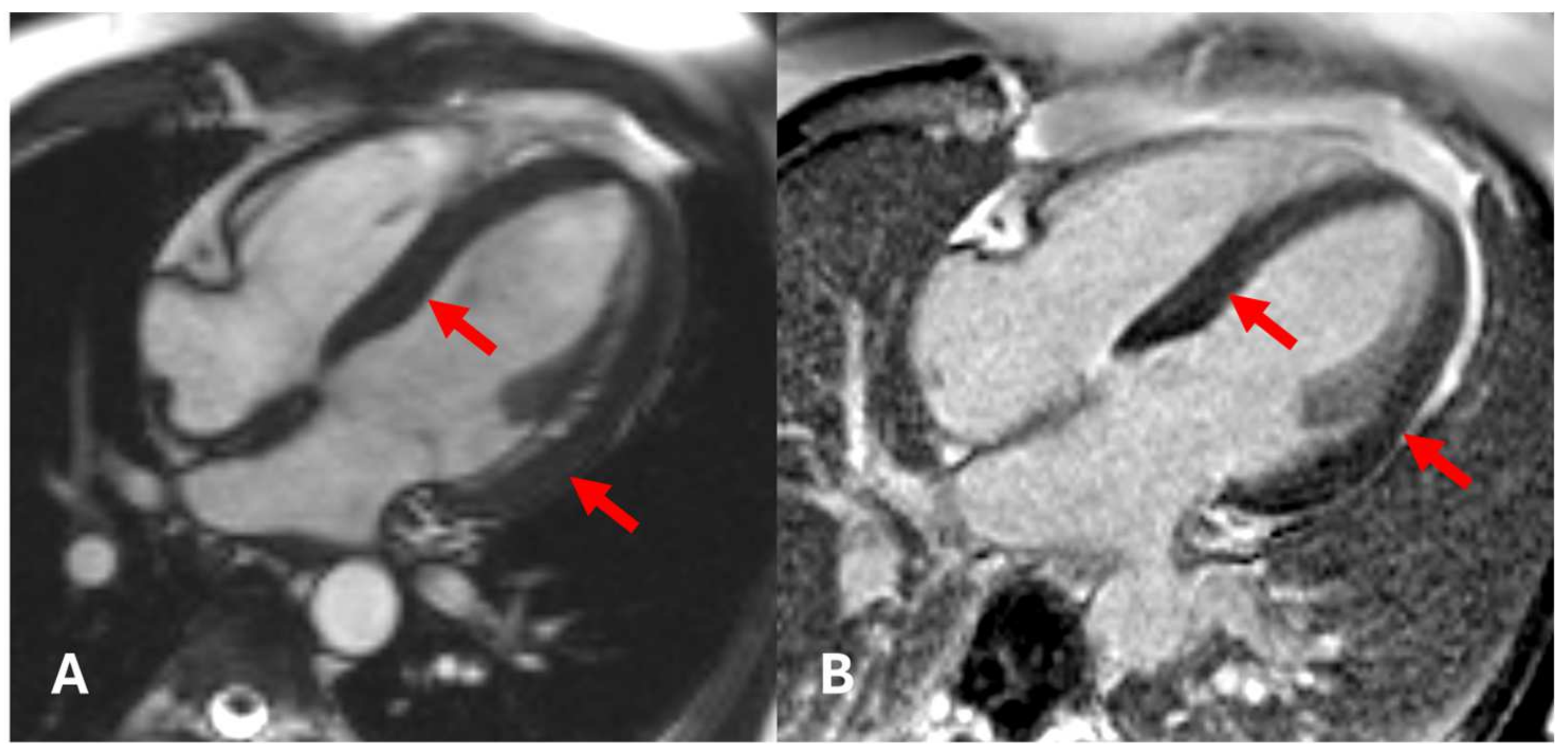
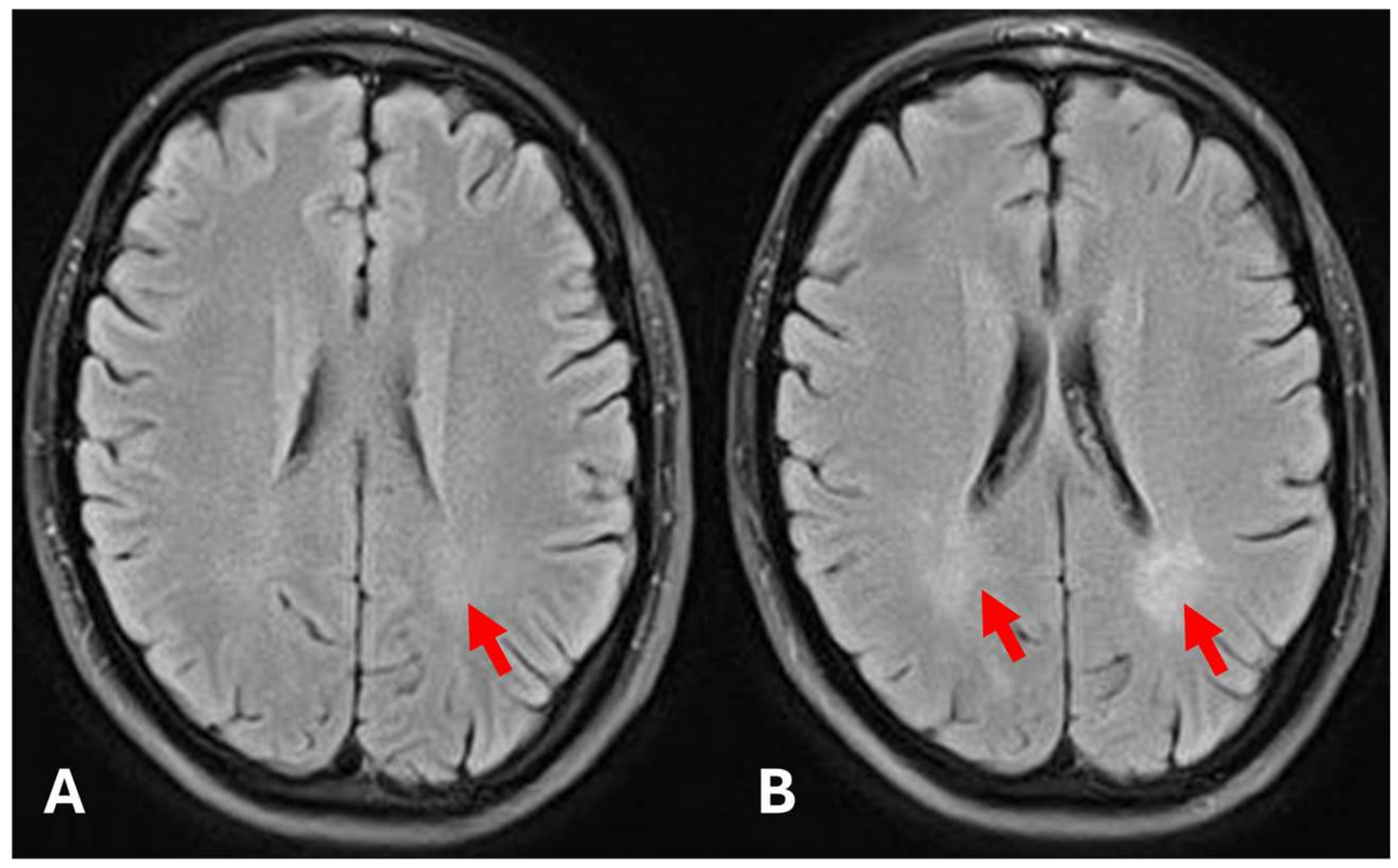
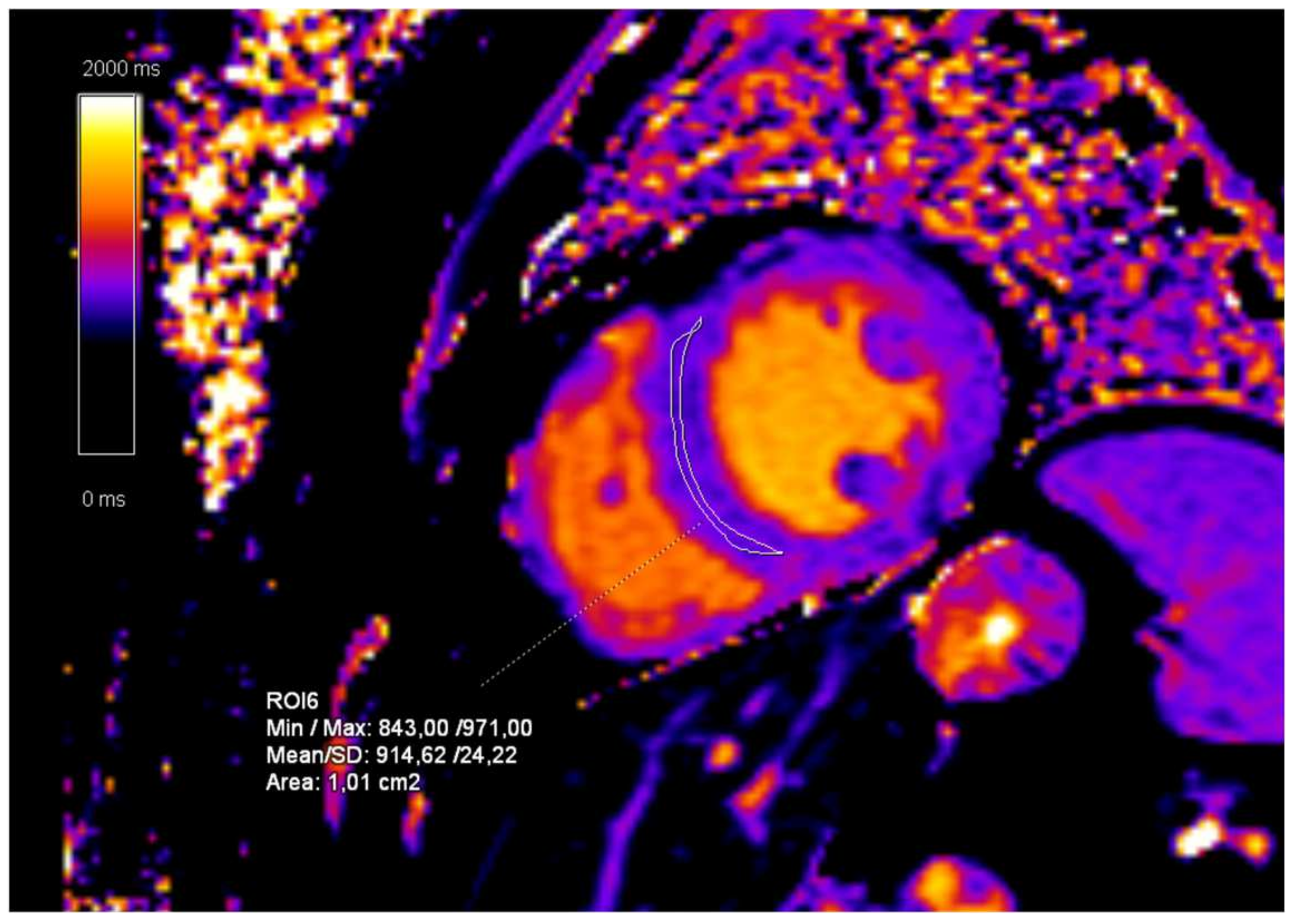
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rajan, J.N.; Ireland, K.; Johnson, R.; Stepien, K.M. Review of Mechanisms, Pharmacological Management, Psychosocial Implications, and Holistic Treatment of Pain in Fabry Disease. J. Clin. Med. 2021, 10, 4168. [Google Scholar] [CrossRef] [PubMed]
- Hagège, A.; Réant, P.; Habib, G.; Damy, T.; Barone-Rochette, G.; Soulat, G.; Donal, E.; Germain, D.P. Fabry disease in cardiology practice: Literature review and expert point of view. Arch. Cardiovasc. Dis. 2019, 112, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, Y.; Fiorini, F.; Azzini, C.; Esposito, P.; De Vito, A.; Granata, A.; Storari, A.; Mignani, R. Deficiency in the Screening Process of Fabry Disease: Analysis of Chronic Kidney Patients Not on Dialysis. Front. Med. 2021, 8, 640876. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Spada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R.J. High Incidence of Later-Onset Fabry Disease Revealed by Newborn Screening. Am. J. Hum. Genet. 2006, 79, 31–40. [Google Scholar] [CrossRef]
- Faro, D.C.; Di Pino, F.L.; Monte, I.P. Inflammation, Oxidative Stress, and Endothelial Dysfunction in the Pathogenesis of Vascular Damage: Unraveling Novel Cardiovascular Risk Factors in Fabry Disease. Int. J. Mol. Sci. 2024, 25, 8273. [Google Scholar] [CrossRef]
- Baig, S.; Edward, N.C.; Kotecha, D.; Liu, B.; Nordin, S.; Kozor, R.; Moon, J.C.; Geberhiwot, T.; Steeds, R.P. Ventricular arrhythmia and sudden cardiac death in Fabry disease: A systematic review of risk factors in clinical practice. EP Eur. 2018, 20, f153–f161. [Google Scholar] [CrossRef]
