
Pneumocystis Jirovecii Pneumonia: The Potential of KEX1, MSG1, and MSG2 as Key Antigens in Cytokine Release Assays

 , , , , and
, , , , and
Abstract
1. Introduction
1.1. Pneumocystis Jirovecii: An Unusual Fungus
1.2. Pneumocystis Jirovecii Pneumonia (PJP) and Epidemiology
1.3. The Challenge of Diagnosing PJP
1.4. Cytokine Release Assays (CRA)
1.5. The Idea of a CRA for Diagnosing PJP
2. Materials and Methods
2.1. Participant Selection
2.2. Flow Cytometric Analysis
2.3. Antigen and Cytokine Selection
2.4. In-Vitro Cell Stimulation and Cytokine Measurement
2.5. Data Analysis
3. Results
3.1. Leukocytes, T-Cells, and CD4+/CD8+ Ratio
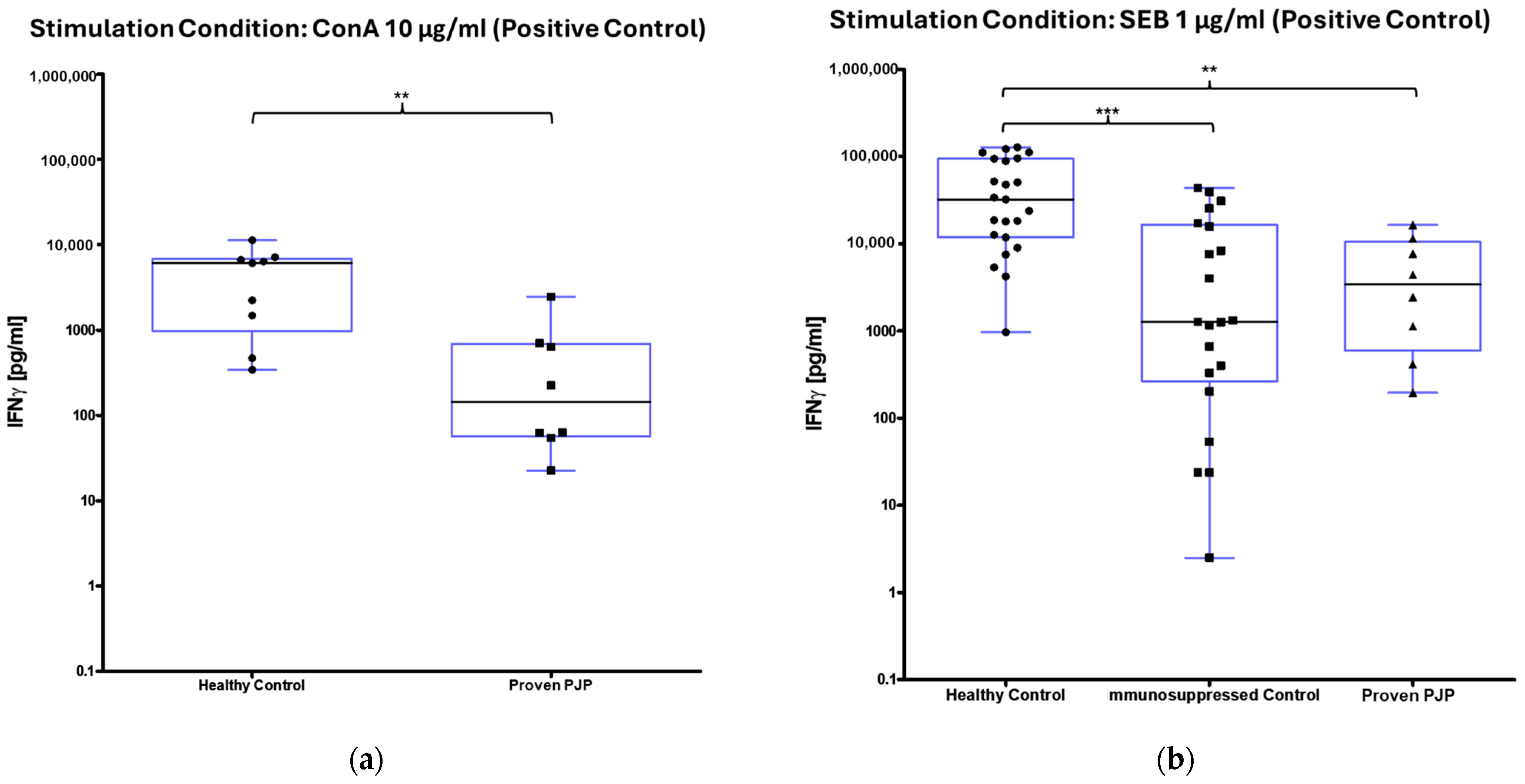
3.2. IFN-γ Release After Stimulation
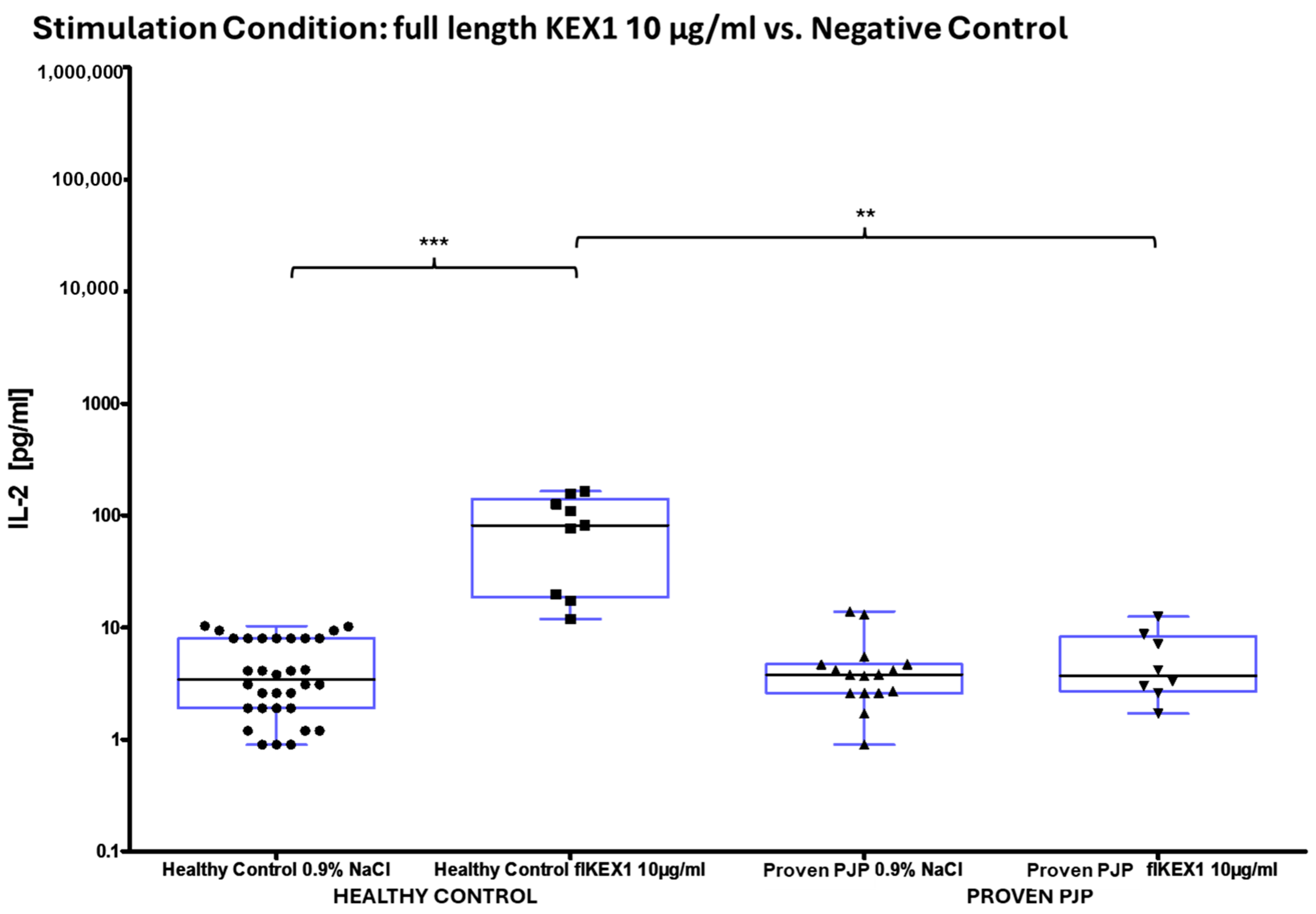
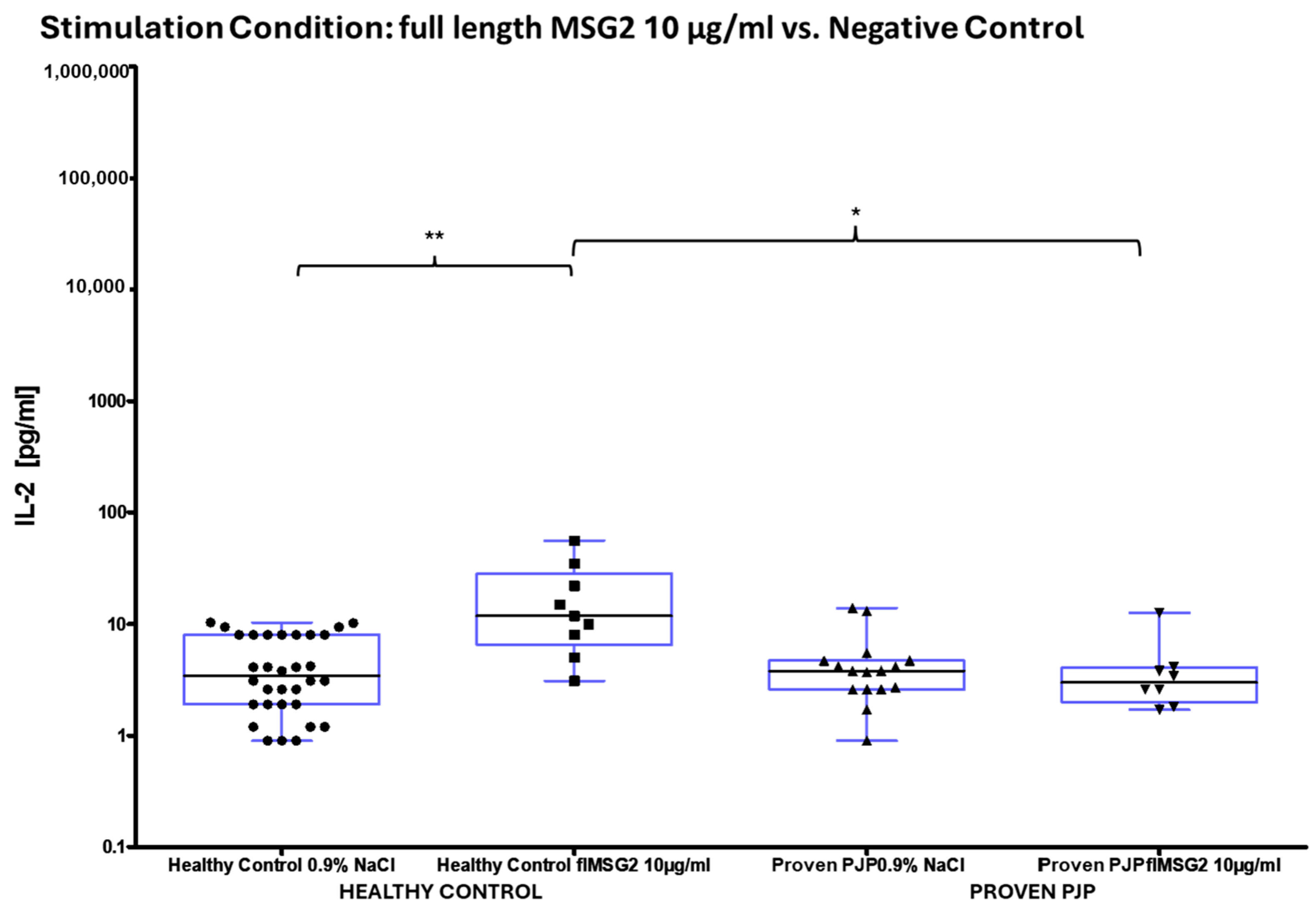
3.3. IL-2 Release After Stimulation
3.4. IL-17A and IL-17F Release After Stimulation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Edman, J.C.; Kovacs, J.A.; Masur, H.; Santi, D.V.; Elwood, H.J.; Sogin, M.L. Ribosomal RNA Sequence Shows Pneumocystis Carinii to Be a Member of the Fungi. Nature 1988, 334, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Stringer, J.R.; Beard, C.B.; Miller, R.F.; Wakefield, A.E. A New Name (Pneumocystis jiroveci) for Pneumocystis from Humans. Emerg. Infect. Dis. 2002, 8, 891–896. [Google Scholar] [CrossRef]
- Schmatz, D.M.; Romancheck, M.A.; Pittarelli, L.A.; Schwartz, R.E.; Fromtling, R.A.; Nollstadt, K.H.; Vanmiddlesworth, F.L.; Wilson, K.E.; Turner, M.J. Treatment of Pneumocystis Carinii Pneumonia with 1,3-Beta-Glucan Synthesis Inhibitors. Proc. Natl. Acad. Sci. USA 1990, 87, 5950–5954. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, M.S.; Queener, S.F.; Shaw, M.M.; Richardson, J.D.; Smith, J.W. Pneumocystis Carinii Is Resistant to Imidazole Antifungal Agents. Antimicrob. Agents Chemother. 1994, 38, 1859–1861. [Google Scholar] [CrossRef]
- Ruhnke, M.; Cornely, O.A.; Schmidt-Hieber, M.; Alakel, N.; Boell, B.; Buchheidt, D.; Christopeit, M.; Hasenkamp, J.; Heinz, W.J.; Hentrich, M.; et al. Treatment of Invasive Fungal Diseases in Cancer Patients—Revised 2019 Recommendations of the Infectious Diseases Working Party (AGIHO) of the German Society of Hematology and Oncology (DGHO). Mycoses 2020, 63, 653–682. [Google Scholar] [CrossRef]
- Ma, L.; Chen, Z.; Huang, D.W.; Kutty, G.; Ishihara, M.; Wang, H.; Abouelleil, A.; Bishop, L.; Davey, E.; Deng, R.; et al. Genome Analysis of Three Pneumocystis Species Reveals Adaptation Mechanisms to Life Exclusively in Mammalian Hosts. Nat. Commun. 2016, 7, 10740. [Google Scholar] [CrossRef] [PubMed]
- Stringer, J.R.; Keely, S.P. Genetics of Surface Antigen Expression in Pneumocystis Carinii. Infect. Immun. 2001, 69, 627–639. [Google Scholar] [CrossRef]
- Cushion, M.T.; Tisdale-Macioce, N.; Sayson, S.G.; Porollo, A. The Persistent Challenge of Pneumocystis Growth Outside the Mammalian Lung: Past and Future Approaches. Front. Microbiol. 2021, 12, 681474. [Google Scholar] [CrossRef]
- Thomas, C.F.; Limper, A.H. Pneumocystis Pneumonia. N. Engl. J. Med. 2004, 350, 2487–2498. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control. Europe, W.H.O.R.O. for HIV/AIDS Surveillance in Europe 2019: 2018 Data; European Centre for Disease Prevention and Control: Solna Sweden; World Health Organization Regional Office for Europe: Geneva, Swtizerland, 2019; ISBN 978-92-890-5460-7. [Google Scholar]
