
Machine Learning-Based Prediction of Elastic Properties Using Reduced Datasets of Accurate Calculations Results


Abstract
:1. Introduction
2. Materials and Methods
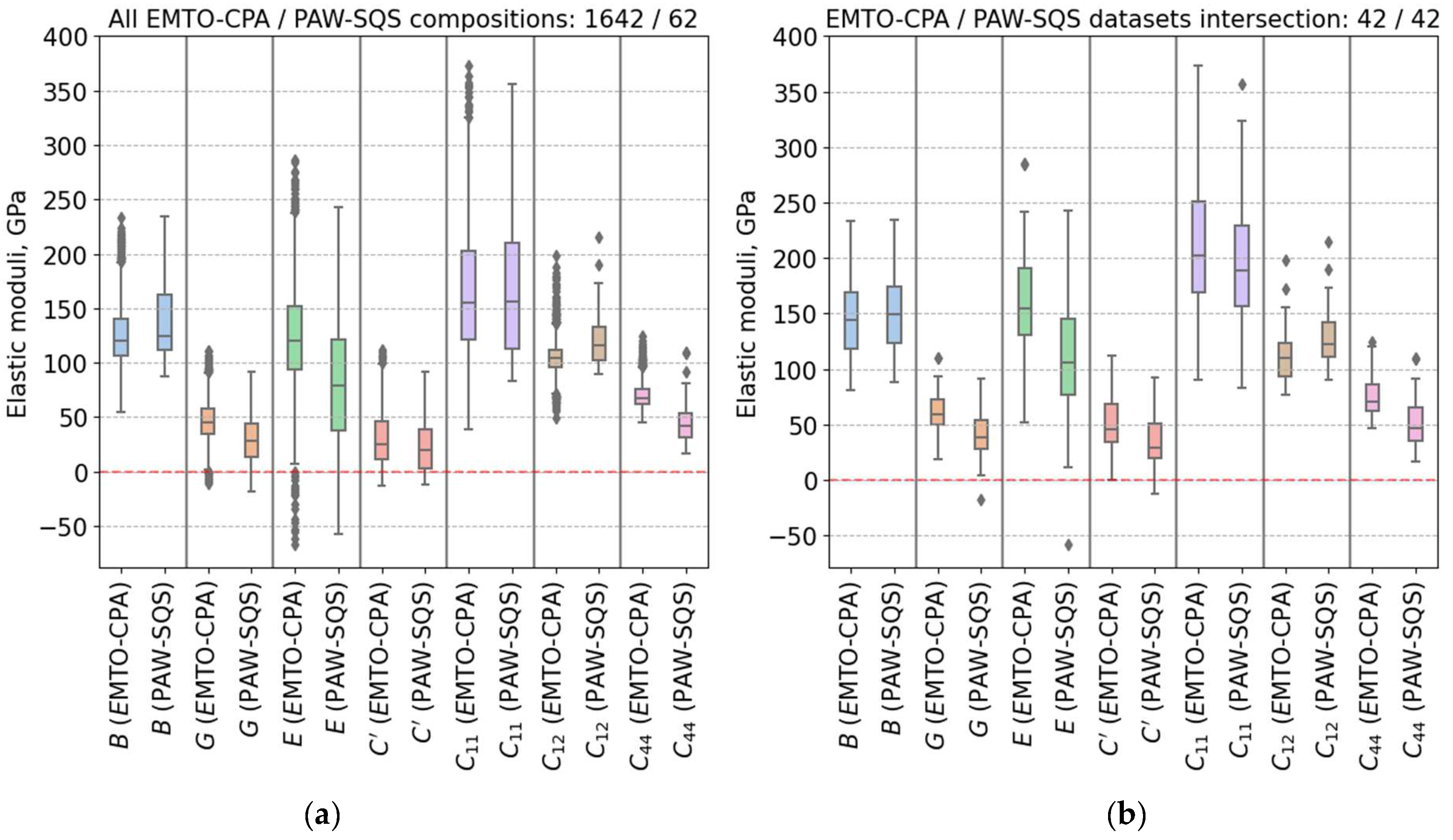
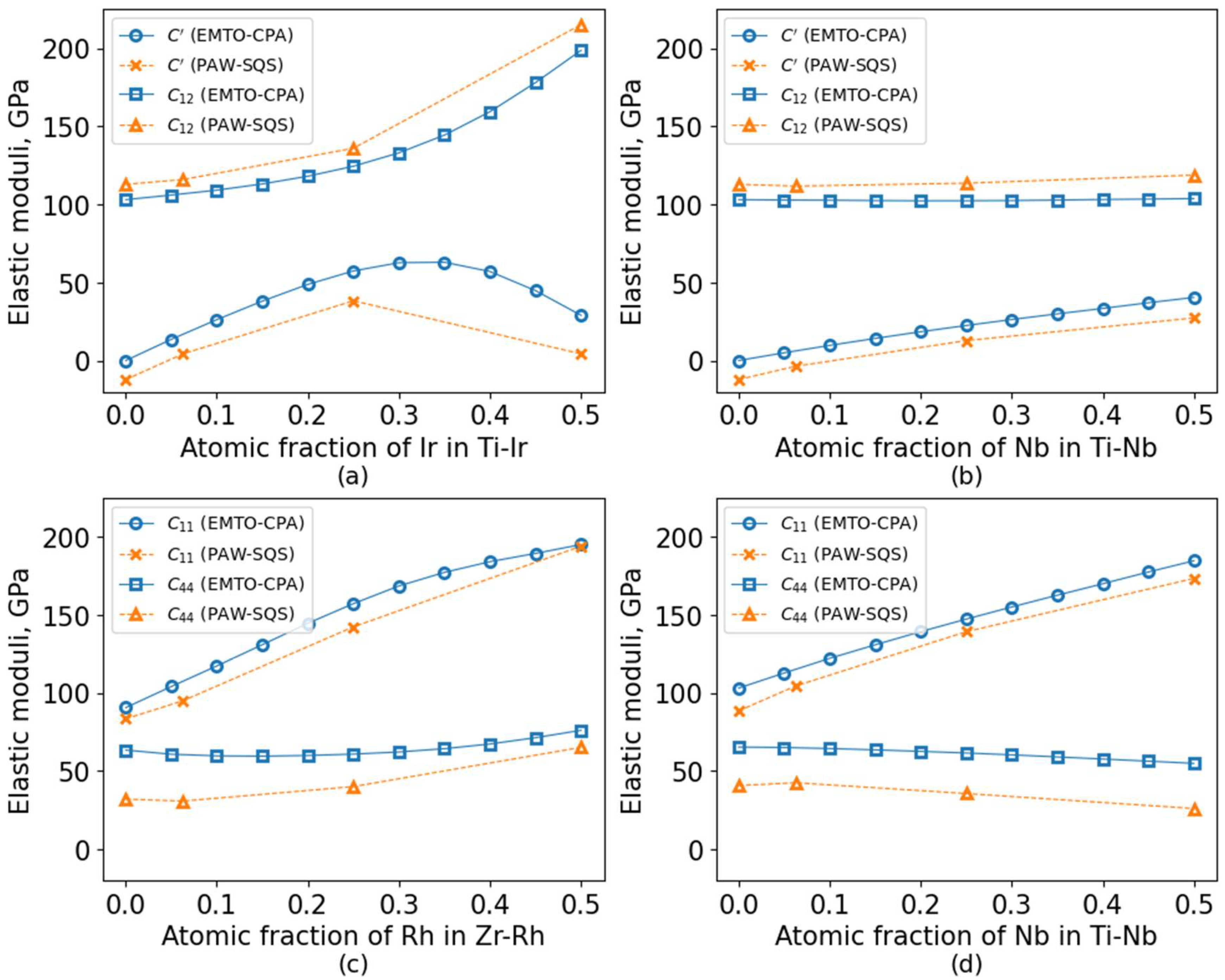
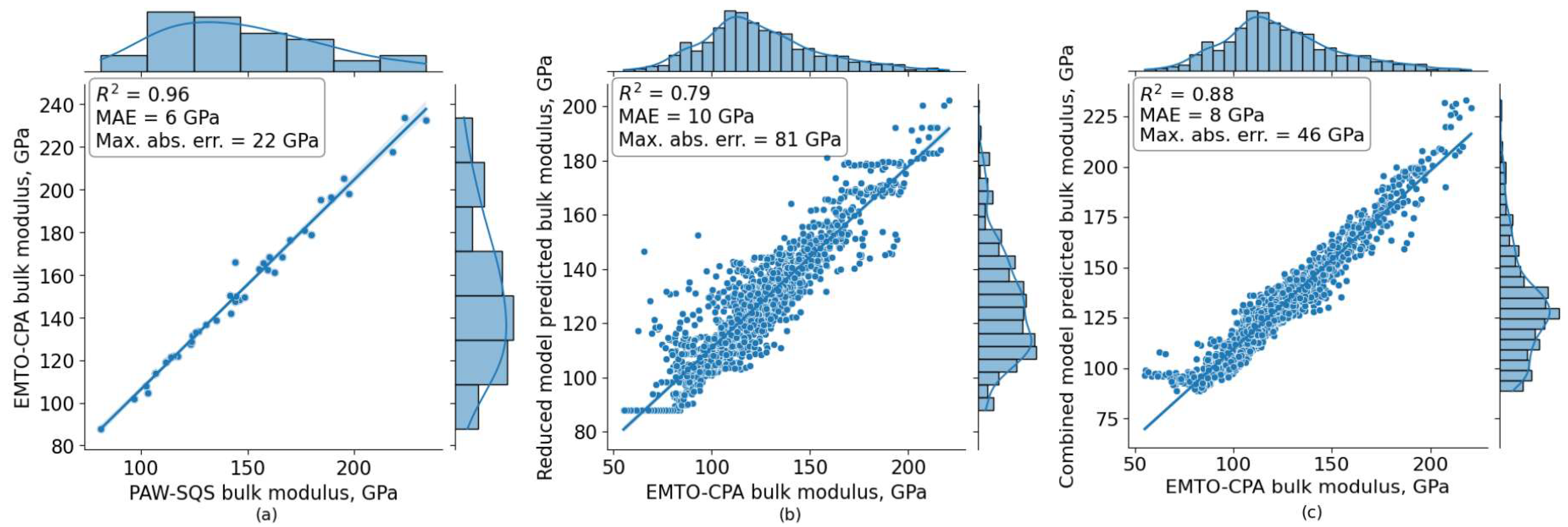
3. Results
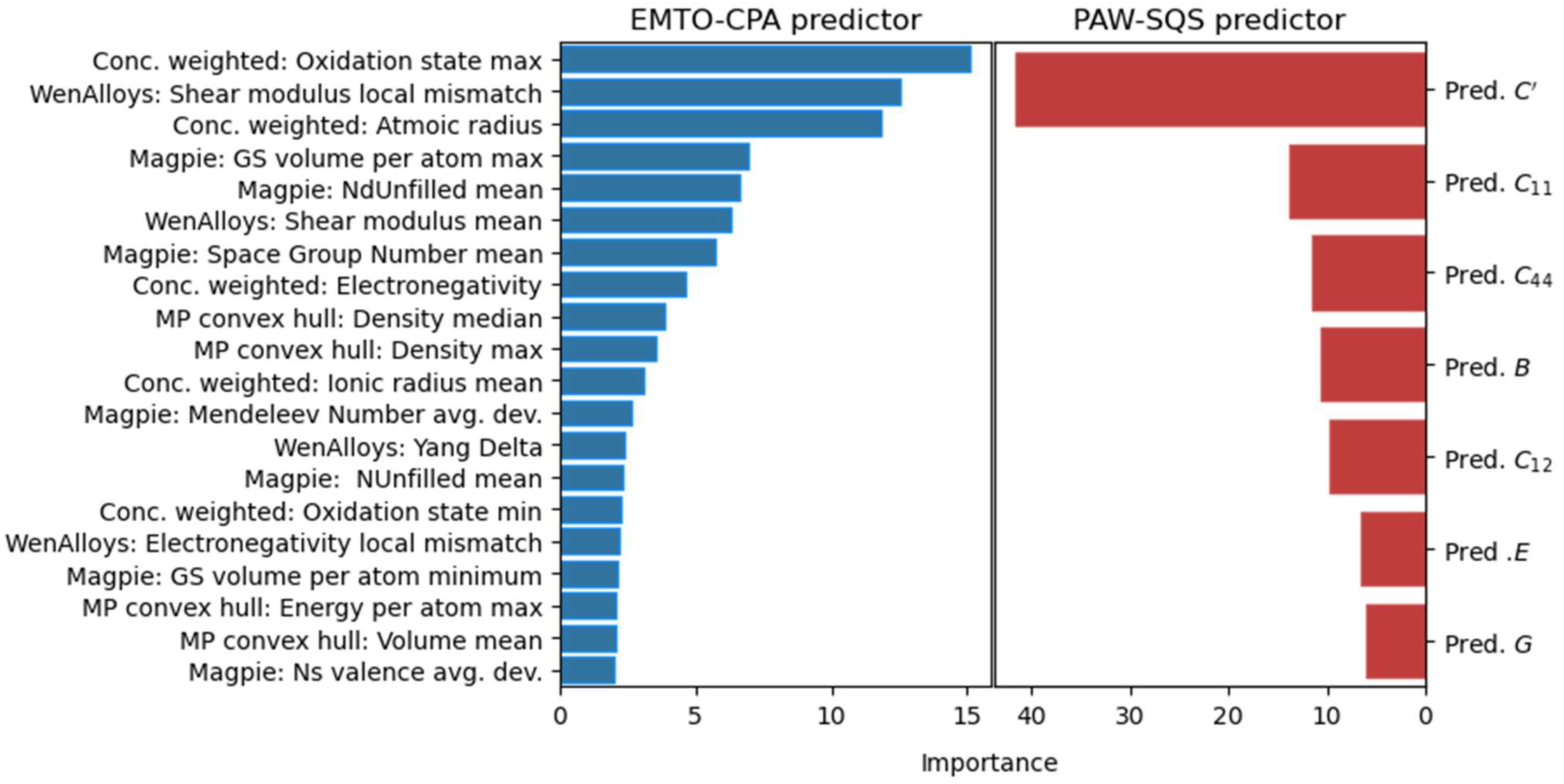
3.1. Selected Features
- Five features containing concentration-weighted properties of each element contained in each composition (“Conc. weighted”) with importance of 37% in total;
- Four features extracted from the “Materials Project” phase diagrams (“MP convex hull feats”) with importance of 11% in total;
- Eleven features from the “Matminer” presets (“Magpie”, “WenAlloys”) with importance of 52% in total;
- Seven features are available only for the PAW-SQS regressor predicting final values based on predictions of the first predictor (“Pred.”).
