
A Combinatorial Single-Molecule Real-Time and Illumina Sequencing Analysis of Postembryonic Gene Expression in the Asian Citrus Psyllid Diaphorina citri

 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. D. citri Rearing, Sample Collection, and Assay of Weight and Length
2.2. RNA Isolation and cDNA Synthesis
2.3. cDNA Library Construction and Sequencing
2.4. Functional Annotation of Full-Length, Non-Redundant Transcripts
2.5. Alternative Splicing (AS) Event, Simple Sequence Repeat (SSR), and Open Reading Frame (ORF) Prediction
2.6. Long Noncoding RNA (LncRNA) Prediction and Transcription Factor (TF) Analysis
2.7. Identification of DEGs and Functional Enrichment Analysis
2.8. dsRNA Synthesis and Microinjection
2.9. Analysis of Expression Levels of DEGs in Different Samples by RT-qPCR
2.10. Statistical Analysis
3. Results
3.1. Detection of Weight and Length of D. citri Fifth-Instar Nymphs and Adults
3.2. PacBio and Illumina Sequencing of D. citri Nymphs and Adults
3.3. Functional Annotation of New Transcripts
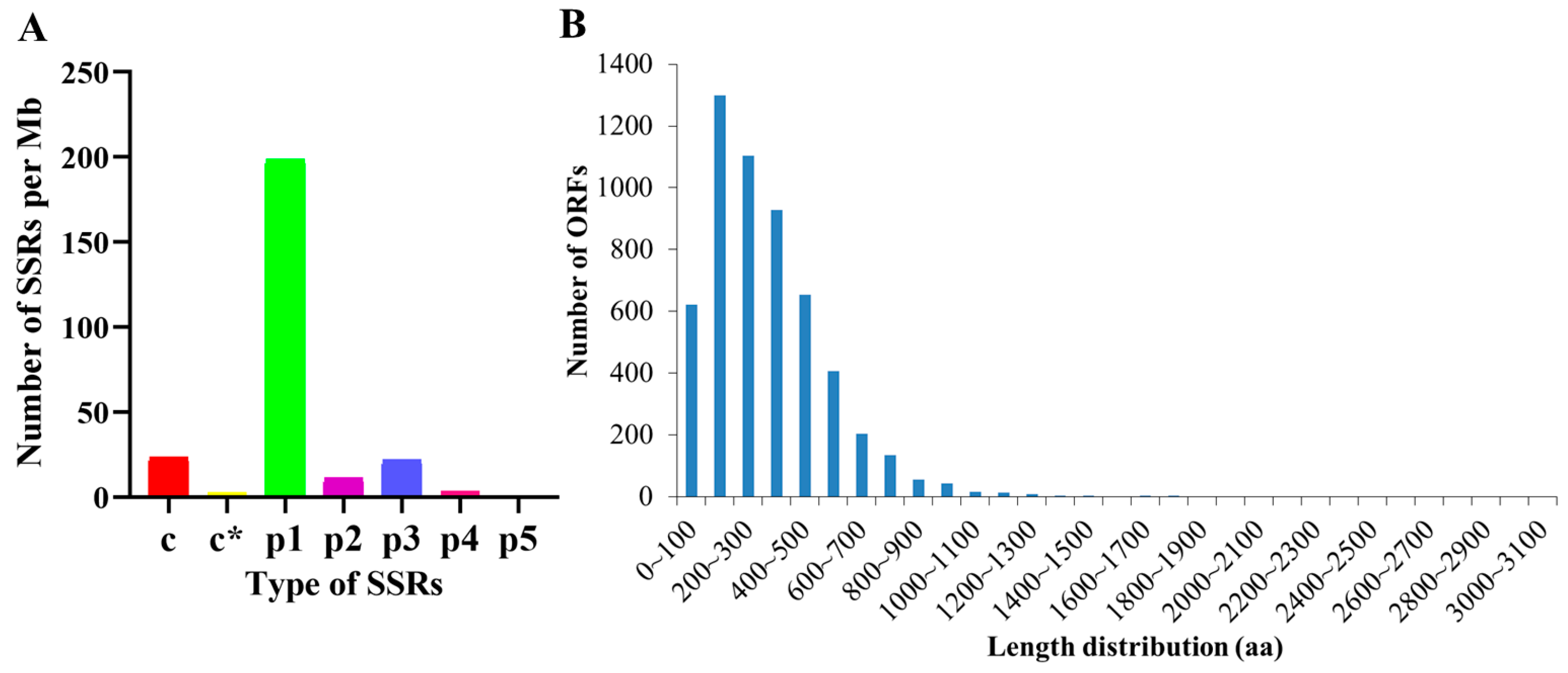
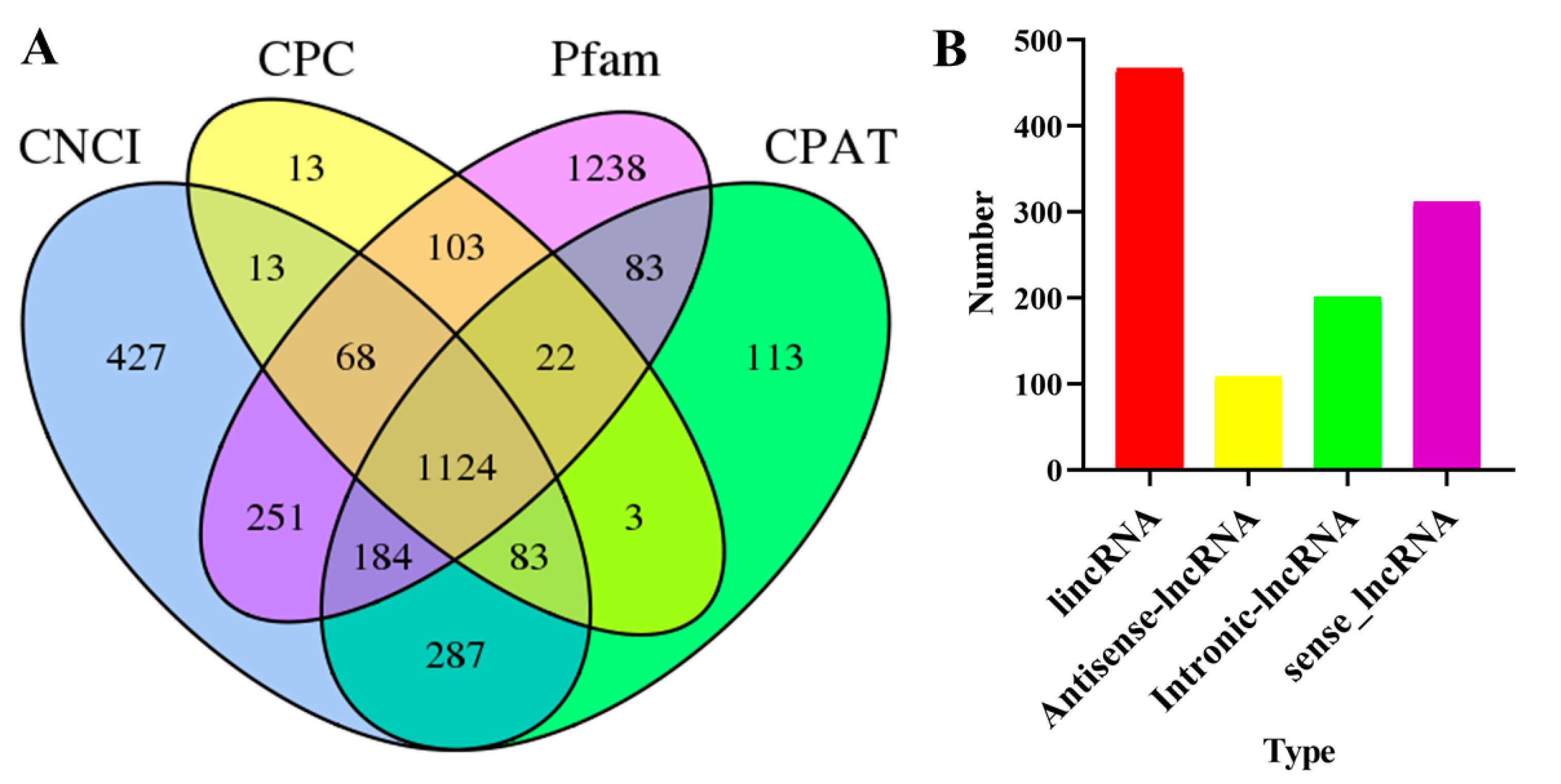
3.4. Analysis of SSRs, ORFs, and LncRNAs
3.5. Prediction of TFs, AS Events, and APA
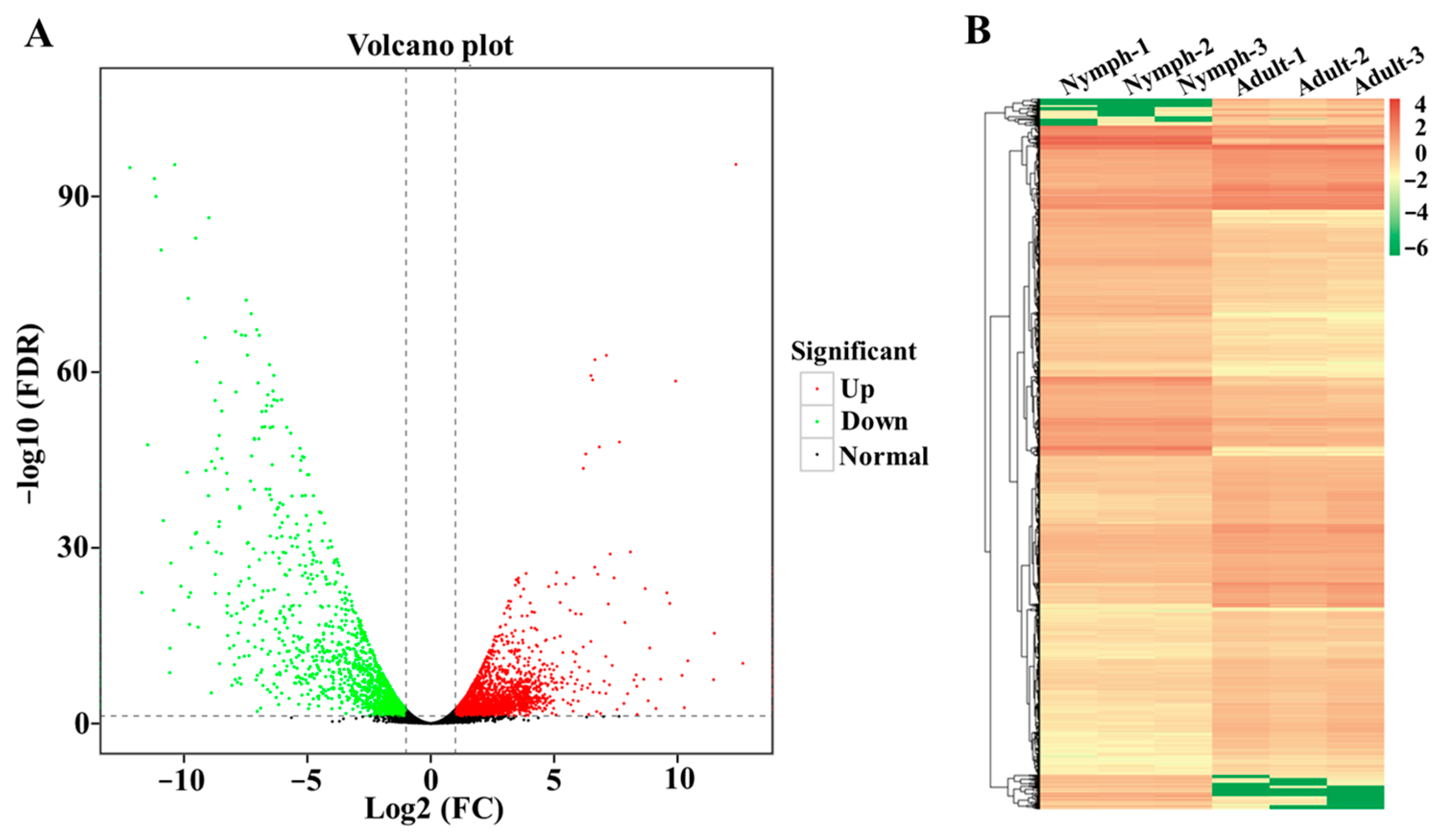
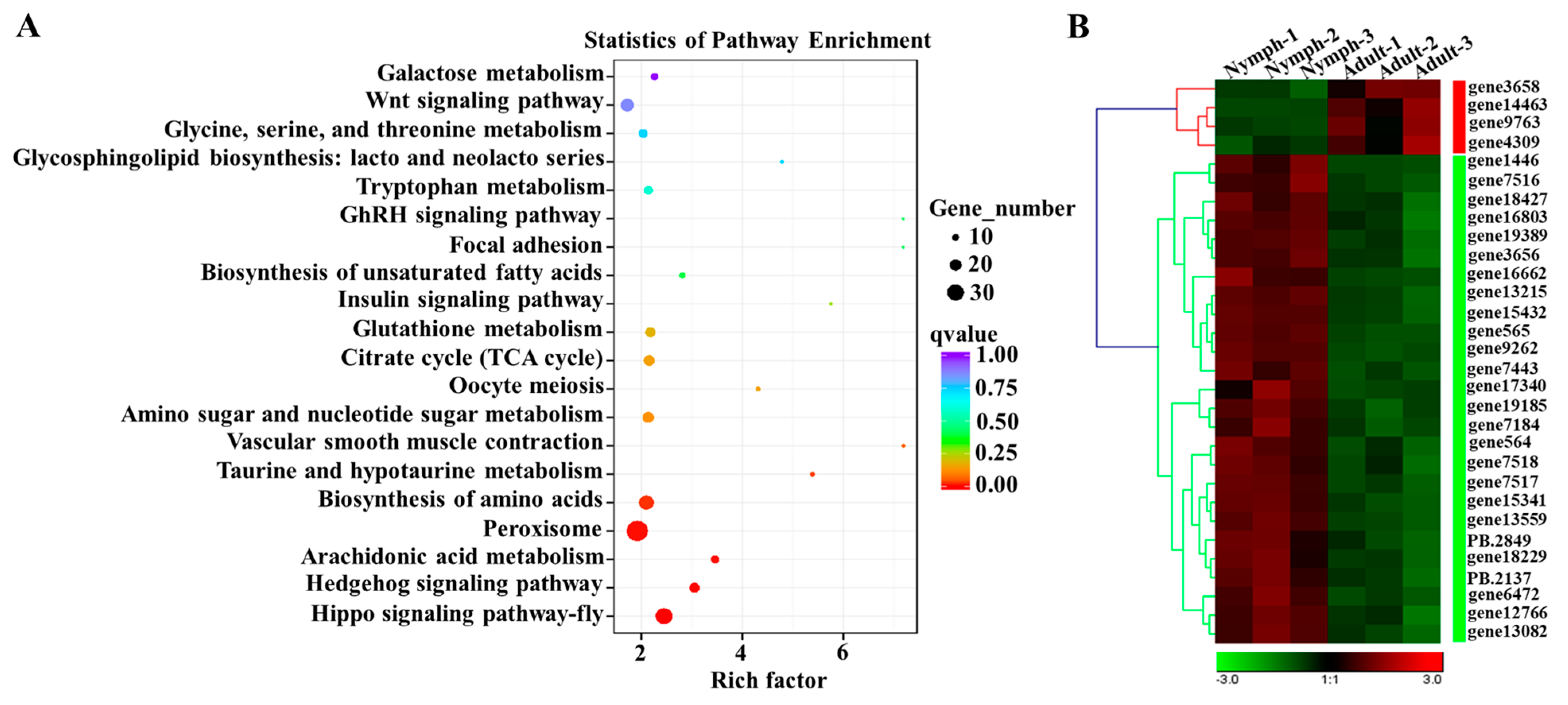
3.6. Identification of Differentially Expressed Genes (DEGs) and Functional Enrichment Analysis
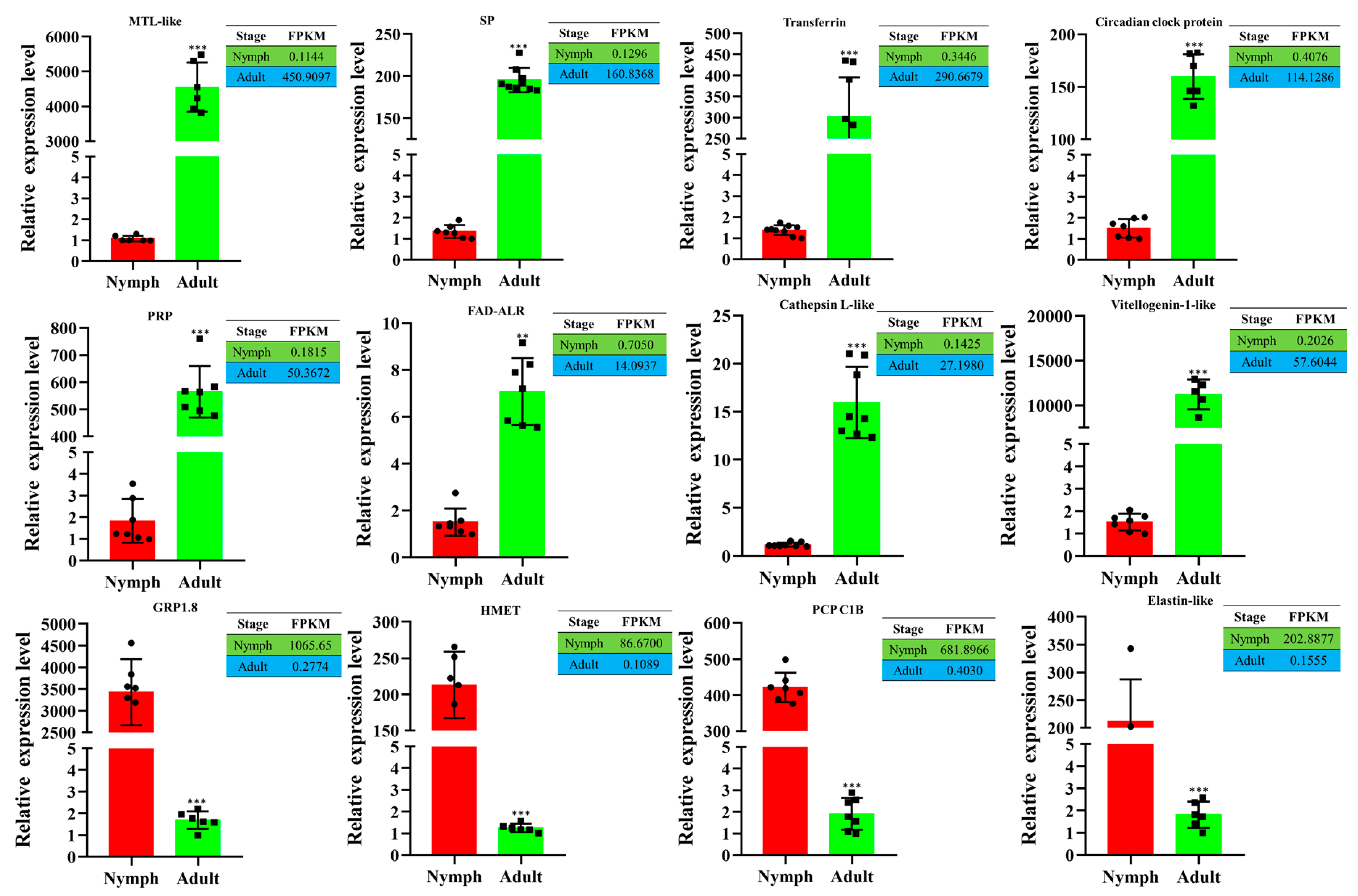
3.7. Validation of DEGs by Reverse Transcription–Quantitative PCR (RT-qPCR)
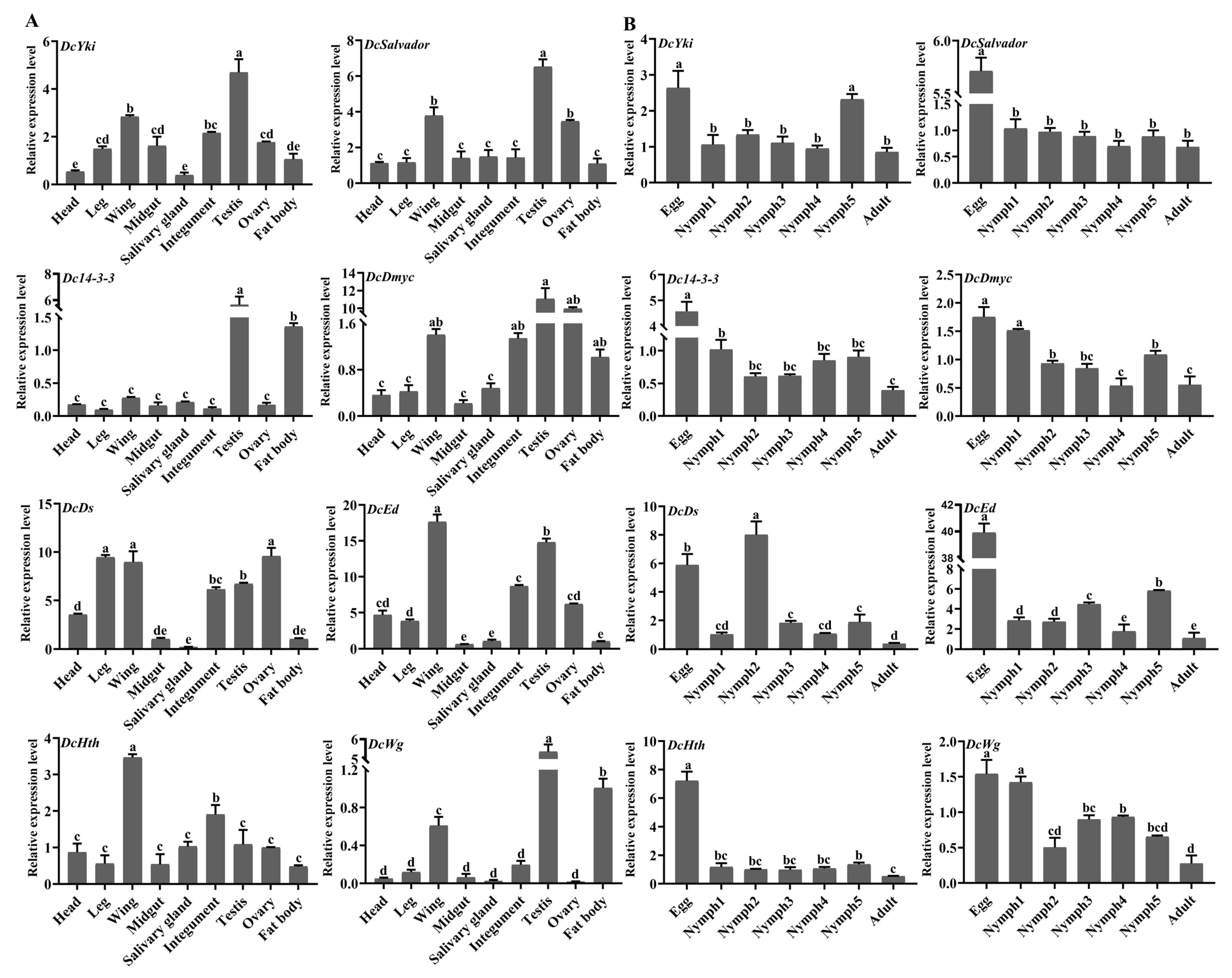
3.8. Analysis of Spatiotemporal Expression Patterns of DEGs Involved in the Hippo Signaling Pathway
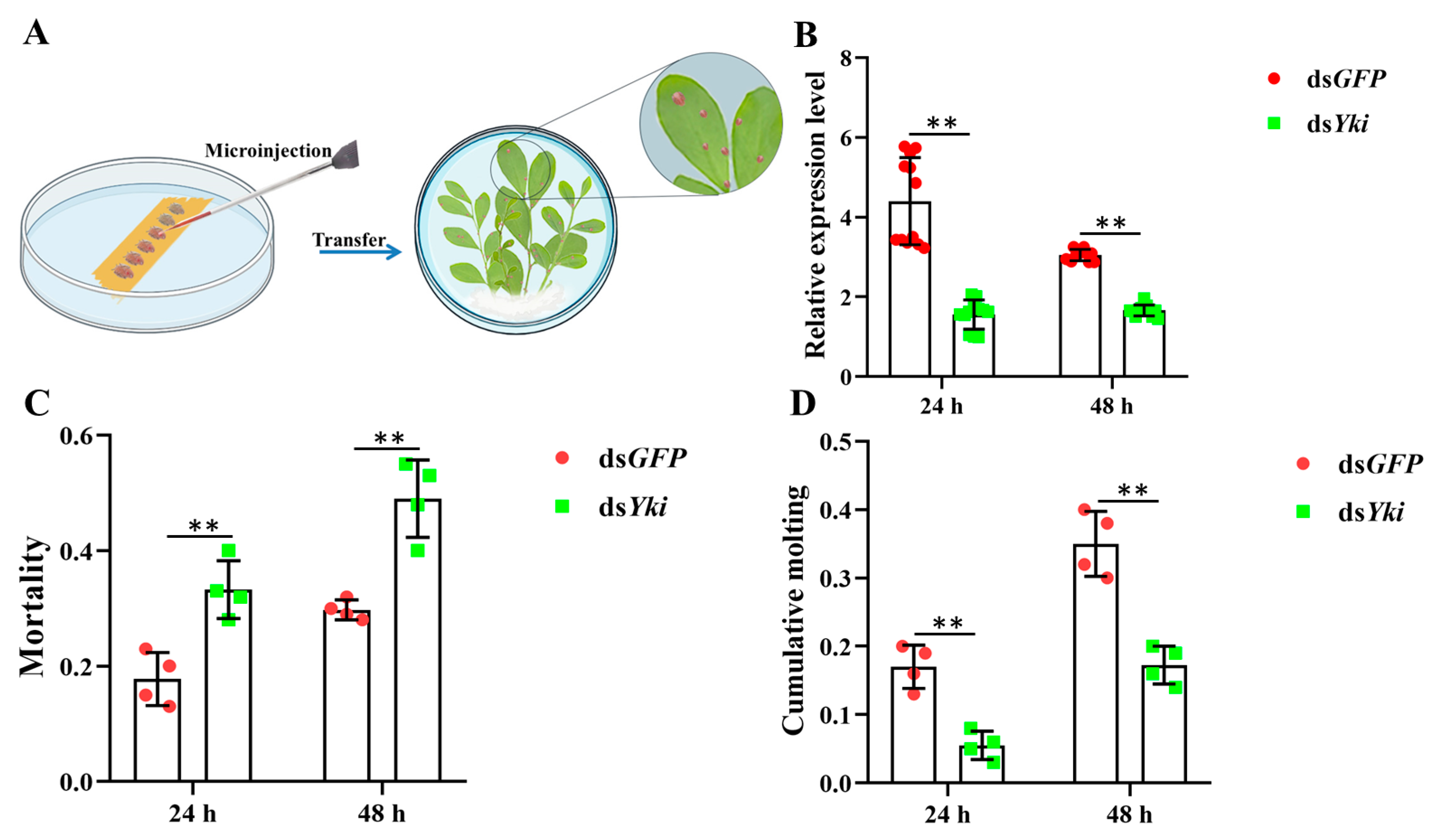
3.9. Silencing of DcYki by RNAi Affects the Development of D. citri