- O’Brien, K.L.; Baggett, H.C.; Brooks, W.A.; Feikin, D.R.; Hammitt, L.L.; Higdon, M.M.; Howie, S.R.C.; Deloria Knoll, M.; Kotloff, K.L.; Levine, O.S.; et al. Causes of Severe Pneumonia Requiring Hospital Admission in Children without HIV Infection from Africa and Asia: The PERCH Multi-Country Case-Control Study. Lancet 2019, 394, 757–779. [Google Scholar] [CrossRef]
- Pereira-Díaz, E.; Moreno-Verdejo, F.; de la Horra, C.; Guerrero, J.A.; Calderón, E.J.; Medrano, F.J. Changing Trends in the Epidemiology and Risk Factors of Pneumocystis Pneumonia in Spain. Front. Public Health 2019, 7, 275. [Google Scholar] [CrossRef]
- Roux, A.; Canet, E.; Valade, S.; Gangneux-Robert, F.; Hamane, S.; Lafabrie, A.; Maubon, D.; Debourgogne, A.; Le Gal, S.; Dalle, F.; et al. Pneumocystis jirovecii Pneumonia in Patients with or without AIDS, France. Emerg. Infect. Dis. 2014, 20, 1490–1497. [Google Scholar] [CrossRef]
- Benedict, K.; Baggs, J.; Gold, J.A.W. Epidemiology, Clinical Characteristics, and Diagnostic Testing Practices for Pneumocystis Pneumonia–Associated Hospitalizations, United States, 2019–2022. Open Forum Infect. Dis. 2024, 11, ofae054. [Google Scholar] [CrossRef]
- Mansharamani, N.G.; Garland, R.; Delaney, D.; Koziel, H. Management and Outcome Patterns for Adult Pneumocystis Carinii Pneumonia, 1985 to 1995: Comparison of HIV-Associated Cases to Other Immunocompromised States. Chest 2000, 118, 704–711. [Google Scholar] [CrossRef]
- Kamel, T.; Boulain, T. Pneumocystis Pneumonia in French Intensive Care Units in 2013–2019: Mortality and Immunocompromised Conditions. Ann. Intensive Care 2024, 14, 80. [Google Scholar] [CrossRef] [PubMed]
- Li, M.-C.; Lee, N.-Y.; Lee, C.-C.; Lee, H.-C.; Chang, C.-M.; Ko, W.-C. Pneumocystis jiroveci Pneumonia in Immunocompromised Patients: Delayed Diagnosis and Poor Outcomes in Non-HIV-Infected Individuals. J. Microbiol. Immunol. Infect. 2014, 47, 42–47. [Google Scholar] [CrossRef]
- Kovacs, J.A.; Masur, H. Evolving Health Effects of Pneumocystis: One Hundred Years of Progress in Diagnosis and Treatment. JAMA 2009, 301, 2578–2585. [Google Scholar] [CrossRef]
- Reinwald, M.; Buchheidt, D.; Hummel, M.; Duerken, M.; Bertz, H.; Schwerdtfeger, R.; Reuter, S.; Kiehl, M.G.; Barreto-Miranda, M.; Hofmann, W.-K.; et al. Diagnostic Performance of an Aspergillus-Specific Nested PCR Assay in Cerebrospinal Fluid Samples of Immunocompromised Patients for Detection of Central Nervous System Aspergillosis. PLoS ONE 2013, 8, e56706. [Google Scholar] [CrossRef] [PubMed]
- Reinwald, M.; Spiess, B.; Heinz, W.J.; Vehreschild, J.J.; Lass-Flörl, C.; Kiehl, M.; Schultheis, B.; Krause, S.W.; Wolf, H.-H.; Bertz, H.; et al. Diagnosing Pulmonary Aspergillosis in Patients with Hematological Malignancies: A Multicenter Prospective Evaluation of an Aspergillus PCR Assay and a Galactomannan ELISA in Bronchoalveolar Lavage Samples. Eur. J. Haematol. 2012, 89, 120–127. [Google Scholar] [CrossRef]
- Fauchier, T.; Hasseine, L.; Gari-Toussaint, M.; Casanova, V.; Marty, P.M.; Pomares, C. Detection of Pneumocystis jirovecii by Quantitative PCR To Differentiate Colonization and Pneumonia in Immunocompromised HIV-Positive and HIV-Negative Patients. J. Clin. Microbiol. 2016, 54, 1487–1495. [Google Scholar] [CrossRef]
- Grønseth, S.; Rogne, T.; Hannula, R.; Åsvold, B.O.; Afset, J.E.; Damås, J.K. Semiquantitative Real-Time PCR to Distinguish Pneumocystis Pneumonia from Colonization in a Heterogeneous Population of HIV-Negative Immunocompromised Patients. Microbiol. Spectr. 2021, 9, e0002621. [Google Scholar] [CrossRef]
