
Targeting and Understanding HIV Latency: The CRISPR System against the Provirus



Abstract
:1. Introduction
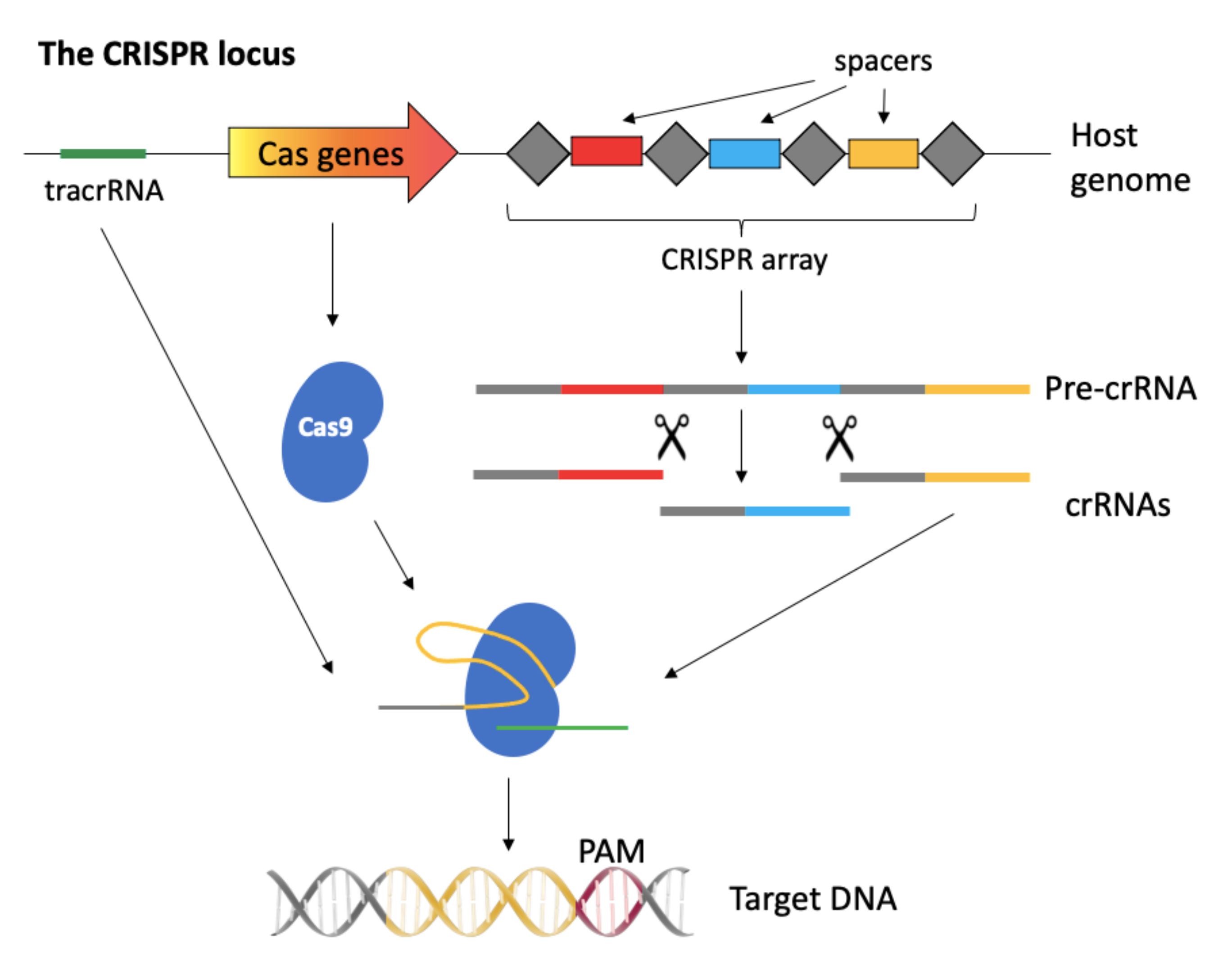
2. A Brief Overview of the CRISPR-Cas System
2.1. The CRISPR-Cas System
2.2. Delivery Systems
2.3. Cas Nuclease Variants