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wu, Z.Z.; Qu, M.Q.; Chen, M.S.; Lin, J.T. Proteomic and transcriptomic analyses of saliva and salivary glands from the Asian citrus psyllid, Diaphorina citri. J. Proteom. 2021, 238, 104036. [Google Scholar] [CrossRef] [PubMed]
- Grafton-Cardwell, E.E.; Stelinski, L.L.; Stansly, P.A. Biology and Management of Asian Citrus Psyllid, Vector of the Huanglongbing Pathogens. Annu. Rev. Entomol. 2013, 58, 413–432. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, J.A.; Stansly, P.A. Integrated approaches for managing the Asian citrus psyllid Diaphorina citri (Homoptera: Psyllidae) in Florida. Proc. Fla. State Hort. Soc. 2007, 120, 110–115. [Google Scholar]
- Naeem, A.; Freed, S.; Jin, F.L.; Akmal, M.; Mehmood, M. Monitoring of insecticide resistance in Diaphorina citri Kuwayama (Hemiptera: Psyllidae) from citrus groves of Punjab, Pakistan. Crop Prot. 2016, 86, 62–68. [Google Scholar] [CrossRef]
- Yu, H.Z.; Huang, Y.L.; Lu, Z.J.; Zhang, Q.; Su, H.N.; Du, Y.M.; Yi, L.; Zhong, B.L.; Chen, C.X. Inhibition of trehalase affects the trehalose and chitin metabolism pathways in Diaphorina citri (Hemiptera: Psyllidae). Insect Sci. 2021, 28, 718–734. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.J.; Huang, Y.L.; Yu, H.Z.; Li, N.Y.; Xie, Y.X.; Zhang, Q.; Zeng, X.D.; Hu, H.; Huang, A.J.; Yi, L.; et al. Silencing of the Chitin Synthase Gene Is Lethal to the Asian Citrus Psyllid, Diaphorina citri. Int. J. Mol. Sci. 2019, 20, 3734. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.B.; Zou, X.J.; Zhang, Q.; Wang, A.Y.; Amir, M.B.; Du, Y.M.; Liu, X.Q.; Chen, W.; Lu, Z.J.; Yu, H.Z. Quantitative ubiquitylome crosstalk with proteome analysis revealed cytoskeleton proteins influence CLas pathogen infection in Diaphorina citri. Int. J. Biol. Macromol. 2023, 31, 123411. [Google Scholar] [CrossRef] [PubMed]