- Al-Qadi, M.O.; Cartin-Ceba, R.; Kashyap, R.; Kaur, S.; Peters, S.G. The Diagnostic Yield, Safety, and Impact of Flexible Bronchoscopy in Non-HIV Immunocompromised Critically Ill Patients in the Intensive Care Unit. Lung 2018, 196, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Bateman, M.; Oladele, R.; Kolls, J.K. Diagnosing Pneumocystis jirovecii Pneumonia: A Review of Current Methods and Novel Approaches. Med. Mycol. 2020, 58, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Tomás, A.L.; Cardoso, F.; de Sousa, B.; Matos, O. Detection of Anti-Pneumocystis jirovecii Antibodies in Human Serum Using a Recombinant Synthetic Multi-Epitope Kexin-Based Antigen. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 2205–2209. [Google Scholar] [CrossRef]
- Pungan, D.; Eddens, T.; Song, K.; Lakey, M.A.; Crovetto, N.S.; Arora, S.K.; Husain, S.; Kolls, J.K. Targeted NGS-Based Analysis of Pneumocystis jirovecii Reveals Novel Genotypes. J. Fungi 2022, 8, 863. [Google Scholar] [CrossRef]
- Maertens, J.; Cesaro, S.; Maschmeyer, G.; Einsele, H.; Donnelly, J.P.; Alanio, A.; Hauser, P.M.; Lagrou, K.; Melchers, W.J.G.; Helweg-Larsen, J.; et al. ECIL Guidelines for Preventing Pneumocystis jirovecii Pneumonia in Patients with Haematological Malignancies and Stem Cell Transplant Recipients. J. Antimicrob. Chemother. 2016, 71, 2397–2404. [Google Scholar] [CrossRef] [PubMed]
- Christopeit, M.; Schmidt-Hieber, M.; Sprute, R.; Buchheidt, D.; Hentrich, M.; Karthaus, M.; Penack, O.; Ruhnke, M.; Weissinger, F.; Cornely, O.A.; et al. Prophylaxis, Diagnosis and Therapy of Infections in Patients Undergoing High-Dose Chemotherapy and Autologous Haematopoietic Stem Cell Transplantation. 2020 Update of the Recommendations of the Infectious Diseases Working Party (AGIHO) of the German Society of Hematology and Medical Oncology (DGHO). Ann. Hematol. 2021, 100, 321–336. [Google Scholar] [CrossRef]
- Classen, A.Y.; Henze, L.; von Lilienfeld-Toal, M.; Maschmeyer, G.; Sandherr, M.; Graeff, L.D.; Alakel, N.; Christopeit, M.; Krause, S.W.; Mayer, K.; et al. Primary Prophylaxis of Bacterial Infections and Pneumocystis jirovecii Pneumonia in Patients with Hematologic Malignancies and Solid Tumors: 2020 Updated Guidelines of the Infectious Diseases Working Party of the German Society of Hematology and Medical Oncology (AGIHO/DGHO). Ann. Hematol. 2021, 100, 1603–1620. [Google Scholar] [CrossRef]
- Goletti, D.; Delogu, G.; Matteelli, A.; Migliori, G.B. The Role of IGRA in the Diagnosis of Tuberculosis Infection, Differentiating from Active Tuberculosis, and Decision Making for Initiating Treatment or Preventive Therapy of Tuberculosis Infection. Int. J. Infect. Dis. 2022, 124, S12–S19. [Google Scholar] [CrossRef]
- Dammermann, W.; Bentzien, F.; Stiel, E.-M.; Kühne, C.; Ullrich, S.; Schulze Zur Wiesch, J.; Lüth, S. Development of a Novel IGRA Assay to Test T Cell Responsiveness to HBV Antigens in Whole Blood of Chronic Hepatitis B Patients. J. Transl. Med. 2015, 13, 157. [Google Scholar] [CrossRef]
- Dammermann, W.; Bochmann, D.; Bentzien, F.; Komorowski, L.; Steinhagen, K.; Ullrich, S.; van Lunzen, J.; Lüth, S. CMV Specific Cytokine Release Assay in Whole Blood Is Optimized by Combining Synthetic CMV Peptides and Toll like Receptor Agonists. J. Immunol. Methods 2014, 414, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, D.; Malkovsky, M.; Reibnegger, G.; Werner, E.R.; Forni, G.; Wachter, H. Endogenous Release of Interferon-Gamma and Diminished Response of Peripheral Blood Mononuclear Cells to Antigenic Stimulation. Immunol. Lett. 1989, 23, 103–108. [Google Scholar] [CrossRef]
