1. Introduction
The mucopolysaccharidoses (MPSs) are a group of rare lysosomal storage disorders caused by the deficiency of specific enzymes that catalyze the stepwise degradation of glycosaminoglycans (GAGs). Currently, 11 enzymes are known to be involved in the catabolism of dermatan sulfate (DS), heparan sulfate (HS), keratan sulfate (KS), chondroitin sulfate (CS), and hyaluronic acid. Specific enzyme deficiency leads to the accumulation of GAGs in cells and eventually causes progressive damage to various tissues and organs [
1]. The clinical presentations of MPS include coarse facial features, developmental delay, corneal clouding, adenotonsillar hypertrophy, hearing loss, upper airway obstruction, pulmonary function impairment, obstructive sleep apnea, cardiovascular disease, hepatosplenomegaly, short stature, joint stiffness, and skeletal deformities (dysostosis multiplex). The clinical signs and symptoms in these patients are chronic and progressive, and may present from early to late childhood or even in early adulthood, and the severity and prognosis vary among different types with a wide spectrum of clinical severity [
2,
3,
4,
5,
6,
7,
8,
9,
10]. All types of MPS exhibit autosomal recessive inheritance except MPS II (Hunter syndrome), which is transmitted in an X-linked recessive mode and thus predominantly affects males. The incidence of MPS in different populations ranges from 1.9 to 4.5 per 100,000 live births. In Taiwan, this has been estimated to be 2.04 per 100,000 live births [
11].
A precise diagnosis of MPS disorders is traditionally made through three consecutive analyses: the quantification of urinary GAGs, two-dimensional electrophoresis (2-D EP) qualitative examination, and leukocyte enzyme activity assay [
12,
13]. Urinary GAG quantitative tests can be used as a diagnostic screening device for MPS; however, they cannot be used to determine a specific MPS type. The dimethylmethylene blue (DMB) spectrophotometry method is broadly used in most biochemical genetics laboratories; however, it involves a non-specific total GAG assay which can cause both false positive and false negative results, especially in patients with MPS III and IV [
13,
14]. Two-dimensional EP is the most commonly used method to identify specific types of MPS; however, it is laborious, and its interpretation is subjective and ambiguous, making the diagnosis unreliable. To resolve the limitations of these first-line screening methods for MPS, the liquid chromatography/tandem mass spectrometry (LC-MS/MS) method has been used to identify MPS subgroups, and it has been shown to be an accurate and reliable method [
15,
16,
17,
18].
The major therapies for MPS disorders include enzyme replacement therapy (ERT) and hematopoietic stem cell transplantation (HSCT). ERT is now available for MPS I, II, IVA, VI, and VII, and it has been demonstrated to remarkably reduce urinary GAG levels and substantially improve endurance, joint mobility, physiological activities, and quality of life [
19,
20,
21,
22,
23,
24,
25,
26,
27,
28]. HSCT is currently the only treatment to prevent progressive neurodegenerative disorders in MPS I, II, VI, and VII [
29]. Some developing therapeutics for MPS disorders are currently in clinical trials, including substrate reduction therapy, chaperone therapy, and gene therapy [
30,
31]. Previous studies have indicated that early treatment may contribute to a better clinical outcome [
32,
33,
34].
Since MPS disorders are rare, multisystemic and progressive diseases with subtle signs and symptoms at the beginning of the natural course, making an early diagnosis can be a challenge for first-line health care professionals. Tracing back the medical history, the patients are usually brought to miscellaneous medical specialists due to diverse manifestations before the confirmative diagnosis of MPS [
35,
36,
37]. In Taiwan, there is insufficient awareness of MPS, which can lead to a delay in the diagnosis or even misdiagnosis with other disorders, and thus these patients often receive inappropriate management. Identifying and understanding the early signs and symptoms of this disease may allow for an early diagnosis and timely appropriate treatment [
38,
39,
40,
41]. Therefore, the purpose of this study was to develop a feasible MPS screening algorithm and establish a cross-specialty collaboration platform between medical geneticists and other medical specialists based on at-risk criteria by measuring urinary GAG fractionation biomarkers using the LC-MS/MS method to allow for an earlier confirmative diagnosis of MPS.