- Linhart, A.; Kampmann, C.; Zamorano, J.L.; Sunder-Plassmann, G.; Beck, M.; Mehta, A.; Elliott, P.M.; on Behalf of European FOS Investigators. Cardiac manifestations of Anderson-Fabry disease: Results from the international Fabry outcome survey. Eur. Heart J. 2007, 28, 1228–1235. [Google Scholar] [CrossRef]
- Hiestand, R.; Nowak, A.; Sokolska, J.M.; Chan, R.; Ruschitzka, F.; Manka, R.; Gruner, C. Clinical and CMR characteristics associated with cardiac events in patients with Fabry disease. Int. J. Cardiol. 2023, 382, 46–51. [Google Scholar] [CrossRef]
- Pieroni, M.; Moon, J.C.; Arbustini, E.; Barriales-Villa, R.; Camporeale, A.; Vujkovac, A.C.; Elliott, P.M.; Hagege, A.; Kuusisto, J.; Linhart, A.; et al. Cardiac Involvement in Fabry Disease: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2021, 77, 922–936. [Google Scholar] [CrossRef]
- Ricci, F.; Bisaccia, G.; Mansour, D.; Molinari, L.V.; Di Mauro, M.; Renda, G.; Khanji, M.Y.; Gallina, S. Prognostic Significance of Late Gadolinium Enhancement in Fabry Disease—A Systematic Review and Meta-Analysis. Am. J. Cardiol. 2023, 202, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Messroghli, D.R.; Moon, J.C.; Ferreira, V.M.; Grosse-Wortmann, L.; He, T.; Kellman, P.; Mascherbauer, J.; Nezafat, R.; Salerno, M.; Schelbert, E.B.; et al. Clinical recommendations for cardiovascular magnetic resonance mapping of T1, T2, T2* and extracellular volume: A consensus statement by the Society for Cardiovascular Magnetic Resonance (SCMR) endorsed by the European Association for Cardiovascular Imaging (EACVI). J. Cardiovasc. Magn. Reson. 2016, 19, 75. [Google Scholar] [CrossRef]