- Rahlwes, K.C.; Dias, B.R.S.; Campos, P.C.; Alvarez-Arguedas, S.; Shiloh, M.U. Pathogenicity and Virulence of Mycobacterium Tuberculosis. Virulence 2023, 14, 2150449. [Google Scholar] [CrossRef]
- Anderson, R.P.; Goel, G.; Hardy, M.Y.; Russell, A.K.; Wang, S.; Szymczak, E.; Zhang, R.; E Goldstein, K.; Neff, K.; E Truitt, K.; et al. Whole Blood Interleukin-2 Release Test to Detect and Characterize Rare Circulating Gluten-specific T Cell Responses in Coeliac Disease. Clin. Exp. Immunol. 2021, 204, 321. [Google Scholar] [CrossRef]
- Dammermann, W.; Dornbrack, J.; Bröker, K.; Bentzien, F.; Lüth, S. CpG Oligonucleotides Increase HBV-Specific Cytokine Responses in Whole Blood and Enhance Cytokine Release Assay Sensitivity. J. Virol. Methods 2017, 248, 195–201. [Google Scholar] [CrossRef]
- Kutty, G.; Kovacs, J.A. A Single-Copy Gene Encodes Kex1, a Serine Endoprotease of Pneumocystis Jiroveci. Infect. Immun. 2003, 71, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Tomás, A.L.; Pinto, M.; Cardoso, F.; de Almeida, M.P.; de Sousa, B.; Pereira, E.; Franco, R.; Matos, O. Application of Synthetic Recombinant Multi-Epitope Antigens and Gold Nanoparticles for a Pneumocystis Pneumonia Rapid Diagnostic Test. Ann. Med. 2019, 51, 92. [Google Scholar] [CrossRef]
- Tomás, A.L.; de Almeida, M.P.; Cardoso, F.; Pinto, M.; Pereira, E.; Franco, R.; Matos, O. Development of a Gold Nanoparticle-Based Lateral-Flow Immunoassay for Pneumocystis Pneumonia Serological Diagnosis at Point-of-Care. Front. Microbiol. 2019, 10, 2917. [Google Scholar] [CrossRef]
- Djawe, K.; Huang, L.; Daly, K.R.; Levin, L.; Koch, J.; Schwartzman, A.; Fong, S.; Roth, B.; Subramanian, A.; Grieco, K.; et al. Serum Antibody Levels to the Pneumocystis jirovecii Major Surface Glycoprotein in the Diagnosis of P. Jirovecii Pneumonia in HIV+ Patients. PLoS ONE 2010, 5, e14259. [Google Scholar] [CrossRef]
- Datta, K.; LaRue, R.; Permpalung, N.; Das, S.; Zhang, S.; Mehta Steinke, S.; Bushkin, Y.; Nosanchuk, J.D.; Marr, K.A. Development of an Interferon-Gamma Release Assay (IGRA) to Aid Diagnosis of Histoplasmosis. J. Clin. Microbiol. 2022, 60, e01128-22. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Karosiene, E.; Edwards, L.; Grifoni, A.; Paul, S.; Andreatta, M.; Weiskopf, D.; Sidney, J.; Nielsen, M.; Peters, B.; et al. Predicting HLA CD4 Immunogenicity in Human Populations. Front. Immunol. 2018, 9, 1369. [Google Scholar] [CrossRef]
- Schwarzlose-Schwarck, S.; Reinwald, M.; Bauer, T.; Hentschel, F.; Kiderlen, T.; Zapf, D.; Herbst, V.; Lüth, S.; Krieger, D.; Dammermann, W. Evaluation of Three Novel Antigens and Costimulatory Agents for Improvement of M. Tuberculosis Specific Interferon Gamma Release Assays. BMC Infect. Dis. 2025, 25, 188. [Google Scholar] [CrossRef]
- Ben-Nun, A.; Yossefi, S. Staphylococcal Enterotoxin B as a Potent Suppressant of T Lymphocytes: Trace Levelssuppress T Lymphocyte Proliferative Responses. Eur. J. Immunol. 1992, 22, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Ioannidou, K.; Baumgaertner, P.; Gannon, P.O.; Speiser, M.F.; Allard, M.; Hebeisen, M.; Rufer, N.; Speiser, D.E. Heterogeneity Assessment of Functional T Cell Avidity. Sci. Rep. 2017, 7, 44320. [Google Scholar] [CrossRef]
- Malek, T.R. The Biology of Interleukin-2. Annu. Rev. Immunol. 2008, 26, 453–479. [Google Scholar] [CrossRef]