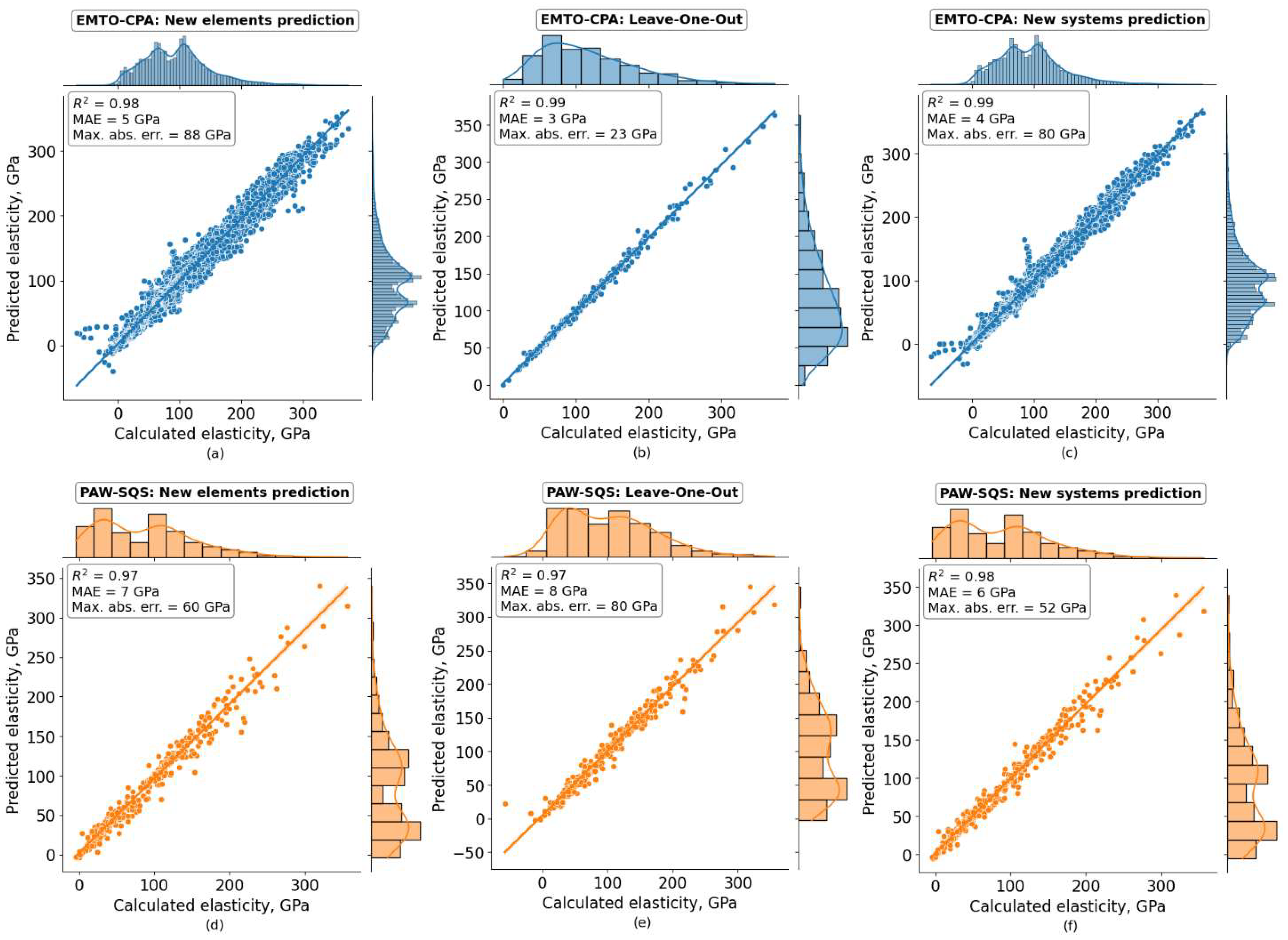
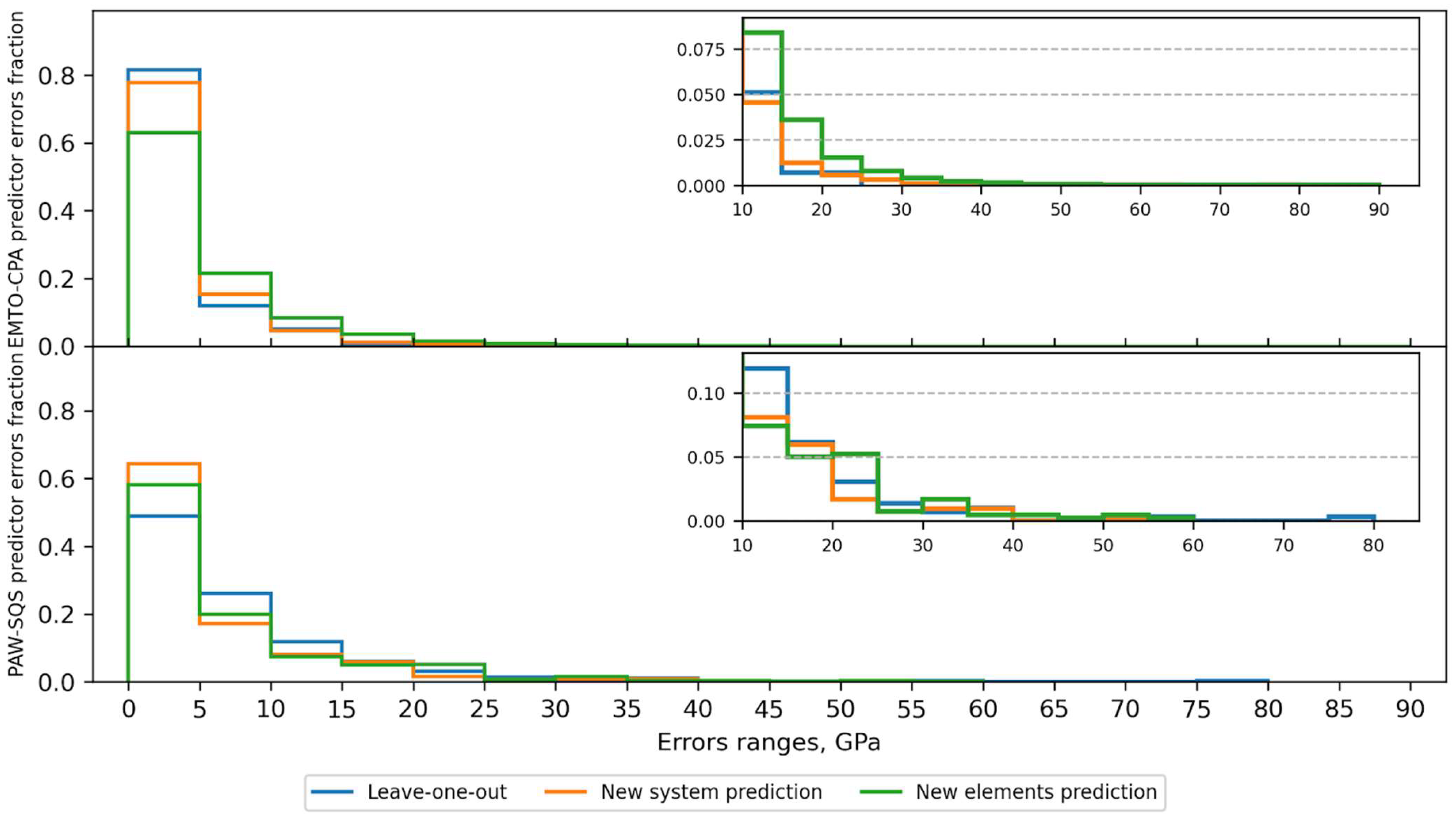
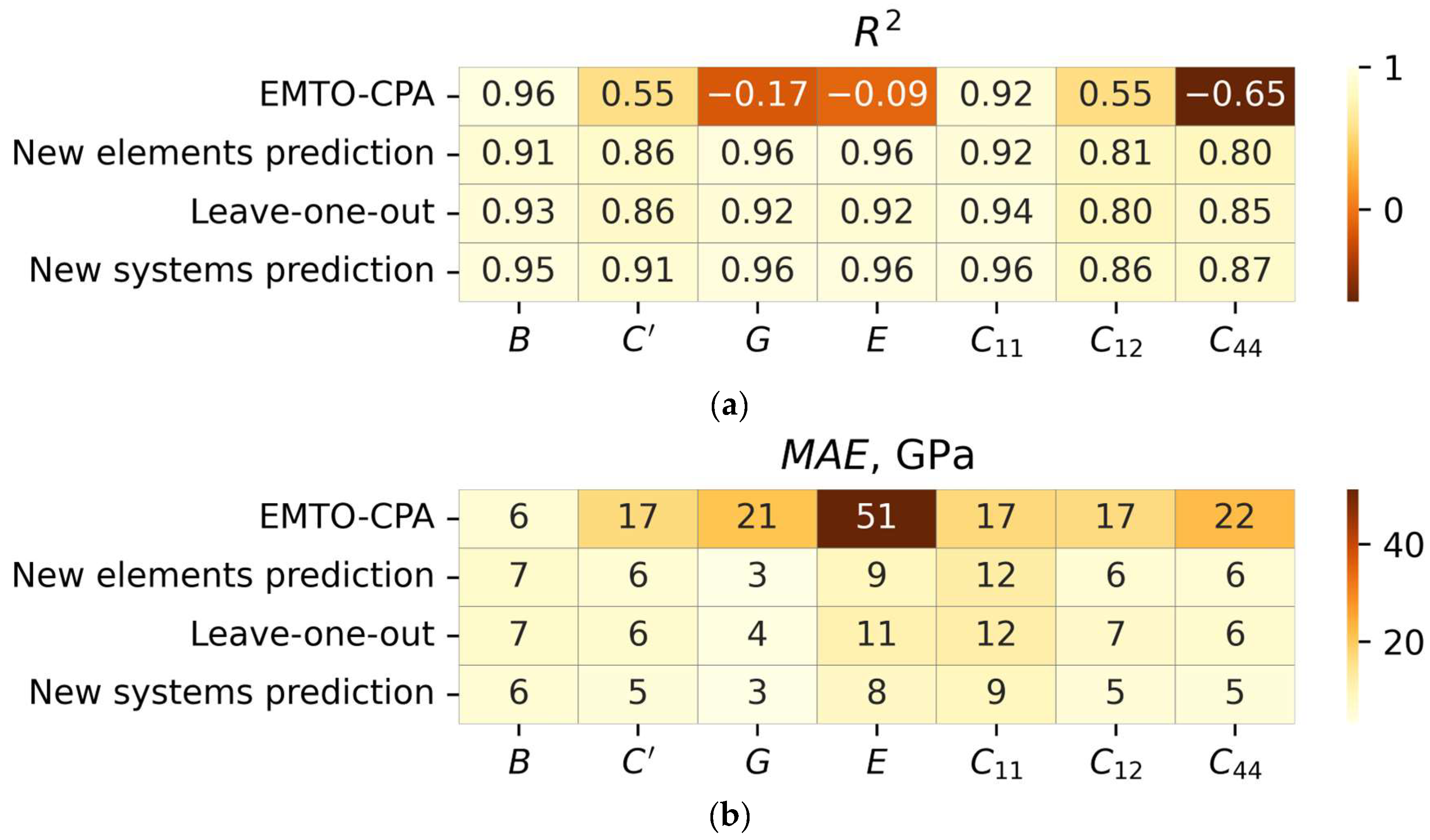
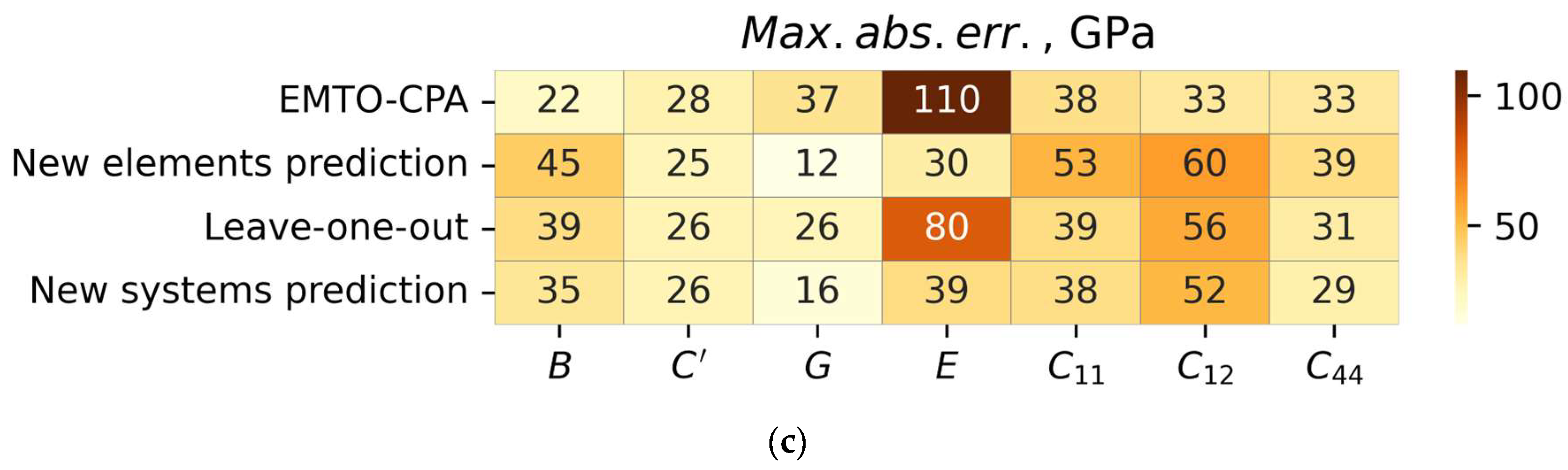
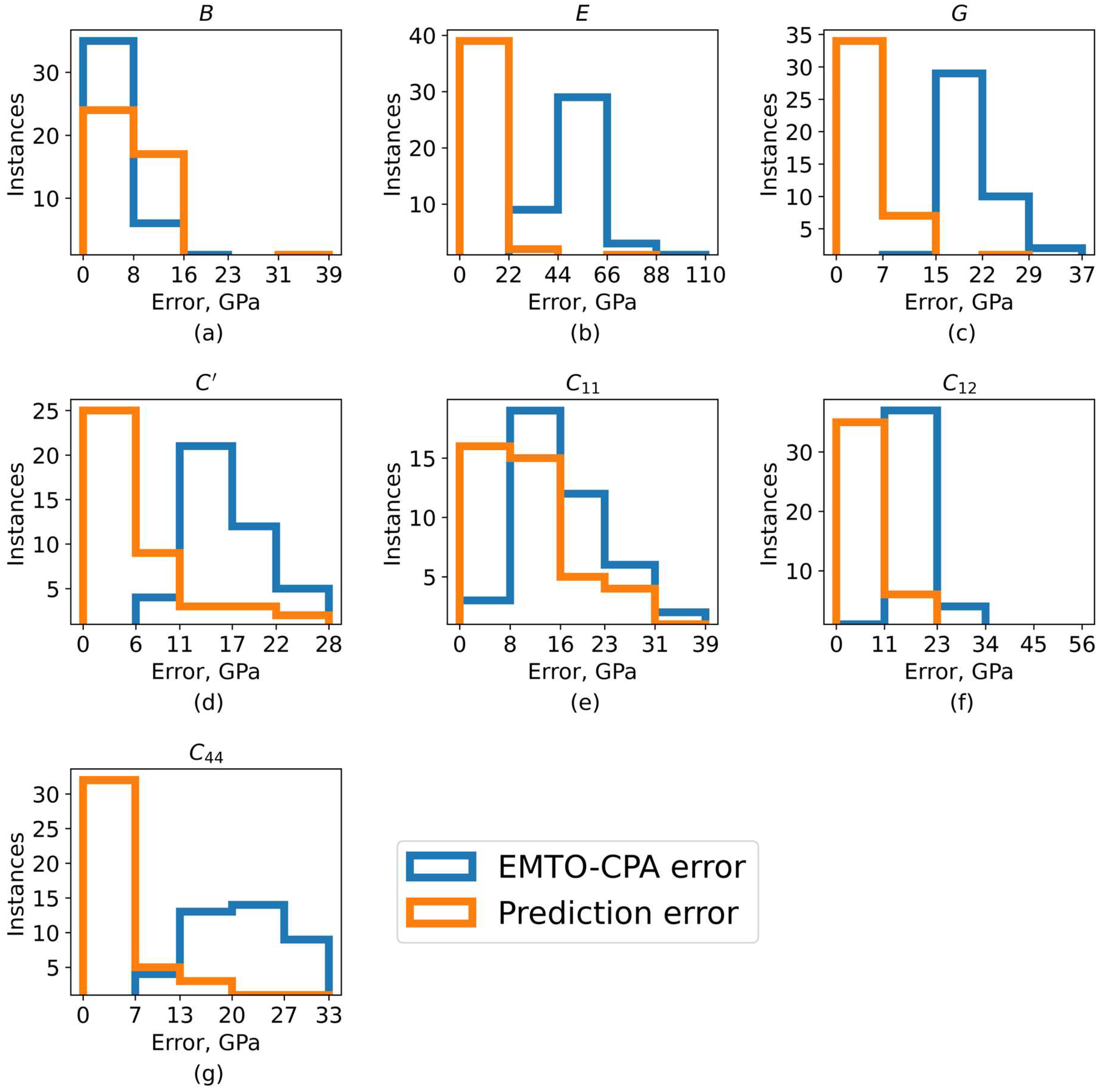
3.2. Cross-Validation Results
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Biesiekierski, A.; Wang, J.; Gepreel, M.A.-H.; Wen, C. A New Look at Biomedical Ti-Based Shape Memory Alloys. Acta Biomater. 2012, 8, 1661–1669. [Google Scholar] [CrossRef]
- Mantripragada, V.P.; Lecka-Czernik, B.; Ebraheim, N.A.; Jayasuriya, A.C. An Overview of Recent Advances in Designing Orthopedic and Craniofacial Implants. J. Biomed. Mater. Res. A 2013, 101, 3349–3364. [Google Scholar] [CrossRef]
- Niinomi, M. Metals for Biomedical Devices, 2nd ed.; Woodhead Publishing: Cambridge, UK, 2019; ISBN 9780081026663. [Google Scholar]
- Li, Y.; Cui, Y.; Zhang, F.; Xu, H. Shape Memory Behavior in Ti–Zr Alloys. Scr. Mater. 2011, 64, 584–587. [Google Scholar] [CrossRef]
- Polmear, I.; StJohn, D.; Nie, J.-F.; Qian, M. Titanium Alloys. In Light Alloys; Elsevier: Amsterdam, The Netherlands, 2017; pp. 369–460. ISBN 9780080994314. [Google Scholar]
- Vitos, L. Computational Quantum Mechanics for Materials Engineers: The EMTO Method and Applications; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2007; ISBN 9781846289514. [Google Scholar]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B Condens. Matter 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Skripnyak, N.V.; Tasnádi, F.; Simak, S.I.; Ponomareva, A.V.; Löfstrand, J.; Berastegui, P.; Jansson, U.; Abrikosov, I.A. Achieving Low Elastic Moduli of Bcc Ti–V Alloys in Vicinity of Mechanical Instability. AIP Adv. 2020, 10, 105322. [Google Scholar] [CrossRef]
- Skripnyak, N.V.; Ponomareva, A.V.; Belov, M.P.; Abrikosov, I.A. Ab Initio Calculations of Elastic Properties of Alloys with Mechanical Instability: Application to BCC Ti-V Alloys. Mater. Des. 2018, 140, 357–365. [Google Scholar] [CrossRef]
- Zunger, A.; Wei, S.; Ferreira, L.G.; Bernard, J.E. Special Quasirandom Structures. Phys. Rev. Lett. 1990, 65, 353–356. [Google Scholar] [CrossRef]
- Smirnova, E.A.; Ponomareva, A.V.; Syzdykova, A.B.; Belov, M.P. Ab Initio Systematic Description of Thermodynamic and Mechanical Properties of Binary Bcc Ti-Based Alloys. Mater. Today Commun. 2022, 31, 103583. [Google Scholar] [CrossRef]
- Hart, G.L.W.; Mueller, T.; Toher, C.; Curtarolo, S. Machine Learning for Alloys. Nat. Rev. Mater. 2021, 6, 730–755. [Google Scholar] [CrossRef]
- Wei, J.; Chu, X.; Sun, X.-Y.; Xu, K.; Deng, H.-X.; Chen, J.; Wei, Z.; Lei, M. Machine Learning in Materials Science. InfoMat 2019, 1, 338–358. [Google Scholar] [CrossRef]