- Pelz-Stelinski, K.S.; Brlansky, R.H.; Ebert, T.A.; Rogers, M.E. Transmission parameters for Candidatus Liberibacter asiaticus by Asian citrus psyllid (Hemiptera: Psyllidae). J. Econ. Entomol. 2010, 103, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.L.; Li, Y.H.; Zhou, Y.T.; Xu, W.M.; Cuthbertson, A.G.S.; Guo, Y.J.; Qiu, B.L. Effects of Candidatus Liberibacter asiaticus on the fitness of the vector Diaphorina citri. J. Appl. Microbiol. 2016, 121, 1718–1726. [Google Scholar] [CrossRef]
- Yu, H.Z.; Yi, L.; Lu, Z.J. Silencing of Chitin-Binding Protein with PYPV-Rich Domain Impairs Cuticle and Wing Development in the Asian Citrus Psyllid, Diaphorina citri. Insects 2022, 13, 353. [Google Scholar] [CrossRef]
- Xu, D.P.; Yang, H.J.; Zhuo, Z.H.; Lu, B.Q.; Hu, J.M.; Yang, F. Characterization and analysis of the transcriptome in Opisina arenosella from different developmental stages using single-molecule real-time transcript sequencing and RNA-seq. Int. J. Biol. Macromol. 2021, 169, 216–227. [Google Scholar] [CrossRef]
- Du, H.Y.; Ge, R.T.; Zhang, L.; Zhang, J.Y.; Chen, K.P.; Li, C.J. Transcriptome-wide identification of development related genes and pathways in Tribolium castaneum. Genomics 2023, 115, 110551. [Google Scholar] [CrossRef]
- Liu, S.N.; Wei, W.; Chu, Y.; Zhang, L.; Shen, J.; An, C.J. De novo transcriptome analysis of wing development-related signaling pathways in Locusta migratoria manilensis and Ostrinia furnacalis (Guenée). PLoS ONE 2014, 9, e106770. [Google Scholar] [CrossRef]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S.N. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.P.; Lu, Z.Y.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef]
