
DNA Methylation Array Analysis Identifies Biological Subgroups of Cutaneous Melanoma and Reveals Extensive Differences with Benign Melanocytic Nevi

 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. Macrodissection and DNA Isolation
2.3. Methylation Array Analysis
2.3.1. Array Preparation and Scanning
2.3.2. Quality Control and Tumor Purity Estimation
2.3.3. Uniform Manifold Approximation and Projection (UMAP) Analysis
2.3.4. Differential Methylation Analysis
2.4. Copy Number Variant (CNV) Detection
2.5. Next Generation Sequencing
2.5.1. Library Preparation and Sequencing
2.5.2. Small Variant Evaluation
2.6. Data Evaluation and Statistics
3. Results
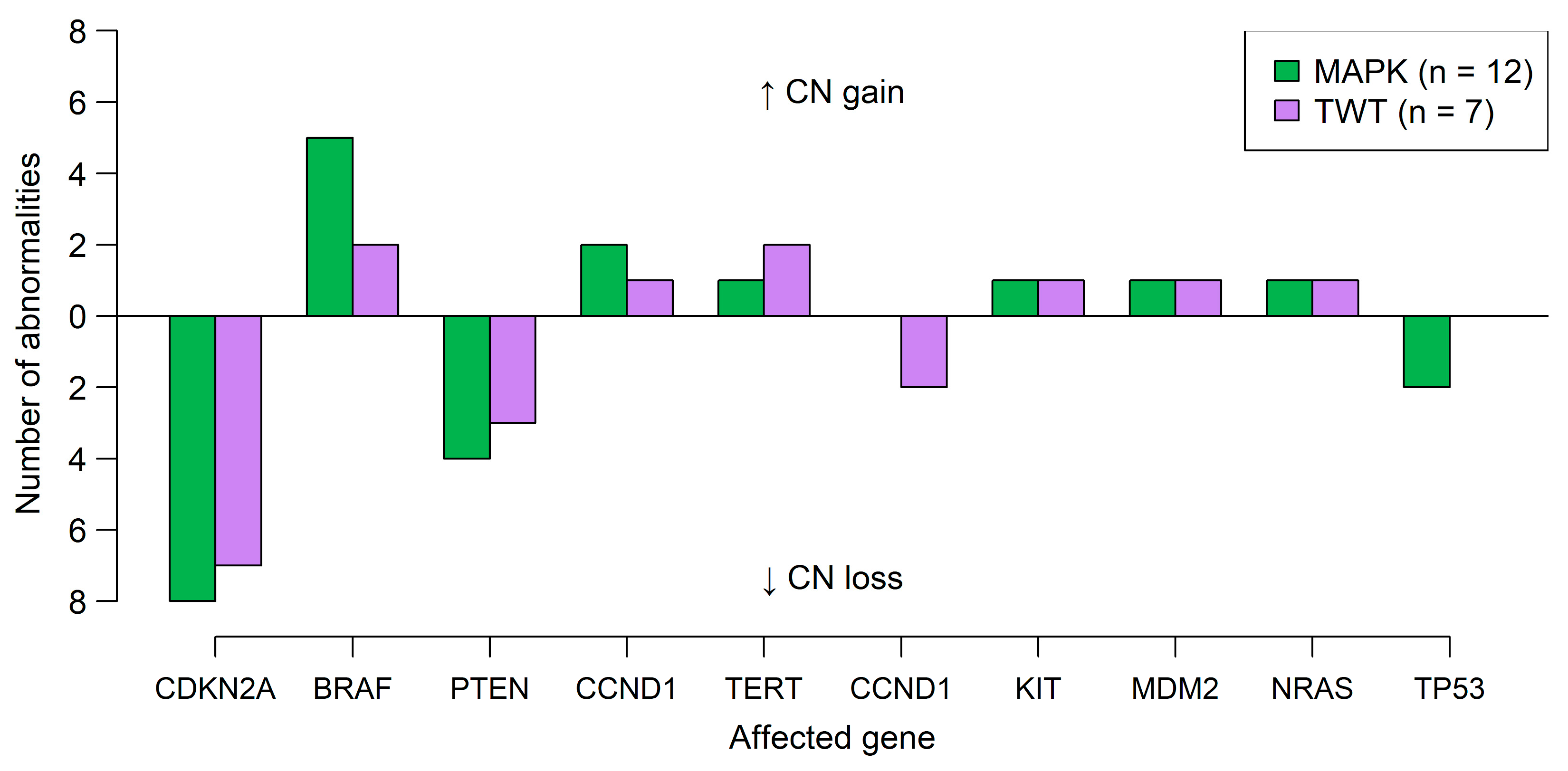
3.1. Genetic Analysis Reveals Distinct Differences Between Cases with Alterations in the MAPK Signaling Pathway and TWT Status
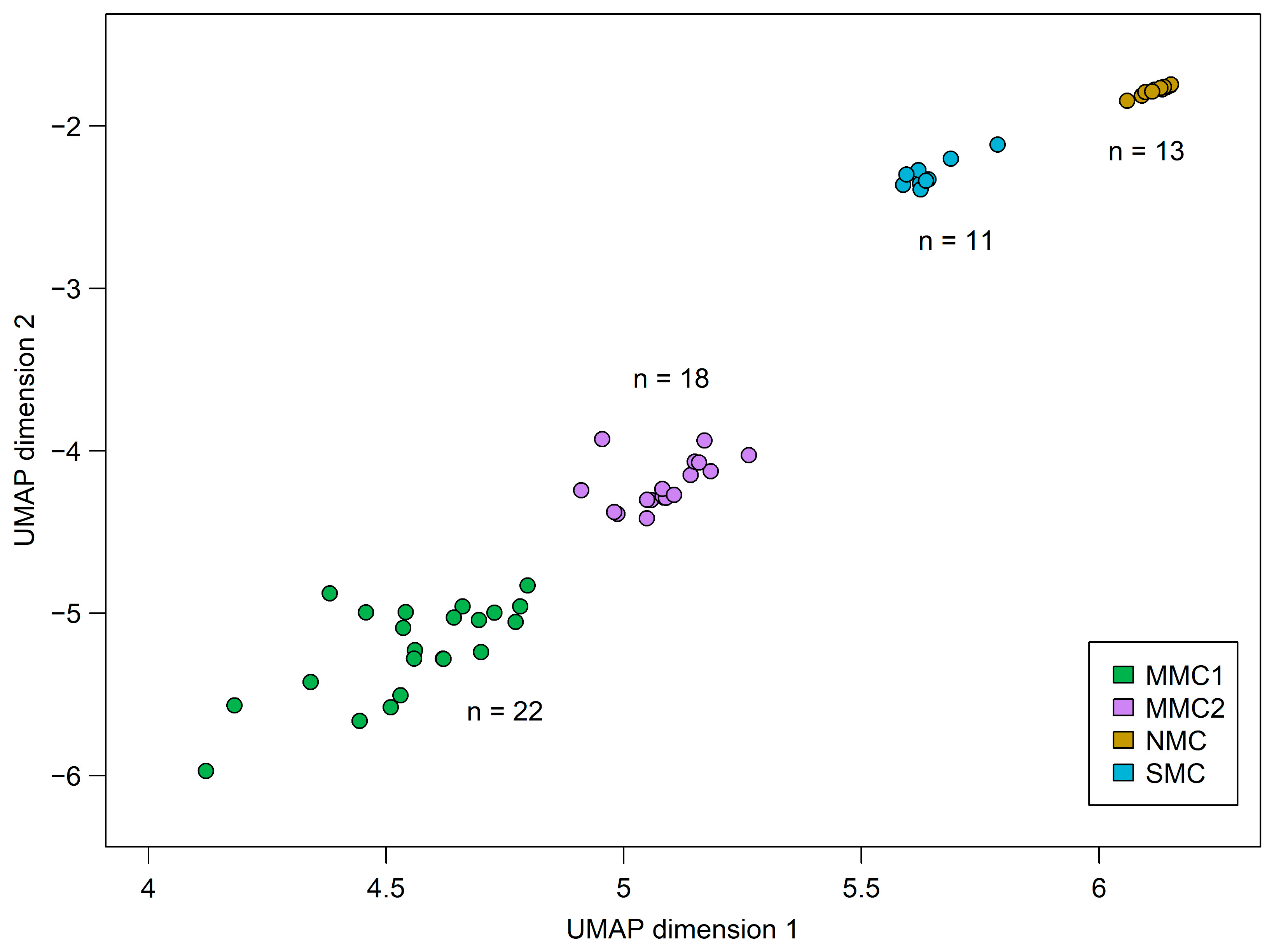
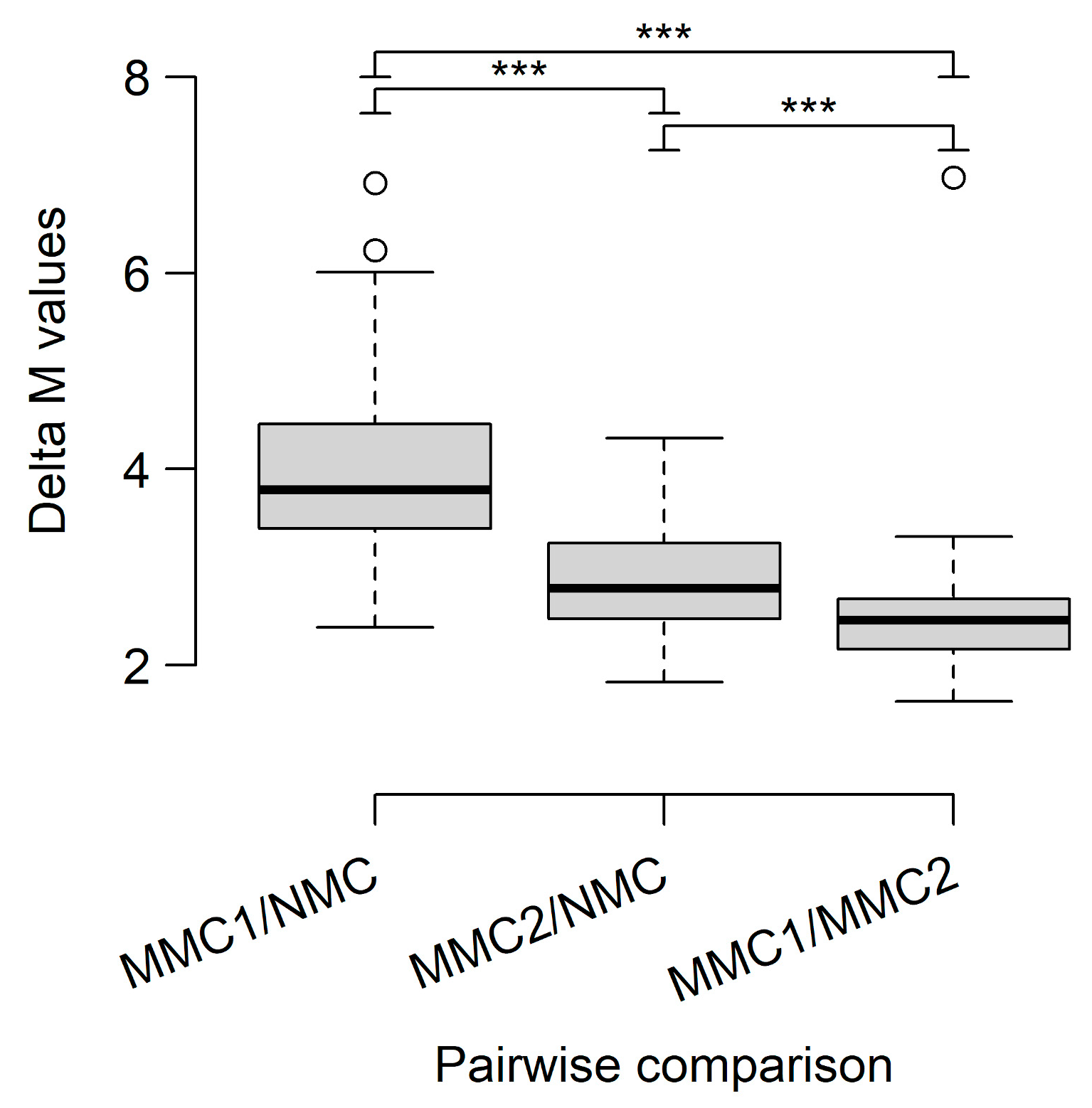
3.2. DNA Methylome Analysis Identifies Differences Between CM and MN and Indicates a Distinct Biological Subtype of CM
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bastian, B.C.; de la Fouchardière, A.; Elder, D.E.; Gerami, P.; Lazar, A.J.; Massi, D.; Nagore, E.; Scolyer, R.A.; Yun, S.J. Genomic landscape of melanoma. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R., Willemze, R., Eds.; IARC: Lyon, France, 2018; pp. 72–75. [Google Scholar]
- The Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.J.; Bastian, B.C.; Duncan, L.M.; Haneke, E.; Uhara, H. Acral melanoma. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R., Willemze, R., Eds.; IARC: Lyon, France, 2018; pp. 116–118. [Google Scholar]
- Pradhan, D.; Jour, G.; Milton, D.; Vasudevaraja, V.; Tetzlaff, M.T.; Nagarajan, P.; Curry, J.L.; Ivan, D.; Long, L.; Ding, Y.; et al. Aberrant DNA methylation predicts melanoma-specific survival in patients with acral melanoma. Cancers 2019, 11, 2031. [Google Scholar] [CrossRef] [PubMed]