- Chen, K.; Kolls, J.K. Interluekin-17A (IL17A). Gene 2017, 614, 8–14. [Google Scholar] [CrossRef]
- Iwakura, Y.; Ishigame, H.; Saijo, S.; Nakae, S. Functional Specialization of Interleukin-17 Family Members. Immunity 2011, 34, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nagase, H.; Tagawa, Y.; Taki, S.; Takamoto, M. Endogenous IFN-γ Facilitates Pneumocystis Infection and Downregulates Carbohydrate Receptors in CD4+ T Cell-Depleted Mice. FEBS Lett. 2024, 598, 1633–1643. [Google Scholar] [CrossRef]
- Kann, G.; Wetzstein, N.; Bielke, H.; Schuettfort, G.; Haberl, A.E.; Wolf, T.; Kuepper-Tetzel, C.P.; Wieters, I.; Kessel, J.; de Leuw, P.; et al. Risk Factors for IRIS in HIV-Associated Pneumocystis-Pneumonia Following ART Initiation. J. Infect. 2021, 83, 347–353. [Google Scholar] [CrossRef]
- Dewasurendra, D.; Kitchen, M.; Gisinger, M.; Heller, I.; Schmuth, M.; Sarcletti, M. Immune Reconstitution Inflammatory Syndrome (IRIS) in an HIV-Positive Patient with Pneumocystis jirovecii Pneumonia (PjP) and Morbus Kaposi. J. Dtsch. Dermatol. Ges. 2020, 18, 1305–1308. [Google Scholar] [CrossRef]
- Bunte, K.; Beikler, T. Th17 Cells and the IL-23/IL-17 Axis in the Pathogenesis of Periodontitis and Immune-Mediated Inflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 3394. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Kaplan, H.J.; Sun, D. Bidirectional Effect of IFN-γ on Th17 Responses in Experimental Autoimmune Uveitis. Front. Ophthalmol. 2022, 2, 831084. [Google Scholar] [CrossRef] [PubMed]
- Bröker, K.; Terzenbach, R.; Bentzien, F.; Lüth, S.; Dammermann, W. Complement Factors C3a and C5a Mimick a Proinflammatory Microenvironment and Increase HBV IGRA Sensitivity. J. Transl. Med. 2019, 17, 6. [Google Scholar] [CrossRef] [PubMed]
- Dammermann, W.; Wollenberg, L.; Bentzien, F.; Lohse, A.; Lüth, S. Toll like Receptor 2 Agonists Lipoteichoic Acid and Peptidoglycan Are Able to Enhance Antigen Specific IFNγ Release in Whole Blood during Recall Antigen Responses. J. Immunol. Methods 2013, 396, 107–115. [Google Scholar] [CrossRef]
- Shen, H.-P.; Tang, Y.-M.; Song, H.; Xu, W.-Q.; Yang, S.-L.; Xu, X.-J. Efficiency of Interleukin 6 and Interferon Gamma in the Differentiation of Invasive Pulmonary Aspergillosis and Pneumocystis Pneumonia in Pediatric Oncology Patients. Int. J. Infect. Dis. 2016, 48, 73–77. [Google Scholar] [CrossRef]
- Karageorgopoulos, D.E.; Qu, J.-M.; Korbila, I.P.; Zhu, Y.-G.; Vasileiou, V.A.; Falagas, M.E. Accuracy of β-D-Glucan for the Diagnosis of Pneumocystis jirovecii Pneumonia: A Meta-Analysis. Clin. Microbiol. Infect. 2013, 19, 39–49. [Google Scholar] [CrossRef]
- Li, W.-J.; Guo, Y.-L.; Liu, T.-J.; Wang, K.; Kong, J.-L. Diagnosis of Pneumocystis Pneumonia Using Serum (1-3)-β-D-Glucan: A Bivariate Meta-Analysis and Systematic Review. J. Thorac. Dis. 2015, 7, 2214–2225. [Google Scholar] [CrossRef]
- Vogel, M.N.; Weissgerber, P.; Goeppert, B.; Hetzel, J.; Vatlach, M.; Claussen, C.D.; Horger, M. Accuracy of Serum LDH Elevation for the Diagnosis of Pneumocystis jiroveci Pneumonia. Swiss Med. Wkly. 2011, 141, w13184. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | ||||
|---|---|---|---|---|
| Healthy Individuals | Immunocompromised Individuals 1 | Individuals with Proven PJP | ||
| Total N = 50 | 22 (44%) | 20 (40%) | 8 (16%) | |
| Male | 12 (55%) | 12 (60%) | 8 (100%) | |
| Female | 10 (45%) | 8 (40%) | - | |
| Age (Years) 2 | 41 ± 17 | 66 ± 12 | 46 ± 23 | |