- Choudhary, K.; DeCost, B.; Chen, C.; Jain, A.; Tavazza, F.; Cohn, R.; Park, C.W.; Choudhary, A.; Agrawal, A.; Billinge, S.J.L.; et al. Recent Advances and Applications of Deep Learning Methods in Materials Science. NPJ Comput. Mater. 2022, 8, 1–26. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The Materials Project: A Materials Genome Approach to Accelerating Materials Innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef]
- Haastrup, S.; Strange, M.; Pandey, M.; Deilmann, T.; Schmidt, P.S.; Hinsche, N.F.; Gjerding, M.N.; Torelli, D.; Larsen, P.M.; Riis-Jensen, A.C.; et al. The Computational 2D Materials Database: High-Throughput Modeling and Discovery of Atomically Thin Crystals. 2D Mater. 2018, 5, 042002. [Google Scholar] [CrossRef]
- Curtarolo, S.; Setyawan, W.; Wang, S.; Xue, J.; Yang, K.; Taylor, R.H.; Nelson, L.J.; Hart, G.L.W.; Sanvito, S.; Buongiorno-Nardelli, M.; et al. AFLOWLIB.ORG: A Distributed Materials Properties Repository from High-Throughput Ab Initio Calculations. Comput. Mater. Sci. 2012, 58, 227–235. [Google Scholar] [CrossRef]
- Saal, J.E.; Kirklin, S.; Aykol, M.; Meredig, B.; Wolverton, C. Materials Design and Discovery with High-Throughput Density Functional Theory: The Open Quantum Materials Database (OQMD). JOM 2013, 65, 1501–1509. [Google Scholar] [CrossRef]
- Draxl, C.; Scheffler, M. The NOMAD Laboratory: From Data Sharing to Artificial Intelligence. J. Phys. Mater. 2019, 2, 036001. [Google Scholar] [CrossRef]
- Tawfik, S.A.; Rashid, M.; Gupta, S.; Russo, S.P.; Walsh, T.R.; Venkatesh, S. Machine Learning-Based Discovery of Vibrationally Stable Materials. NPJ Comput. Mater. 2023, 9, 1–6. [Google Scholar] [CrossRef]
- Tawfik, S.A.; Russo, S.P. Naturally-Meaningful and Efficient Descriptors: Machine Learning of Material Properties Based on Robust One-Shot Ab Initio Descriptors. J. Cheminform. 2022, 14, 78. [Google Scholar] [CrossRef]
- Paz Soldan Palma, J.; Chong, X.; Wang, Y.; Shang, S.-L.; Liu, Z.-K. Thermodynamic Re-Modeling of the Yb-Sb System Aided by First-Principles Calculations. Calphad 2023, 81, 102541. [Google Scholar] [CrossRef]
- Kruthika, G.; Ravindran, P. Discerning the Crystal Structure and Engineering the Optoelectronic Properties through Substitution of Divalent Cations (M= Zn, N = Ge) in C3H3MNI3 for Solar Cell Applications. Mater. Sci. Semicond. Process. 2023, 160, 107449. [Google Scholar] [CrossRef]
- Roy, A.; Senor, D.J.; Casella, A.M.; Devanathan, R. Molecular Dynamics Simulations of Radiation Response of LiAlO2 and LiAl5O8. J. Nucl. Mater. 2023, 576, 154280. [Google Scholar] [CrossRef]