3. Results
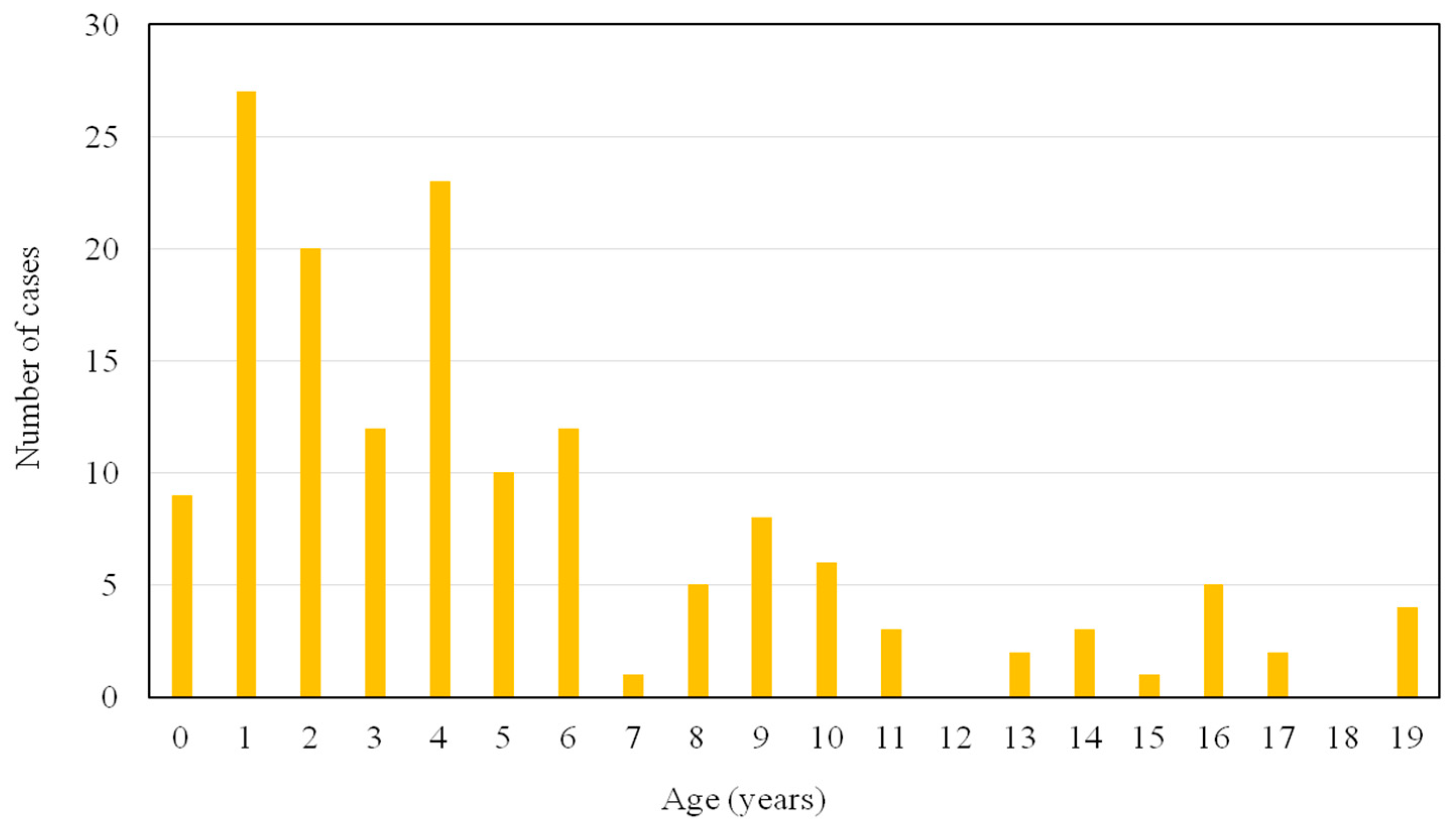
Figure 1 shows the age distribution of the 153 subjects who were enrolled in this study. One hundred and thirteen subjects (74%) were between 0–6 years of age. Forty urine samples sent to our laboratory for at-risk screening for MPS were delivered by pediatric endocrinologists (26%), followed by pediatric neurologists (25%) and general pediatricians (22%) (
Table 2). The major signs and symptoms according to the involved system recorded by health care professionals at the time of the decision to test were the musculoskeletal system (55%), followed by the neurological system (45%) and coarse facial features (39%) (
Table 3). Twenty-two subjects had elevated urinary GAG levels and abnormal GAG disaccharide pattern by 2-D EP, and elevated urinary DS, HS, or KS levels, and they were recalled for specific enzyme activity assays in leukocytes for a confirmative diagnosis. The patients with an enzyme activity level <5% of the mean of normal population underwent genetic testing. Among them, 13 patients had a confirmative diagnosis of MPS (nine males and four females; age range, 0.6 to 10.9 years; median age, 2.9 years; mean age, 3.4 ± 2.9 years; three with MPS I, four with MPS II, five with MPS IIIB, and one with MPS IVA). Three patients (23%) were identified before 1 year of age, and seven patients (54%) were diagnosed before 3 years of age. The false negative rate of the urinary DMB/creatinine ratio for these 13 patients was 31%, including one MPS I, two MPS IIIB, and one MPS IVA. However, there were no false negative results observed from urinary DS, HS and KS quantifications (
Table 4). All 13 patients with MPS had musculoskeletal system involvement, followed by 85% with coarse facial features, 54% with otorhinolaryngological system involvement, 38% with visceromegaly, 38% with hernia, and 38% with neurological system involvement. Two patients diagnosed with MPS II and IVA, respectively, did not have coarse facial features. All five patients with MPS IIIB had speech delay and/or attention deficit. Seven patients (54%) had received surgical procedures before the diagnosis of MPS, including five hernia repairs and three ventilation tube insertions. Six MPS patients were suspected and referred by pediatric neurologists (46%), followed by three patients referred by pediatric orthopedists (23%), and one patient was referred by a pediatric cardiologist, neonatologist, geneticist, and general pediatrician, respectively (
Table 5). Intravenous ERT was started in eight patients (Nos. 1–7 and No. 13) following the diagnosis. Patient No. 1 also received HSCT at 1.6 years of age. Patient No. 8 was enrolled in a phase I/II trial of intracerebroventricular enzymatic therapy for MPS IIIB.
4. Discussion