- Moran, B.; Silva, R.; Perry, A.S.; Gallagher, W.M. Epigenetics of malignant melanoma. Semin. Cancer Biol. 2018, 51, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Micevic, G.; Theodosakis, N.; Bosenberg, M. Aberrant DNA methylation in melanoma: Biomarker and therapeutic opportunities. Clin. Epigenetics 2017, 9, 34. [Google Scholar] [CrossRef]
- Koroknai, V.; Szász, I.; Hernandez-Vargas, H.; Fernandez-Jimenez, N.; Cuenin, C.; Herceg, Z.; Vízkeleti, L.; Ádány, R.; Ecsedi, S.; Balázs, M. DNA hypermethylation is associated with invasive phenotype of malignant melanoma. Exp. Dermatol. 2020, 29, 39–50. [Google Scholar] [CrossRef]
- Tanemura, A.; Terando, A.M.; Sim, M.-S.; van Hoesel, A.Q.; de Maat, M.F.G.; Morton, D.L.; Hoon, D.S.B. CpG Island Methylator Phenotype predicts progression of malignant melanoma. Clin. Cancer Res. 2009, 15, 1801–1807. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Matsusaka, K.; Fukuyo, M.; Rahmutulla, B.; Matsue, H.; Kaneda, A. Higher methylation subtype of malignant melanoma and its correlation with thicker progression and worse prognosis. Cancer Med. 2020, 9, 7194–7204. [Google Scholar] [CrossRef]
- Conway, K.; Tsai, Y.S.; Edmiston, S.N.; Parker, J.S.; Parrish, E.A.; Hao, H.; Kuan, P.F.; Scott, G.A.; Frank, J.S.; Googe, P.; et al. Characterization of the CpG island hypermethylated phenotype subclass in primary melanomas. J. Investig. Dermatol. 2022, 142, 1869–1881. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Maksimovic, J.; Phipson, B.; Oshlack, A. A cross-package Bioconductor workflow for analysing methylation array data. F1000Res 2016, 5, 1281. [Google Scholar] [CrossRef] [PubMed]
- Johann, P.D.; Jäger, N.; Pfister, S.M.; Sill, M. RF_Purify: A novel tool for comprehensive analysis of tumor-purity in methylation array data based on random forest regression. BMC Bioinform. 2019, 20, 428. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.; Creighton, C.J.; Davis, C.A.; Donehower, L.; Drummond, J.; Wheeler, D.; Ally, A.; Balasundaram, M.; Birol, I.; Butterfield, Y.S.N.; et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Peters, T.J.; Buckley, M.J.; Statham, A.L.; Pidsley, R.; Samaras, K.; Lord, R.V.; Clark, S.J.; Molloy, P.L. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin 2015, 8, 6. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, 2017, bax028. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef] [PubMed]
- Holt, M.E.; Mittendorf, K.F.; LeNoue-Newton, M.; Jain, N.M.; Anderson, I.; Lovly, C.M.; Osterman, T.; Micheel, C.; Levy, M. My Cancer Genome: Coevolution of precision oncology and a molecular oncology knowledgebase. JCO Clin. Cancer Inform. 2021, 5, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Nagore, E.; Rachakonda, S.; Kumar, R. TERT promoter mutations in melanoma survival. Oncotarget 2019, 10, 1546–1548. [Google Scholar] [CrossRef]
- Hugdahl, E.; Kalvenes, M.B.; Mannelqvist, M.; Ladstein, R.G.; Akslen, L.A. Prognostic impact and concordance of TERT promoter mutation and protein expression in matched primary and metastatic cutaneous melanoma. Br. J. Cancer 2018, 118, 98–105. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- De Martino, E.; Brunetti, D.; Canzonieri, V.; Conforti, C.; Eisendle, K.; Mazzoleni, G.; Nobile, C.; Rao, F.; Zschocke, J.; Jukic, E.; et al. The association of residential altitude on the molecular profile and survival of melanoma: Results of an Interreg study. Cancers 2020, 12, 2796. [Google Scholar] [CrossRef]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.-H.; Aiba, S.; Bröcker, E.-B.; LeBoit, P.E.; et al. Distinct sets of genetic alterations in melanoma. NEJM 2005, 353, 2135–2147. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Saul, N.; Großberger, L. UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw. 2018, 3, 861. [Google Scholar] [CrossRef]