| Condition Number | Stimulation Condition | Composition | Final Antigen Concentration in Blood [µg/mL] |
|---|---|---|---|
| 1 | Negative control (Baseline): 0.9% sodium chloride (NaCl) | 500 µL whole blood +50 µL 0.9% NaCl solution +5 µL 1 mg/mL glucose solution | - |
| 2 | Positive control: SEB | 500 µL whole blood +50 µL SEB 1 µg/mL +5 µL 1 mg/mL glucose solution | 1 |
| 3 | Positive control: Concanavalin A | 500 µL whole blood | 10 |
| 4 | KEX1 partial (aa 291–380) 100 µg/mL | 500 µL whole blood +50 µL KEX1 partial +5 µL 1 mg/mL glucose solution | 100 |
| 5 | KEX1 partial (aa 291–380) 50 µg/mL | 500 µL whole blood +50 µL KEX1 partial +5 µL 1 mg/mL glucose solution | 50 |
| 6 | KEX1 partial (aa 291–380) 10 µg/mL | 500 µL whole blood +50 µL KEX1 partial +5 µL 1 mg/mL glucose solution | 10 |
| 7 | KEX1 partial (aa 291–380) 5 µg/mL | 500 µL whole blood +50 µL KEX1 partial +5 µL 1 mg/mL glucose solution | 5 |
| 8 | KEX1 partial (aa 291–380) 1 µg/mL | 500 µL whole blood +50 µL KEX1 partial +5 µL 1 mg/mL glucose solution | 1 |
| 9 | KEX1 partial (aa 291–380) 0.5 µg/mL | 500 µL whole blood +50 µL KEX1 partial +5 µL 1 mg/mL glucose solution | 0.5 |
| 10 | KEX1 partial (aa 291–380) 0.1 µg/mL | 500 µL whole blood +50 µL KEX1 partial +5 µL 1 mg/mL glucose solution | 0.1 |
| 11 | MSG2 partial (aa 32–72, 170–208, 264–305) 100 µg/mL | 500 µL whole blood +50 µL MSG2 partial +5 µL 1 mg/mL glucose solution | 100 |
| 12 | MSG2 partial (aa 32–72, 170–208, 264–305) 50 µg/mL | 500 µL whole blood +50 µL MSG2 partial +5 µL 1 mg/mL glucose solution | 50 |
| 13 | MSG2 partial (aa 32–72, 170–208, 264–305) 10 µg/mL | 500 µL whole blood +50 µL MSG2 partial +5 µL 1 mg/mL glucose solution | 10 |
| 14 | MSG2 partial (aa 32–72, 170–208, 264–305) 5 µg/mL | 500 µL whole blood +50 µL MSG2 partial +5 µL 1 mg/mL glucose solution | 5 |
| 15 | MSG2 partial (aa 32–72, 170–208,264–305) 1 µg/mL | 500 µL whole blood +50 µL MSG2 partial +5 µL 1 mg/mL glucose solution | 1 |
| 16 | MSG2 partial (aa 32–72, 170–208, 264–305) 0.5 µg/mL | 500 µL whole blood +50 µL MSG2 partial +5 µL 1 mg/mL glucose solution | 0.5 |
| 17 | MSG2 partial (aa 32–72, 170–208, 264–305) 0.1 µg/mL | 500 µL whole blood +50 µL MSG2 partial +5 µL 1 mg/mL glucose solution | 0.1 |
| 18 | KEX1 full length 10 µg/mL (EUROIMMUN AG) | 500 µL whole blood | 10 |
| 19 | MSG 1 full length 10 µg/mL (EUROIMMUN AG) | 500 µL whole blood | 10 |
| 20 | MSG 2 full length 10 µg/mL (EUROIMMUN AG) | 500 µL whole blood | 10 |
| 21 | PJ-Mix full length 10 µg/mL (EUROIMMUN AG) | 500 µL whole blood | 10 |
| Leucocytes | T-Lymphocytes | ||||
|---|---|---|---|---|---|
| N | Leucocytes (CD45+) [109/L] | T-Lymphocytes (CD45+, CD3+) [Cells/µL] | Th-Lymphocytes (CD4+) [Cells/µL] | CD4+/CD8+–Ratio | |
| All | 25 | 7 ± 1 (6) | 1109 ± 151 (1087) | 553 ± 122 (549) | 2 ± 0 (2) |
| Healthy control | 10 | 7 ± 0 (7) | 1524 ± 176 (1391) | 938 ± 139 (799) | 2 ± 0 (2) |
| Immunocompromised control | 7 | 10 ± 2 (9) | N/A 1 | N/A 1 | N/A 1 |
| Proven PJP | 8 | 5 ± 1 (4) | 701 ± 140 (800) | 118 ± 58 (57) | 0 ± 0 (0) |
| IFN-γ [pg/mL] | Negative Control | Positive Controls | ||
|---|---|---|---|---|
| N | NaCl | SEB | ConA 1 | |
| All | 50 | 12 ± 4 (3) | 25,337 ± 4896 (8995) | 2583 ± 800 (670) |
| Healthy control | 22 | 3 ± 1 (3) | 47,760 ± 9046 (32,220) | 4683 ± 1247 (6104) |
| Immunocompromised control | 20 | 9 ± 3 (3) | 9470 ± 3033 (1277) | N/A 2 |
| Proven PJP | 8 | 54 ± 15 (27) | 5508 ± 2084 (3408) | 528 ± 292 (145) |