To the best of our knowledge, this is the first study to report screening for MPS in an at-risk population in Taiwan by measuring urinary GAG fractionation biomarkers using the LC-MS/MS method, which has been shown to be a powerful and reliable tool for MPS high-risk screening and diagnostic purposes. We established this screening platform with the participation of medical geneticists and other medical specialists to increase awareness and enable an early diagnosis by detecting MPS at the initial onset of clinical symptoms. In this study, three patients (23%) were identified before 1 year of age, and seven patients (54%) were diagnosed before 3 years of age, emphasizing the value of this sign and symptom-based screening program in making an early diagnosis of MPS by raising clinical awareness and educating health care professionals. Colón et al. [
57] reported that they diagnosed eight patients with different types of MPS (one with MPS I, one with MPS II, two with MPS IIIA, one with MPS IIIB, two with MPS IVA, and one with MPS VI) by performing a selective screening program for the early detection of MPS from 2014 to 2016. In their cohort, two cases were identified before 1 year of age, and six cases were detected before 3 years of age, which corresponds well with our results. However, compared to their measurement of urinary GAG levels using only the DMB spectrophotometry method and abnormal GAG disaccharide pattern by 2-D EP, we also measured urinary DS, HS, and KS levels using the LC-MS/MS method at the same time. If any of the data were abnormally elevated, the subjects were recalled for specific enzymatic activity assays in leukocytes by fluorometry for a confirmative diagnosis. In this study, the false negative rate of the urinary DMB ratio using the spectrophotometric method for the 13 MPS patients was 31%, including one MPS I, two MPS IIIB, and one MPS IVA. Nevertheless, there were no false negative results of urinary DS, HS and KS using the LC-MS/MS method. Auray-Blais et al. [
15] reported that the DMB spectrophotometric method can lead to false negative results because of aggregated formation through electrostatic interactions with albumin, glycoproteins, collagen, and other serum proteins which may modify the physicochemical properties of GAGs, especially in MPS III and IV [
14]. In their study, the DMB spectrophotometric method missed the identification of MPS in 30% of their patients (7/23) (one with MPS II, one with MPS III, four with MPS IVA, and one with MPS VI), revealing that this method is relatively unreliable for screening MPS patients, which also corresponds well with our results. Therefore, we recommend that measuring GAG fractionation biomarkers with the LC-MS/MS method is an accurate and reliable method to concurrently quantify urinary levels of DS, HS and KS, and that these biomarkers are more sensitive compared to the traditional DMB ratio using the spectrophotometric method to diagnose MPS, identify subgroups, and screen high-risk populations.