- Hench, J.; Vlajnic, T.; Soysal, S.D.; Obermann, E.C.; Frank, S.; Muenst, S. An integrated epigenomic and genomic view on Phyllodes and Phyllodes-like breast tumors. Cancers 2022, 14, 667. [Google Scholar] [CrossRef] [PubMed]
- Hench, J.; Tolnay, M.; Frank, S. Die DNA-Methylomanalyse. Swiss Med. Forum 2020, 20, 150–154. [Google Scholar] [CrossRef]
- Marx, V. Method of the year: Long-read sequencing. Nat. Methods 2023, 20, 6–11. [Google Scholar] [CrossRef]
- Yasumizu, Y.; Hagiwara, M.; Umezu, Y.; Fuji, H.; Iwaisako, K.; Asagiri, M.; Uemoto, S.; Nakamura, Y.; Thul, S.; Ueyama, A.; et al. Neural-net-based cell deconvolution from DNA methylation reveals tumor microenvironment associated with cancer prognosis. NAR Cancer 2024, 6, zcae022. [Google Scholar] [CrossRef]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.-M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef]
- Bai, X.; Kong, Y.; Chi, Z.; Sheng, X.; Cui, C.; Wang, X.; Mao, L.; Tang, B.; Li, S.; Lian, B.; et al. MAPK Pathway and TERT promoter gene mutation pattern and its prognostic value in melanoma patients: A retrospective study of 2,793 cases. Clin. Cancer Res. 2017, 23, 6120–6127. [Google Scholar] [CrossRef]
- Liau, J.Y.; Tsai, J.H.; Jeng, Y.M.; Chu, C.Y.; Kuo, K.T.; Liang, C.W. TERT promoter mutation is uncommon in acral lentiginous melanoma. J. Cutan. Pathol. 2014, 41, 504–508. [Google Scholar] [CrossRef]
- de Lima Vazquez, V.; Vicente, A.L.; Carloni, A.; Berardinelli, G.; Soares, P.; Scapulatempo, C.; Martinho, O.; Reis, R.M. Molecular profiling, including TERT promoter mutations, of acral lentiginous melanomas. Melanoma Res. 2016, 26, 93–99. [Google Scholar] [CrossRef]
- Williams, E.A.; Montesion, M.; Shah, N.; Sharaf, R.; Pavlick, D.C.; Sokol, E.S.; Alexander, B.; Venstrom, J.; Elvin, J.A.; Ross, J.S.; et al. Melanoma with in-frame deletion of MAP2K1: A distinct molecular subtype of cutaneous melanoma mutually exclusive from BRAF, NRAS, and NF1 mutations. Mod. Pathol. 2020, 33, 2397–2406. [Google Scholar] [CrossRef] [PubMed]