- Mukhamedov, B.O.; Karavaev, K.V.; Abrikosov, I.A. Machine Learning Prediction of Thermodynamic and Mechanical Properties of Multicomponent Fe-Cr-Based Alloys. Phys. Rev. Mater. 2021, 5, 104407. [Google Scholar] [CrossRef]
- Hayashi, G.; Suzuki, K.; Terai, T.; Fujii, H.; Ogura, M.; Sato, K. Prediction Model of Elastic Constants of BCC High-Entropy Alloys Based on First-Principles Calculations and Machine Learning Techniques. Sci. Technol. Adv. Mater. Methods 2022, 2, 381–391. [Google Scholar] [CrossRef]
- Kim, G.; Diao, H.; Lee, C.; Samaei, A.T.; Phan, T.; de Jong, M.; An, K.; Ma, D.; Liaw, P.K.; Chen, W. First-Principles and Machine Learning Predictions of Elasticity in Severely Lattice-Distorted High-Entropy Alloys with Experimental Validation. Acta Mater. 2019, 181, 124–138. [Google Scholar] [CrossRef]
- Frohlich, H.; Chapelle, O.; Scholkopf, B. Feature Selection for Support Vector Machines by Means of Genetic Algorithm. In Proceedings of the 15th IEEE International Conference on Tools with Artificial Intelligence, Sacramento, CA, USA, 5 November 2003; IEEE Computer Society: New York, NY, USA, 2004. [Google Scholar]
- Deb, K.; Pratap, A.; Agarwal, S.; Meyarivan, T. A Fast and Elitist Multiobjective Genetic Algorithm: NSGA-II. IEEE Trans. Evol. Comput. 2002, 6, 182–197. [Google Scholar] [CrossRef]
- Ward, L.; Dunn, A.; Faghaninia, A.; Zimmermann, N.E.R.; Bajaj, S.; Wang, Q.; Montoya, J.; Chen, J.; Bystrom, K.; Dylla, M.; et al. Matminer: An Open Source Toolkit for Materials Data Mining. Comput. Mater. Sci. 2018, 152, 60–69. [Google Scholar] [CrossRef]
- Xu, P.; Ji, X.; Li, M.; Lu, W. Small Data Machine Learning in Materials Science. NPJ Comput. Mater. 2023, 9, 1–15. [Google Scholar] [CrossRef]
- Ranaweera, M.; Mahmoud, Q.H. Virtual to Real-World Transfer Learning: A Systematic Review. Electronics 2021, 10, 1491. [Google Scholar] [CrossRef]
- Zhuang, F.; Qi, Z.; Duan, K.; Xi, D.; Zhu, Y.; Zhu, H.; Xiong, H.; He, Q. A Comprehensive Survey on Transfer Learning. Proc. IEEE Inst. Electr. Electron. Eng. 2021, 109, 43–76. [Google Scholar] [CrossRef]
- Lee, J.; Asahi, R. Transfer Learning for Materials Informatics Using Crystal Graph Convolutional Neural Network. Comput. Mater. Sci. 2021, 190, 110314. [Google Scholar] [CrossRef]
- Chen, H.; Shang, Z.; Lu, W.; Li, M.; Tan, F. A Property-Driven Stepwise Design Strategy for Multiple Low-Melting Alloys via Machine Learning. Adv. Eng. Mater. 2021, 23, 2100612. [Google Scholar] [CrossRef]
- Lu, T.; Li, H.; Li, M.; Wang, S.; Lu, W. Inverse Design of Hybrid Organic–Inorganic Perovskites with Suitable Bandgaps via Proactive Searching Progress. ACS Omega 2022, 7, 21583–21594. [Google Scholar] [CrossRef]