It is well-known that urine GAG is different from blood GAG. The limitation of uKS in its use as a biomarker to prove therapeutic efficacy is distinct. A large amount of literature has revealed that uGAG is not an appropriate biomarker for monitoring therapeutic effects [
58]. According to the experimental findings reported by Saville J.T. et al., the concentration of oligosaccharides and the ratio of HS:DS in urine were similar to those observed in the kidney, suggesting that the oligosaccharide storage pattern in urine is a reflection of that in the kidney. Although serum, liver and brain had a similar ratio of HS:DS, which was lower than that seen in the urine and kidney, a distribution of oligosaccharides which ranked from most to least abundant between serum, liver and brain was observed, suggesting that serum more closely reflects the oligosaccharides of the brain and liver and may therefore be a more informative measurement of disease burden than urine [
59]. In addition, Kahn et al. [
47] and Fujitsuka et al. [
60] also stated that blood KS remained high while uKS was reduced during ERT. The fact of the lack of a reduction of blood KS should be notable because this is critical evidence that uKS is ineffective as a biomarker to prove therapeutic efficacy.
Different types of MPS have many similar clinical features and some type-specific manifestations. The severe forms of MPS I and II present with both somatic and cognitive involvement and are characterized by coarse facial features, vision and hearing impairment, recurrent respiratory infections, decreased pulmonary function, obstructive sleep apnea, cardiac disease, inguinal and umbilical hernias, hepatosplenomegaly, spinal cord compression, communicating hydrocephalus, and dysostosis multiplex [
56,
61]. MPS III manifests as neurological and cognitive impairment and mild somatic involvement [
62]. MPS IV presents as short stature, odontoid hypoplasia, ligamentous laxity, joint hypermobility, and skeletal dysplasia [
63]. MPS VI is characterized by a purely somatic manifestation similar to MPS I and II without cognitive involvement [
1,
2,
3]. In our study, at the time of diagnosis, all 13 MPS patients had musculoskeletal system involvement, followed by 85% with coarse facial features, 54% with otorhinolaryngological system involvement, 38% with visceromegaly, 38% with hernia, and 38% with neurological system involvement. All five MPS IIIB patients had a speech delay and/or attention deficit. None of the patients had cornea clouding, and one MPS II patient and one MPS IVA patient did not have coarse facial features when they were diagnosed.
Using data from an MPS I registry (
n = 544), Arn et al. [
64] reported that at least one surgery preceded the diagnosis in 36%, 46%, and 63% of the patients with Hurler, Hurler–Scheie, and Scheie syndromes, respectively. In addition, more than one-third (39%) of all patients had hernia repair surgery before they were diagnosed with MPS I. Using data from the Hunter Outcome Survey (
n = 389), Mendelsohn et al. [
65] reported that the majority of patients (57%) underwent at least one surgical intervention before the diagnosis of MPS II, and that 55% and 45% of the patients underwent hernia repair and tympanostomy, respectively, before MPS II was diagnosed. In our cohort, seven patients (54%) received surgical procedures before the diagnosis of MPS, including five with hernia repair and three with ventilation tube insertion, which is consistent with Mendelsohn et al.’s study. These findings indicate that health care professionals need to be aware of the surgical burden of MPS as well as its presenting signs and symptoms and the importance of timely referral for MPS diagnostic testing.
A delayed diagnosis of MPS is often due to referrals from one physician to another. This is mainly because of the rare nature of the disorder, phenotypic heterogeneity, and the broad range of nonspecific early signs and symptoms [
35]. As a consequence, in addition to medical geneticists, it is important that general health care professionals are aware of the early signs and symptoms of MPS. In this prospective study, we established an at-risk population screening program for MPS that involved the participation of medical geneticists and other medical specialists, including pediatric endocrinologists, pediatric neurologists, general pediatricians, pediatric rheumatologists, neonatologists, pediatric orthopedists, pediatric cardiologists, and pediatric surgeons. Among the 13 patients diagnosed with MPS, 46% were referred by pediatric neurologists, followed by pediatric orthopedists (23%). Other patients were referred by pediatric cardiologists, neonatologists, geneticists, and a general pediatrician.
Although ERT and HSCT cannot cure MPS disorders, they can improve or lessen the natural progression, and better outcomes may be related to commencing these treatments at a younger age [
32,
33,
34]. The increasing clinical awareness of MPS disease and increased ability to make a confirmative diagnosis has made an earlier diagnosis possible. In this study, eight of the 13 patients diagnosed with MPS started to receive ERT, for which the payments were reimbursed by the Taiwanese National Health Insurance program following international standards. In addition, one MPS I patient also received HSCT, and one MPS IIIB patient enrolled in a phase I/II clinical trial.

 ,
, 




{kind=link}