- Ponsiglione, A.; Gambardella, M.; Green, R.; Cantoni, V.; Nappi, C.; Ascione, R.; De Giorgi, M.; Cuocolo, R.; Pisani, A.; Petretta, M.; et al. Cardiovascular magnetic resonance native T1 mapping in Anderson-Fabry disease: A systematic review and meta-analysis. J. Cardiovasc. Magn. Reson. 2022, 24, 31. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Scalia, S.; Eichler, S.; Pockrandt, A.M.; Dehn, N.; Cozma, C.; Giese, A.K.; Rolfs, A. Functional and Clinical Consequences of Novel α-Galactosidase A Mutations in Fabry Disease. Hum. Mutat. 2016, 37, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Reisin, R.; Perrin, A.; García-Pavía, P. Time delays in the diagnosis and treatment of Fabry disease. Int. J. Clin. Pract. 2017, 71, e12914. [Google Scholar] [CrossRef]
- Azevedo, O.; Gago, M.; Miltenberger-Miltenyi, G.; Gaspar, P.; Sousa, N.; Cunha, D. Mild Left Ventricular Hypertrophy Unravels a Novel Nonsense Mutation of the GLA Gene Associated with the Classical Phenotype of Fabry Disease. Cardiology 2017, 137, 67–73. [Google Scholar] [CrossRef]
- Doheny, D.; Srinivasan, R.; Pagant, S.; Chen, B.; Yasuda, M.; Desnick, R.J. Fabry Disease: Prevalence of affected males and heterozygotes with pathogenic GLA mutations identified by screening renal, cardiac and stroke clinics, 1995–2017. J. Med. Genet. 2018, 55, 261–268. [Google Scholar] [CrossRef]
- Thompson, R.B.; Chow, K.; Khan, A.; Chan, A.; Shanks, M.; Paterson, I.; Oudit, G.Y. T1 Mapping With Cardiovascular MRI Is Highly Sensitive for Fabry Disease Independent of Hypertrophy and Sex. Circ. Cardiovasc. Imaging 2013, 6, 637–645. [Google Scholar] [CrossRef]