- Pham, D.D.M.; Guhan, S.; Tsao, H. KIT and melanoma: Biological insights and clinical implications. Yonsei Med. J. 2020, 61, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S.; Wang, K.; Chmielecki, J.; Gay, L.; Johnson, A.; Chudnovsky, J.; Yelensky, R.; Lipson, D.; Ali, S.M.; Elvin, J.A.; et al. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int. J. Cancer 2016, 138, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Jurmeister, P.; Wrede, N.; Hoffmann, I.; Vollbrecht, C.; Heim, D.; Hummel, M.; Wolkenstein, P.; Koch, I.; Heynol, V.; Schmitt, W.D.; et al. Mucosal melanomas of different anatomic sites share a common global DNA methylation profile with cutaneous melanoma but show location-dependent patterns of genetic and epigenetic alterations. J. Pathol. 2022, 256, 61–70. [Google Scholar] [CrossRef]
- Lezcano, C.; Jungbluth, A.A.; Nehal, K.S.; Hollmann, T.J.; Busam, K.J. PRAME Expression in Melanocytic Tumors. Am J Surg Pathol. 2018, 42, 1456–1465. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | n (%) or Mean ± SD |
|---|---|
| Gender | |
| Cutaneous melanoma | |
| Female | 10 (53%) |
| Male | 9 (47%) |
| Melanocytic nevi | |
| Female | 5 (45%) |
| Male | 6 (55%) |
| Skin | |
| Female | 6 (55%) |
| Male | 5 (45%) |
| Age at diagnosis (years, mean ± SD) | |
| Cutaneous melanoma | 59.8 ± 23.8 (31–96) |
| Melanocytic nevi | 34.1 ± 11.9 (16–52) |
| Altitude of residence (cutaneous melanoma) | |
| <1000 m | 16 (84%) |
| >1000 m | 3 (16%) |
| Localization | |
| Cutaneous melanoma | |
| Head or neck | 3 (15%) |
| Trunk | 4 (21%) |
| Extremities | 6 (32%) |
| Acral | 6 (32%) |
| Melanocytic nevi | |
| Head or neck | 3 (27%) |
| Trunk | 6 (55%) |
| Extremities | 2 (18%) |
| Histological subtype | |
| Cutaneous melanoma | |
| Desmoplastic melanoma | 1 (5.25%) |
| Acral lentiginous melanoma | 4 (21%) |
| Lentigo maligna melanoma | 1 (5.25%) |
| Nodular melanoma | 8 (42%) |
| Spindle cell melanoma | 2 (11%) |
| Superficial spreading melanoma | 3 (16%) |