- Xin, R.; Siriwardane, E.M.D.; Song, Y.; Zhao, Y.; Louis, S.-Y.; Nasiri, A.; Hu, J. Active-Learning-Based Generative Design for the Discovery of Wide-Band-Gap Materials. J. Phys. Chem. C 2021, 125, 16118–16128. [Google Scholar] [CrossRef]
- Wanchen, Z.; Chen, Z.; Bin, X.; Xing, L.; Lu, L.; Tongxin, Y.; Yanjie, L.; Ziqiang, D.; Yi, L.; Ce, Z.; et al. Composition Refinement of 6061 Aluminum Alloy Using Active Machine Learning Model Based on Bayesian Optimization Sampling. Acta Metall. Sinica 2020, 57, 797–810. [Google Scholar]
- Kaufmann, K.; Maryanovsky, D.; Mellor, W.M.; Zhu, C.; Rosengarten, A.S.; Harrington, T.J.; Oses, C.; Toher, C.; Curtarolo, S.; Vecchio, K.S. Discovery of High-Entropy Ceramics via Machine Learning. NPJ Comput. Mater. 2020, 6, 1–9. [Google Scholar] [CrossRef]
- CatBoost—State-of-the-Art Open-Source Gradient Boosting Library with Categorical Features Support. Available online: https://catboost.ai (accessed on 20 February 2024).
- Duan, J.; Asteris, P.G.; Nguyen, H.; Bui, X.-N.; Moayedi, H. A Novel Artificial Intelligence Technique to Predict Compressive Strength of Recycled Aggregate Concrete Using ICA-XGBoost Model. Eng. Comput. 2020, 37, 3329–3346. [Google Scholar] [CrossRef]
- Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.-Y. LightGBM: A highly efficient gradient boosting decision tree. In Proceedings of the 31st International Conference on Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017; NIPS’17. pp. 3148–3157. [Google Scholar]
- Friedman, J.H. Greedy Function Approximation: A Gradient Boosting Machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Kailiang, L.; Dongping, C.; Xiaobo, J.; Minjie, L.; Wencong, L. Machine Learning Aided Discovery of the Layered Double Hydroxides with the Largest Basal Spacing for Super-Capacitors. Int. J. Electrochem. Sci. 2021, 16, 211146. [Google Scholar]
- Natekin, A.; Knoll, A. Gradient Boosting Machines, a Tutorial. Front. Neurorobot. 2013, 7, 21. [Google Scholar] [CrossRef]
- Friedman, J.H. Stochastic Gradient Boosting. Comput. Stat. Data Anal. 2002, 38, 367–378. [Google Scholar] [CrossRef]
- Vitos, L.; Abrikosov, I.A.; Johansson, B. Anisotropic Lattice Distortions in Random Alloys from First-Principles Theory. Phys. Rev. Lett. 2001, 87, 156401. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B Condens. Matter 1999, 59, 1758. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Steneteg, P.; Hellman, O.; Vekilova, O.Y.; Shulumba, N.; Tasnádi, F.; Abrikosov, I.A. Temperature Dependence of TiN Elastic Constants from ab Initio Molecular Dynamics Simulations. Phys. Rev. B Condens. Matter 2013, 87, 094114. [Google Scholar] [CrossRef]