| IFN-γ [pg/mL] | N | Full Length KEX1 10 µg/mL | Full Length MSG1 10 µg/mL | Full Length MSG2 10 µg/mL | Full Length PJ-MIX 10 µg/mL |
|---|---|---|---|---|---|
| All | 17 1 | 42 ± 12 (20) | 52 ± 18 (17) | 34 ± 11 (16) | 67 ± 19 (23) |
| Healthy control | 9 | 31 ± 14 (15) | 52 ± 30 (14) | 17 ± 9 (3) | 82 ± 32 (26) |
| Proven PJP | 8 | 60 ± 22 (33) | 59 ± 22 (32) | 58 ± 20 (40) | 59 ± 23 (27) |
| IFN-γ [pg/mL] | ||||||||
|---|---|---|---|---|---|---|---|---|
| Partial KEX1 | N | 100 µg/mL | 50 µg/mL | 10 µg/mL | 5 µg/mL | 1 µg/mL | 0.5 µg/mL | 0.1 µg/mL |
| All | 35 | 17 ± 7 (3) | 17 ± 7 (3) | 13 ± 4 (3) | 17 ± 7 (3) | 15 ± 6 (3) | 16 ± 7 (3) | 15 ± 6 (3) |
| Healthy control | 14 | 3 ± 1 (3) | 3 ± 1 (3) | 3 ± 1 (3) | 2 ± 0 (3) | 2 ± 1 (3) | 3 ± 1 (3) | 2 ± 0 (3) |
| Immunocompromised control | 13 | 6 ± 3 (3) | 6 ± 3 (3) | 9 ± 4 (3) | 6 ± 3 (3) | 7 ± 3 (3) | 6 ± 3 (3) | 6 ± 3 (3) |
| Proven PJP | 8 | 53 ± 23 (24) | 53 ± 22 (26) | 52 ± 22 (24) | 54 ± 23 (29) | 47 ± 20 (30) | 52 ± 21 (32) | 48 ± 20 (24) |
| IFN-γ [pg/mL] | ||||||||
|---|---|---|---|---|---|---|---|---|
| Partial MSG2 | N | 100 µg/mL | 50 µg/mL | 10µg/mL | 5 µg/mL | 1 µg/mL | 0.5 µg/mL | 0.1 µg/mL |
| All | 30 | 23 ± 9 (5) | 23 ± 8 (6) | 19 ± 6 (4) | 21 ± 8 (6) | 23 ± 8 (7) | 21 ± 7 (6) | 23 ± 8 (4) |
| Healthy control | 9 | N/A 1 | N/A 1 | 4 ± 1 (3) | N/A 1 | N/A 1 | N/A 1 | N/A 1 |
| Immunocompromised control | 13 | 11 ± 4 (4) | 12 ± 5 (5) | 11 ± 4 (4) | 10 ± 4 (5) | 11 ± 4 (6) | 11 ± 5 (3) | 11 ± 5 (3) |
| Proven PJP | 8 | 53 ± 23 (24) | 52 ± 22 (27) | 49 ± 20 (24) | 48 ± 20 (24) | 51 ± 22 (24) | 46 ± 18 (24) | 53 ± 22 (26) |
| IL-2 [pg/mL] | Negative Control | Positive Controls | ||
|---|---|---|---|---|
| N | NaCl | SEB | ConA 1 | |
| All | 47 | 4 ± 1 (3) | 36,404 ± 7046 (15,291) | 2146 ± 1266 (93) |
| Healthy Control | 18 | 5 ± 1 | 57,300 ± 14,420 (25,910) | 6815 ± 3212 (5.007) |
| Immunocompromised Control | 21 | 4 ± 1 | 23,860 ± 7645 (11,800) | N/A 2 |
| Proven PJP | 8 | 5 ± 1 | 4766 ± 1947 (2542) | 79 ± 42 (17) |
| IL-2 [pg/mL] | N | Full Length KEX1 10 µg/mL | Full Length MSG1 10 µg/mL | Full Length MSG2 10 µg/mL | Full Length PJ-MIX 10 µg/mL |
|---|---|---|---|---|---|
| All | 17 1 | 45 ± 14 (12) | 31 ± 9 (14) | 11 ± 3 (5) | 63 ± 21 (19) |
| Healthy Control | 9 | 85 ± 20 (82) | 58 ± 14 (52) | 18 ± 6 (12) | 131 ± 32 (136) |
| Proven PJP | 8 | 5 ± 1 (4) | 8 ± 2 (6) | 4 ± 1 (3) | 10 ± 3 (8) |
| IL-2 [pg/mL] | ||||||||
|---|---|---|---|---|---|---|---|---|
| Partial KEX1 | N | 100 µg/mL | 50 µg/mL | 10 µg/mL | 5 µg/mL | 1 µg/mL | 0.5 µg/mL | 0.1 µg/mL |
| All | 35 | 4 ± 1 (3) | 5 ± 1 (3) | 4 ± 1 (3) | 4 ± 1 (3) | 4 ± 1 (3) | 6 ± 3 (3) | 4 ± 1 (2) |
| Healthy Control | 14 | 3 ± 1 (2) | 4 ± 2 (2) | 4 ± 2 (3) | 2 ± 0 (2) | 2 ± 0 (2) | 7 ± 5 (2) | 2 ± 0 (2) |
| Immunocompromised Control | 13 | 6 ± 2 (2) | 6 ± 2 (4) | 4 ± 1 (2) | 6 ± 2 (2) | 6 ±2 (3) | 6 ± 2 (2) | 5 ± 2 (2) |
| Proven PJP | 8 | 4 ± 1 (4) | 4 ± 1 (4) | 5 ± 1 (3) | 4 ± 1 (4) | 5 ± 1 (4) | 5 ± 1 (3) | 5 ± 1 (4) |
| IL-2 [pg/mL] | ||||||||
|---|---|---|---|---|---|---|---|---|
| Partial MSG2 | N | 100 µg/mL | 50 µg/mL | 10 µg/mL | 5 µg/mL | 1 µg/mL | 0.5 µg/mL | 0.1 µg/mL |
| All | 30 | 3 ± 0 (3) | 3 ± 1 (3) | 5 ± 1 (3) | 3 ± 1 (3) | 3 ± 1 (3) | 3 ± 1 (3) | 3 ± 1 (3) |
| Healthy Control | 9 | N/A 1 | N/A 1 | 8 ± 2 (8) | N/A 1 | N/A 1 | N/A 1 | N/A 1 |
| Immunocompromised Control | 13 | 4 ± 1 (2) | 4 ± 1 (2) | 3 ± 1 (2) | 3 ± 1 (2) | 3 ± 1 (2) | 3 ± 1 (2) | 3 ± 1 (2) |
| Proven PJP | 8 | 4 ± 1 (4) | 4 ± 1 (4) | 5 ± 1 (4) | 4 ± 1 (4) | 4 ± 1 (4) | 5 ± 1 (4) | 5 ± 1 (4) |