- Yang, C.X.; Ou, D.; Guo, W.; Lu, J.; Guo, C.F.; Qiu, B.L.; Pen, H.P. De Novo Assembly of the Asian Citrus Psyllid Diaphorina citri (Hemiptera: Psyllidae) Transcriptome across Developmental Stages. Int. J. Mol. Sci. 2020, 21, 4974. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.Z.; Li, N.Y.; Zeng, X.D.; Song, J.C.; Yu, X.D.; Su, H.N.; Chen, C.X.; Yi, L.; Lu, Z.J. Transcriptome Analyses of Diaphorina citri Midgut Responses to Candidatus Liberibacter Asiaticus Infection. Insects 2020, 11, 171. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.Z.; Pu, X.H.; Shu, B.S.; Bin, S.Y.; Lin, J.T. Transcriptome analysis of putative detoxification genes in the Asian citrus psyllid, Diaphorina citri. Pest Manag. Sci. 2020, 76, 3857–3870. [Google Scholar] [CrossRef]
- Xiong, Y.; Liu, X.Q.; Xiao, P.A.; Tang, G.H.; Liu, S.H.; Lou, B.H.; Wang, J.J.; Jiang, H.B. Comparative transcriptome analysis reveals differentially expressed genes in the Asian citrus psyllid (Diaphorina citri) upon heat shock. Comp. Biochem. Physiol. Part D Genom. Proteom. 2019, 30, 256–261. [Google Scholar] [CrossRef]
- Saha, S.; Hosmani, P.S.; Villalobos-Ayala, K.; Miller, S.; Shippy, T.; Flores, M.; Rosendale, A.; Cordola, C.; Bell, T.; Mann, H.; et al. Improved annotation of the insect vector of citrus greening disease: Biocuration by a diverse genomics community. Database 2019, 1, baz035. [Google Scholar] [CrossRef]
- Hosmani, P.S.; Flores-Gonzalez, M.; Shippy, T.; Vosburg, C.; Massimino, C.; Tank, W.; Reynolds, M.; Tamayo, B.; Miller, S.; Norus, J.; et al. Chromosomal length reference assembly for Diaphorina citri using singlemolecule sequencing and Hi-C proximity ligation with manually curated genes in developmental, structural and immune pathways. bioRxiv 2019. [Google Scholar] [CrossRef]