- Kozor, R.; Grieve, S.M.; Tchan, M.C.; Callaghan, F.; Hamilton-Craig, C.; Denaro, C.; Moon, J.C.; Figtree, G.A. Cardiac involvement in genotype-positive Fabry disease patients assessed by cardiovascular MR. Heart 2016, 102, 298–302. [Google Scholar] [CrossRef]
- Al-Arnawoot, A.; O’Brien, C.; Karur, G.R.; Nguyen, E.T.; Wasim, S.; Iwanochko, R.M.; Morel, C.F.M.; Hanneman, K.M. Clinical Significance of Papillary Muscles on Left Ventricular Mass Quantification Using Cardiac Magnetic Resonance Imaging. J. Thorac. Imaging 2021, 36, 242–247. [Google Scholar] [CrossRef]
- Coughlan, J.J.; Elkholy, K.; O’Brien, J.; Kiernan, T. Atypical patterns of cardiac involvement in Fabry disease. BMJ Case Rep. 2016, 2016, bcr2015213819. [Google Scholar] [CrossRef] [PubMed]
- Nojiri, A.; Anan, I.; Morimoto, S.; Kawai, M.; Sakuma, T.; Kobayashi, M.; Kobayashi, H.; Ida, H.; Ohashi, T.; Eto, Y.; et al. Clinical findings of gadolinium-enhanced cardiac magnetic resonance in Fabry patients. J. Cardiol. 2020, 75, 27–33. [Google Scholar] [CrossRef]
- Burton, J.O.; Jefferies, H.J.; Selby, N.M.; McIntyre, C.W. Hemodialysis-Induced Cardiac Injury: Determinants and Associated Outcomes. Clin. J. Am. Soc. Nephrol. 2009, 4, 914–920. [Google Scholar] [CrossRef]
- Pavlu, L.; Kocourkova, L.; Taborsky, M.; Petrkova, J. Ventricular tachycardia: A presentation of Fabry disease case report. Eur. Heart J. Case Rep. 2019, 3, yty154. [Google Scholar] [CrossRef]
- Nickander, J.; Cole, B.; Nordin, S.; Vijapurapu, R.; Steeds, R.P.; Moon, J.C.; Kellman, P.; Ugander, M.; Kozor, R. Increased cardiac involvement in Fabry disease using blood-corrected native T1 mapping. Sci. Rep. 2023, 13, 4420. [Google Scholar] [CrossRef]