| Melanocytic nevi | |
| Compound nevus | 7 (64%) |
| Dermal nevus | 4 (36%) |
| Tumor stage (cutaneous melanoma) | |
| IIA | 1 (5.25%) |
| IIB | 3 (16%) |
| IIC | 4 (21%) |
| III | 1 (5.25%) |
| IIIA | 1 (5.25%) |
| IIIB | 1 (5.25%) |
| IIIC | 5 (26%) |
| IV | 3 (16%) |
| Location | Range | Length | Meth. Status | DNA Elements of Interest |
|---|---|---|---|---|
| MMC1 vs. NMC | ||||
| chr2 | 223,163,573–223,172,329 | 8757 | + | CCDC140, PAX3 |
| chr14 | 10,1505,130–101,515,879 | 10,750 | − | microRNA cluster |
| chr3 | 147,122,664–147,131,860 | 9197 | + | ZIC4, ZIC1, GH03J147407 |
| chr14 | 101,487,756–101,493,252 | 5497 | − | microRNA cluster |
| chr2 | 200,328,645–200,336,146 | 7502 | + | ATB2, SAT2B, GH02J199454 |
| chr6 | 29,520,527–29,521,803 | 1277 | + | OR2I1P, GH06J029552 |
| chr22 | 22,898,356–22,902,665 | 4310 | − | PRAME |
| chr3 | 157,812,018–157,817,678 | 5661 | + | SHOX2, GH03J158097 |
| chr6 | 31,650,735–31,651,676 | 942 | + | GH06J031682 |
| chr14 | 60,972,853–60,978,852 | 6000 | + | SIX6 |
| MMC2 vs. NMC | ||||
| chr3 | 46,446,998–46,449,636 | 2639 | − | CCR5AS, CCRL2, GH03J046404 |
| chr1 | 160,680,856–160,682,655 | 1800 | − | CD48, GH01J160703 |
| chr1 | 233,248,709–233,249,314 | 606 | − | - |
| chr13 | 102,568,345–102,570,482 | 2138 | − | FGF14 |
| MMC2 vs. NMC (continued) | ||||
| chr2 | 176,963,315–176,965,729 | 2415 | + | HOXD12 |
| chr1 | 203,320,190–203,321,087 | 898 | − | GH01J203319 |
| chr14 | 61,108,227–61,110,649 | 2423 | + | SIX1, GH14J060640 |
| chr18 | 53,068,921–53,070,851 | 1931 | − | TCF4, GH18J055398 |
| chr11 | 2,846,681–2,848,492 | 1812 | − | GH11J002824 |
| chr1 | 234,907,722–234,908,514 | 793 | − | GH01J234766 |
| MMC1 vs. MMC2 | ||||
| chr14 | 101,487,756–101,493,252 | 5497 | − | microRNA cluster |
| chr2 | 166,649,910–166,651,571 | 1662 | + | GALTN3, GH02J165791 |
| chr14 | 101,518,766–101,522,431 | 3666 | − | microRNA cluster |
| chr7 | 157,527,573–157,534,758 | 7186 | − | - |