- Ogihara, Y.; Mochida, K.; Kawaura, K.; Murai, K.; Seki, M.; Kamiya, A.; Shinozaki, K.; Carninci, P.; Hayashizaki, Y.; Shin-I, T.; et al. Constructions of a full-length cDNA library from yong spikelets of hexaploid wheat and its characterization by large-scale sequencing of expressed sequence tags. Genes Genet Syst. 2004, 79, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.J.; Carneiro, M.O.; Schatz, M.C. The advantages of SMRT sequencing. Genome Biol. 2013, 14, 405. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.; Lu, X.Y.; Luo, J.; Tang, F. SMRT sequencing of the full-length transcriptome of Odontotermes formosanus (Shiraki) under Serratia marcescens treatment. Sci. Rep. 2020, 10, 15909. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Wang, Y.X.; Liu, Y.H.; Hu, J.; Guo, Y.Q.; Gao, L.L.; Ma, R.Y. SMRT sequencing of full-length transcriptome of flea beetle Agasicles hygrophila (Selman and Vogt). Sci. Rep. 2018, 8, 2197. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.P.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.T.; Bu, D.C.; Zhao, G.G.; Yu, K.T.; Zhang, C.H.; Liu, Y.N.; Chen, R.S.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Wang, L.G.; Park, H.J.; Dassari, S.; Wang, S.Q.; Kocher, J.; Li, W. CPAT: Using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Foissac, S.; Sammeth, M. Analysis of alternative splicing events in custom gene datasets by AStalavista. Methods Mol. Biol. 2015, 1269, 379–392. [Google Scholar] [PubMed]
- Wang, L.J.; Zhu, P.; Mo, Q.L.; Luo, W.; Du, Z.J.; Jiang, J.; Yang, S.; Zhao, L.L.; Gong, Q.; Wang, Y. Comprehensive analysis of full-length transcriptomes of Schizothorax prenanti by single-molecule long-read sequencing. Genomics 2022, 114, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinf. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.; Yu, S.J.; Pan, Q.; Ding, L.L.; Li, S.C.; Cheng, L.Y.; Wang, S.Q.; Lou, B.H.; He, J.; Lei, C.Y.; et al. Chromosome-level genome assembly of the Asian citrus psyllid, Diaphorina citri. Insect Sci. 2024, 31, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Sharma, S.; Meghwanshi, K.K.; Patel, S.; Mehta, P.; Shukla, N.; Do, D.N.; Rajpurohit, S.; Suravajhala, P.; Shukla, J.N. Long Non-Coding RNAs in Insects. Animals 2021, 11, 1118. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.Q.; Cheng, T.C.; Liu, C.; Liu, D.L.; Zhang, Q.; Long, R.W.; Zhao, P.; Xia, Q.Y. Systematic Identification and Characterization of Long Non-Coding RNAs in the Silkworm, Bombyx mori. PLoS ONE 2016, 11, e0147147. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhang, Y.; Zhang, X.; Jia, S.L.; Chen, S.; Kang, L. Genome-wide identification and developmental expression profiling of long noncoding RNAs during Drosophila metamorphosis. Sci. Rep. 2016, 6, 23330. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, J.F.; Zheng, Y.; Zhang, J.Y.; Chen, S.; Zhao, F.Q. Comprehensive identification of internal structure and alternative splicing events in circular RNAs. Nat. Commun. 2016, 7, 12060. [Google Scholar] [CrossRef] [PubMed]
- Tilgner, H.; Grubert, F.; Sharon, D.; Snyder, M.P. Defining a personal, allele-specific, and single-molecule long-read transcriptome. Proc. Natl. Acad. Sci. USA 2014, 111, 9869–9874. [Google Scholar] [CrossRef]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhong, W.M.; He, W.Y.; Li, Y.Y.; Li, Y.Q.; Li, T.P.; Vasseur, L.; You, M.S. Genome-wide profiling of the alternative splicing provides insights into development in Plutella xylostella. BMC Genom. 2019, 20, 463. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.T.; Lin, C.X.; Fang, Z.Y.; Li, H.D. Intron Retention as a Mode for RNA-Seq Data Analysis. Front. Genet. 