- Pica, S.; Sado, D.M.; Maestrini, V.; Fontana, M.; White, S.K.; Treibel, T.; Captur, G.; Anderson, S.; Piechnik, S.K.; Robson, M.D.; et al. Reproducibility of native myocardial T1 mapping in the assessment of Fabry disease and its role in early detection of cardiac involvement by cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reson. 2014, 16, 99. [Google Scholar] [CrossRef]
- Ponsiglione, A.; De Giorgi, M.; Ascione, R.; Nappi, C.; Sanduzzi, L.; Pisani, A.; Dell’aversana, S.; Cuocolo, A.; Imbriaco, M. Advanced CMR Techniques in Anderson-Fabry Disease: State of the Art. Diagnostics 2023, 13, 2598. [Google Scholar] [CrossRef]
- Camporeale, A.; Moroni, F.; Lazzeroni, D.; Garibaldi, S.; Pieroni, M.; Pieruzzi, F.; Lusardi, P.; Spada, M.; Mignani, R.; Burlina, A.; et al. Trabecular complexity as an early marker of cardiac involvement in Fabry disease. Eur. Heart J. Cardiovasc. Imaging 2022, 23, 200–208. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovačić, S.; Nadarević, T.; Žauhar, P.; Vujičić, B.; Žuža, I. Cardiac Manifestations in Fabry Disease: A Case Report on Two Siblings. Diagnostics 2025, 15, 340. https://doi.org/10.3390/diagnostics15030340
Kovačić S, Nadarević T, Žauhar P, Vujičić B, Žuža I. Cardiac Manifestations in Fabry Disease: A Case Report on Two Siblings. Diagnostics. 2025; 15(3):340. https://doi.org/10.3390/diagnostics15030340
Chicago/Turabian StyleKovačić, Slavica, Tin Nadarević, Petar Žauhar, Božidar Vujičić, and Iva Žuža. 2025. "Cardiac Manifestations in Fabry Disease: A Case Report on Two Siblings" Diagnostics 15, no. 3: 340. https://doi.org/10.3390/diagnostics15030340
APA StyleKovačić, S., Nadarević, T., Žauhar, P., Vujičić, B., & Žuža, I. (2025). Cardiac Manifestations in Fabry Disease: A Case Report on Two Siblings. Diagnostics, 15(3), 340. https://doi.org/10.3390/diagnostics15030340