| IL-17A [pg/mL] | Negative Control | Positive Controls | Partial Antigens | |||
|---|---|---|---|---|---|---|
| N | NaCl 1 | SEB | ConA | Partial KEX1 10 µg/mL | Partial MSG2 10 µg/mL | |
| All | 4 1 | 4 ± 2 (3) | 253 ± 0 (253) | 46 ± 17 (31) | 3 ± 0 (3) | 5 ± 2 (3) |
| Healthy Control | 2 | 3 ± 0 (3) | 253 ± 0 (253) | 67 ± 30 (67) | 3 ± 0 (3) | 3 ± 0 (3) |
| Immunocompromised Control | 1 | 3 ± 0 (3) | 253 ± 0 (253) | 25 ± 0 (25) | 3 ± 0 (3) | 3 ± 0 (3) |
| Proven PJP | 1 | 6 ± 2 (6) | 253 ± 0 (253) | 25 ± 0 (25) | 3 ± 0 (3) | 11 ± 0 (11) |
| IL-17A [pg/mL] | N | Full Length KEX1 10 µg/mL | Full Length MSG1 10 µg/mL | Full Length MSG2 10 µg/mL | Full Length PJ-MIX 10 µg/mL |
|---|---|---|---|---|---|
| All | 4 1 | 4 ± 2 (3) | 5 ± 3 (3) | 3 ± 1 (3) | 3 ± 1 (3) |
| Healthy Control | 2 | 3 ± 0 (3) | 2 ± 0 (2) | 3 ± 0 (3) | 3 ± 0 (3) |
| Immunocompromised Control | 1 | 3 ± 0 (3) | 2 ± 0 (2) | 3 ± 0 (3) | 3 ± 0 (3) |
| Proven PJP | 1 | 10 ± 0 (10) | 13 ± 0 (13) | 6 ± 0 (6) | 5 ± 0 (5) |
| IL-17F [pg/mL] | Negative Control | Positive Controls | Partial Antigenes | |||
|---|---|---|---|---|---|---|
| N | NaCl | SEB | ConA | Partial KEX1 10 µg/mL | Partial MSG2 10 µg/mL | |
| All | 4 1 | 3 ± 0 (3) | 6 ± 2 (5) | 13 ± 10 (5) | 3 ± 0 (3) | 3 ± 0 (3) |
| Healthy Control | 2 | 3 ± 0 (3) | 9 ± 4 (9) | 33 ± 0 (33) | 3 ± 0 (3) | 3 ± 0 (3) |
| Immunocompromised Control | 1 | 3 ± 0 (3) | 5 ± 0 (5) | 5 ± 0 (5) | 3 ± 0 (3) | 3 ± 0 (3) |
| Proven PJP | 2 | 3 ± 0 (3) | 4 ± 1 (4) | 3 ± 0 (3) | 3 ± 0 (3) | 3 ± 0 (3) |
| IL-17F [pg/mL] | N | Full Length KEX1 10 µg/mL | Full Length MSG1 10 µg/mL | Full Length MSG2 10 µg/mL | Full Length PJ-MIX 10 µg/mL |
|---|---|---|---|---|---|
| All | 3 1 | 3 ± 0 (3) | 3 ± 0 (3) | 3 ± 0 (3) | 3 ± 0 (3) |
| Healthy Control | 1 | 3 ± 0 (3) | 3 ± 0 (3) | 3 ± 0 (3) | 3 ± 0 (3) |
| Immunocompromised Control | 1 | 3 ± 0 (3) | 3 ± 0 (3) | 3 ± 0 (3) | 3 ± 0 (3) |
| Proven PJP | 1 | 3 ± 0 (3) | 3 ± 0 (3) | 3 ± 0 (3) | 3 ± 0 (3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neumann, F.A.O.; Müller, M.; Mattert, G.; Liebig, S.; Herbst, V.; Zapf, D.; Kiderlen, T.R.; Linke, C.; Arp, F.; Deckert, P.M.; et al. Pneumocystis Jirovecii Pneumonia: The Potential of KEX1, MSG1, and MSG2 as Key Antigens in Cytokine Release Assays. Diagnostics 2025, 15, 793. https://doi.org/10.3390/diagnostics15070793
Neumann FAO, Müller M, Mattert G, Liebig S, Herbst V, Zapf D, Kiderlen TR, Linke C, Arp F, Deckert PM, et al. Pneumocystis Jirovecii Pneumonia: The Potential of KEX1, MSG1, and MSG2 as Key Antigens in Cytokine Release Assays. Diagnostics. 2025; 15(7):793. https://doi.org/10.3390/diagnostics15070793
Chicago/Turabian StyleNeumann, F. A. Ottilie, Markus Müller, Gregor Mattert, Sven Liebig, Victor Herbst, Dorinja Zapf, Til R. Kiderlen, Christian Linke, Franziska Arp, P. Markus Deckert, and et al. 2025. "Pneumocystis Jirovecii Pneumonia: The Potential of KEX1, MSG1, and MSG2 as Key Antigens in Cytokine Release Assays" Diagnostics 15, no. 7: 793. https://doi.org/10.3390/diagnostics15070793
APA StyleNeumann, F. A. O., Müller, M., Mattert, G., Liebig, S., Herbst, V., Zapf, D., Kiderlen, T. R., Linke, C., Arp, F., Deckert, P. M., Lüth, S., Schwarzlose-Schwarck, S., Dammermann, W., & Reinwald, M. (2025). Pneumocystis Jirovecii Pneumonia: The Potential of KEX1, MSG1, and MSG2 as Key Antigens in Cytokine Release Assays. Diagnostics, 15(7), 793. https://doi.org/10.3390/diagnostics15070793