2020, 11, 586. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Xia, T.; Wang, A.Y.; Liu, Y.; Li, N.Y.; Yi, L.; Lu, Z.J.; Yu, H.Z. Alternative splicing of chitin deacetylase 2 regulates chitin and fatty acid metabolism in Asian citrus psyllid, Diaphorina citri. Arch. Insect Biochem. Physiol. 2023, 114, e22050. [Google Scholar] [CrossRef]
- Ding, R.; Weynans, K.; Bossing, T.; Barros, C.S.; Berger, C. The Hippo signalling pathway maintains quiescence in Drosophila neural stem cells. Nat. Commun. 2016, 7, 10510. [Google Scholar] [CrossRef] [PubMed]
- Li, N.N.; Tong, X.L.; Zeng, J.; Meng, G.; Sun, F.Z.; Hu, H.; Song, J.B.; Lu, C.; Dai, F.Y. Hippo pathway regulates somatic development and cell proliferation of silkworm. Genomics 2019, 111, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Zhang, J.; Li, T.; Sun, X.; Qin, S.; Hou, C.X.; Zhang, G.Z.; Li, M.W. BmSd gene regulates the silkworm wing size by affecting the Hippo pathway. Insect Sci. 2020, 27, 655–664. [Google Scholar] [CrossRef]
- Hsu, T.H.; Yang, C.Y.; Yeh, T.H.; Huang, Y.C.; Wang, T.W.; Yu, J.Y. The Hippo pathway acts downstream of the Hedgehog signaling to regulate follicle stem cell maintenance in the Drosophila ovary. Sci. Rep. 2017, 7, 4480. [Google Scholar] [CrossRef]
- Wang, D.; Li, X.R.; Dong, D.J.; Huang, H.; Wang, J.X.; Zhao, X.F. The steroid hormone 20-hydroxyecdysone promotes the cytoplasmic localization of Yorkie to suppress cell proliferation and induce apoptosis. J. Biol. Chem. 2016, 291, 21761–21770. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Statistics | Nymph (Number) | Adult (Number) |
|---|---|---|
| SMRT cells | 3 | 1 |
| cDNA size | 1–6 K | 1–6 K |
| Data size (G) | 20.17 | 23.97 |