- Hill, R. The Elastic Behaviour of a Crystalline Aggregate. Proc. Phys. Soc. A 1952, 65, 349. [Google Scholar] [CrossRef]
- Grimvall, G. Thermophysical Properties of Materials, 1st ed.; Elsevier Science: Amsterdam, The Netherlands, 1999; Enlarged and revised edition; p. 52. ISBN 0444827943. [Google Scholar]
- Mouhat, F.; Coudert, F.-X. Necessary and Sufficient Elastic Stability Conditions in Various Crystal Systems. Phys. Rev. B Condens. Matter 2014, 90, 224104. [Google Scholar] [CrossRef]
- Ong, S.P.; Richards, W.D.; Jain, A.; Hautier, G.; Kocher, M.; Cholia, S.; Gunter, D.; Chevrier, V.L.; Persson, K.A.; Ceder, G. Python Materials Genomics (pymatgen): A Robust, Open-Source Python Library for Materials Analysis. Comput. Mater. Sci. 2013, 68, 314–319. [Google Scholar] [CrossRef]
- Ward, L.; Agrawal, A.; Choudhary, A.; Wolverton, C. A General-Purpose Machine Learning Framework for Predicting Properties of Inorganic Materials. NPJ Comput. Mater. 2016, 2, 16028. [Google Scholar] [CrossRef]
- Wen, C.; Zhang, Y.; Wang, C.; Xue, D.; Bai, Y.; Antonov, S.; Dai, L.; Lookman, T.; Su, Y. Machine Learning Assisted Design of High Entropy Alloys with Desired Property. Acta Mater. 2019, 170, 109–117. [Google Scholar] [CrossRef]
- Zhang, R.F.; Zhang, S.H.; He, Z.J.; Jing, J.; Sheng, S.H. Miedema Calculator: A Thermodynamic Platform for Predicting Formation Enthalpies of Alloys within Framework of Miedema’s Theory. Comput. Phys. Commun. 2016, 209, 58–69. [Google Scholar] [CrossRef]
- Schmidt, J.; Pettersson, L.; Verdozzi, C.; Botti, S.; Marques, M.A.L. Crystal Graph Attention Networks for the Prediction of Stable Materials. Sci. Adv. 2021, 7, 7948. [Google Scholar] [CrossRef] [PubMed]
- Prokhorenkova, L.; Gusev, G.; Vorobev, A.; Dorogush, A.V.; Gulin, A. CatBoost: Unbiased boosting with categorical features. In Proceedings of the 32nd International Conference on Neural Information Processing Systems (NIPS’18), Montréal, QB, Canada, 3–8 December 2018; Curran Associates Inc.: Red Hook, NY, USA, 2018; pp. 6639–6649. Available online: https://dl.acm.org/doi/10.5555/3327757.3327770 (accessed on 16 February 2024).
- Rajan, K. Machine Learning Elastic Constants of Multi-Component Alloys. Comput. Mater. Sci. 2021, 198, 110671. [Google Scholar]
- Levämäki, H.; Tasnádi, F.; Sangiovanni, D.G.; Johnson, L.J.S.; Armiento, R.; Abrikosov, I.A. Predicting Elastic Properties of Hard-Coating Alloys Using Ab-Initio and Machine Learning Methods. NPJ Comput. Mater. 2022, 8, 2–10. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Predictor | CV Type | Property | R2 | MAE, GPa | Max. Abs. Err., GPa |