| chr1 | 203,320,190–203,321,854 | 1665 | + | FMOD, GH01J203349 |
| chr10 | 106,027,915–106,029,358 | 1444 | + | MIR4428, STO2, GSTO2, GH10J104267 |
| chr12 | 120,241,287–120,242,513 | 1227 | + | GH12J119803 |
| chr2 | 54,784,402–54,786,148 | 1747 | + | SPTBN1, GH02J05455 |
| chr1 | 234,667,087–234,668,366 | 1280 | + | LINC01354, GH01J234527 |
| chr1 | 91,300,215–91,302,117 | 1903 | + | LINC02609 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwendinger, S.; Jaschke, W.; Walder, T.; Hench, J.; Vogi, V.; Frank, S.; Hoffmann, P.; Herms, S.; Zschocke, J.; Nguyen, V.A.; et al. DNA Methylation Array Analysis Identifies Biological Subgroups of Cutaneous Melanoma and Reveals Extensive Differences with Benign Melanocytic Nevi. Diagnostics 2025, 15, 531. https://doi.org/10.3390/diagnostics15050531
Schwendinger S, Jaschke W, Walder T, Hench J, Vogi V, Frank S, Hoffmann P, Herms S, Zschocke J, Nguyen VA, et al. DNA Methylation Array Analysis Identifies Biological Subgroups of Cutaneous Melanoma and Reveals Extensive Differences with Benign Melanocytic Nevi. Diagnostics. 2025; 15(5):531. https://doi.org/10.3390/diagnostics15050531
Chicago/Turabian StyleSchwendinger, Simon, Wolfram Jaschke, Theresa Walder, Jürgen Hench, Verena Vogi, Stephan Frank, Per Hoffmann, Stefan Herms, Johannes Zschocke, Van Anh Nguyen, and et al. 2025. "DNA Methylation Array Analysis Identifies Biological Subgroups of Cutaneous Melanoma and Reveals Extensive Differences with Benign Melanocytic Nevi" Diagnostics 15, no. 5: 531. https://doi.org/10.3390/diagnostics15050531
APA StyleSchwendinger, S., Jaschke, W., Walder, T., Hench, J., Vogi, V., Frank, S., Hoffmann, P., Herms, S., Zschocke, J., Nguyen, V. A., Schmuth, M., & Jukic, E. (2025). DNA Methylation Array Analysis Identifies Biological Subgroups of Cutaneous Melanoma and Reveals Extensive Differences with Benign Melanocytic Nevi. Diagnostics, 15(5), 531. https://doi.org/10.3390/diagnostics15050531