| Number of CCSs | 306,224 | 347,641 |
| Read bases of CCSs | 806,848,990 | 638,839,227 |
| Mean read length of CCSs | 2634 | 1837 |
| Mean number of passes | 28 | 44 |
| Number of undesired primer reads | 77,099 | 78,527 |
| Number of filtered short reads | 49 | 89 |
| Number of full-length, non-chimeric reads | 218,559 | 256,717 |
| Full-length, non-chimeric percentage (FLNC %) | 71.37% | 73.85% |
| Number of consensus isoforms | 16,119 | 20,157 |
| Average consensus isoform read length | 2559 | 1778 |
| Number of polished, high-quality isoforms | 15,992 | 20,048 |
| Number of polished, low-quality isoforms | 126 | 105 |
| Number of full-length, non-redundant transcripts | 4307 | 6334 |
| Percentage of polished, high-quality isoforms | 99.22% | 99.48% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Zhang, C.; Zhong, H.; He, Q.; Xia, Z.-Y.; Hu, Y.; Liao, Y.-X.; Yi, L.; Lu, Z.-J.; Yu, H.-Z. A Combinatorial Single-Molecule Real-Time and Illumina Sequencing Analysis of Postembryonic Gene Expression in the Asian Citrus Psyllid Diaphorina citri. Insects 2024, 15, 391. https://doi.org/10.3390/insects15060391
Zhang Q, Zhang C, Zhong H, He Q, Xia Z-Y, Hu Y, Liao Y-X, Yi L, Lu Z-J, Yu H-Z. A Combinatorial Single-Molecule Real-Time and Illumina Sequencing Analysis of Postembryonic Gene Expression in the Asian Citrus Psyllid Diaphorina citri. Insects. 2024; 15(6):391. https://doi.org/10.3390/insects15060391
Chicago/Turabian StyleZhang, Qin, Can Zhang, Hong Zhong, Qing He, Zhao-Ying Xia, Yu Hu, Yu-Xin Liao, Long Yi, Zhan-Jun Lu, and Hai-Zhong Yu. 2024. "A Combinatorial Single-Molecule Real-Time and Illumina Sequencing Analysis of Postembryonic Gene Expression in the Asian Citrus Psyllid Diaphorina citri" Insects 15, no. 6: 391. https://doi.org/10.3390/insects15060391