|---|---|---|---|---|---|
| EMTO-CPA | New elements prediction | B | 0.93 | 5.1 | 43 |
| C’ | 0.94 | 4.0 | 44 | ||
| E | 0.93 | 8.2 | 86 | ||
| G | 0.93 | 3.2 | 21 | ||
| C11 | 0.96 | 8.9 | 88 | ||
| C12 | 0.82 | 4.4 | 53 | ||
| C44 | 0.74 | 4.1 | 30 | ||
| Leave-one-out | B | 0.99 | 3.0 | 11 | |
| C’ | 0.98 | 2.8 | 14 | ||
| E | 0.98 | 4.4 | 22 | ||
| G | 0.98 | 1.8 | 9 | ||
| C11 | 0.99 | 5.5 | 23 | ||
| C12 | 0.97 | 2.9 | 16 | ||
| C44 | 0.97 | 2.2 | 10 | ||
| New system prediction | B | 0.97 | 3.6 | 41 | |
| C’ | 0.97 | 2.5 | 33 | ||
| E | 0.97 | 5.2 | 53 | ||
| G | 0.98 | 2.0 | 14 | ||
| C11 | 0.98 | 6.1 | 80 | ||
| C12 | 0.92 | 3.2 | 31 | ||
| C44 | 0.91 | 2.6 | 18 | ||
| PAW-SQS | New elements prediction | B | 0.91 | 7.2 | 45 |
| C’ | 0.86 | 6.1 | 25 | ||
| E | 0.96 | 8.9 | 30 | ||
| G | 0.96 | 3.4 | 12 | ||
| C11 | 0.92 | 12.3 | 53 | ||
| C12 | 0.81 | 6.1 | 60 | ||
| C44 | 0.80 | 6.0 | 39 | ||
| Leave-one-out | B | 0.93 | 7.2 | 39 | |
| C’ | 0.86 | 6.3 | 26 | ||
| E | 0.92 | 10.8 | 80 | ||
| G | 0.92 | 4.1 | 26 | ||
| C11 | 0.94 | 11.8 | 39 | ||
| C12 | 0.80 | 7.4 | 56 | ||
| C44 | 0.85 | 6.4 | 31 | ||
| New system prediction | B | 0.95 | 5.7 | 35 | |
| C’ | 0.91 | 4.8 | 26 | ||
| E | 0.96 | 8.1 | 39 | ||
| G | 0.96 | 3.1 | 16 | ||
| C11 | 0.96 | 9.0 | 38 | ||
| C12 | 0.86 | 5.2 | 52 | ||
| C44 | 0.87 | 5.0 | 29 |
| Predictor | CV Type | Errors < 5 GPa, % | Errors < 20 GPa, % |
|---|---|---|---|
| EMTO-CPA | New elements prediction | 63 | 97 |
| Leave-one-out | 82 | 99 | |
| New system prediction | 78 | 99 | |
| PAW-SQS | New elements prediction | 58 | 90 |
| Leave-one-out | 49 | 93 | |
| New system prediction | 64 | 95 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sidnov, K.; Konov, D.; Smirnova, E.A.; Ponomareva, A.V.; Belov, M.P. Machine Learning-Based Prediction of Elastic Properties Using Reduced Datasets of Accurate Calculations Results. Metals 2024, 14, 438. https://doi.org/10.3390/met14040438
Sidnov K, Konov D, Smirnova EA, Ponomareva AV, Belov MP. Machine Learning-Based Prediction of Elastic Properties Using Reduced Datasets of Accurate Calculations Results. Metals. 2024; 14(4):438. https://doi.org/10.3390/met14040438
Chicago/Turabian StyleSidnov, Kirill, Denis Konov, Ekaterina A. Smirnova, Alena V. Ponomareva, and Maxim P. Belov. 2024. "Machine Learning-Based Prediction of Elastic Properties Using Reduced Datasets of Accurate Calculations Results" Metals 14, no. 4: 438. https://doi.org/10.3390/met14040438