2.4. Considerations on the Selection of the HIV-1 Targets
2.5. Considerations on the Design of the gRNAs
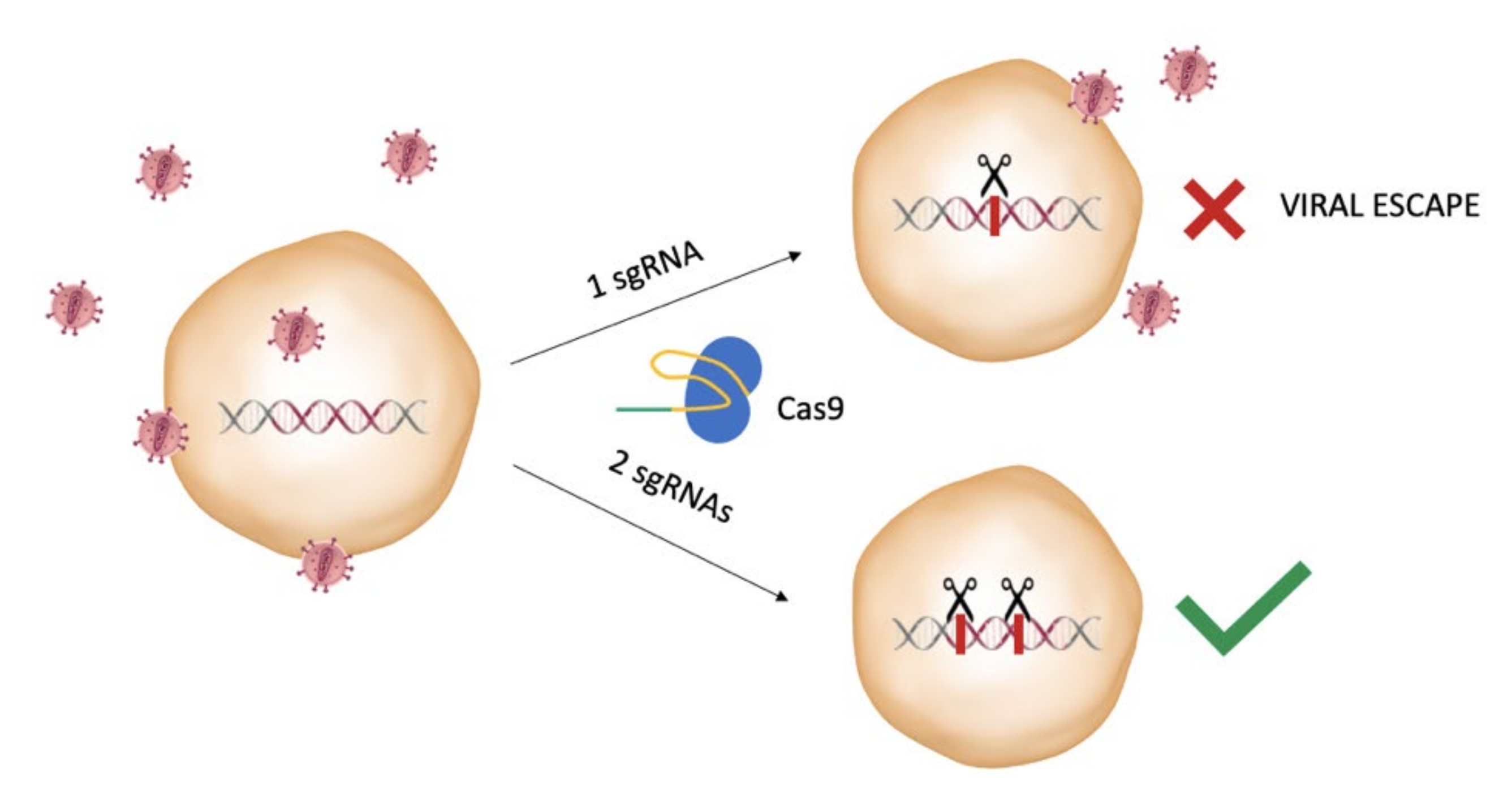
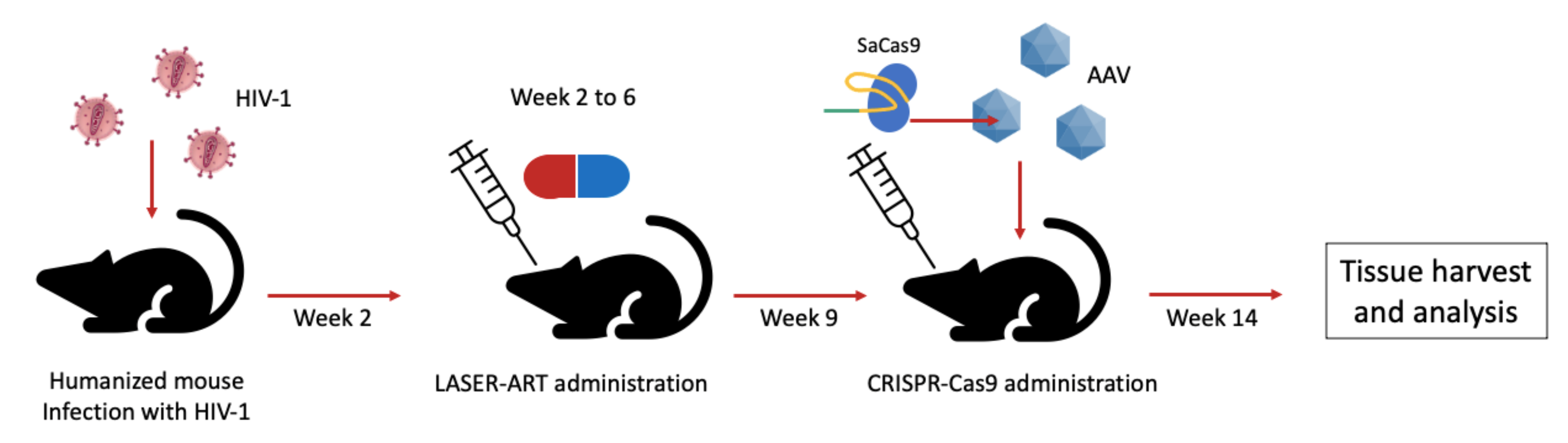
3. The CRISPR System for Editing of HIV Sequence in Latency Models
4. The Catalytically Inactive Cas9 as a Modulator of Provirus Transcription
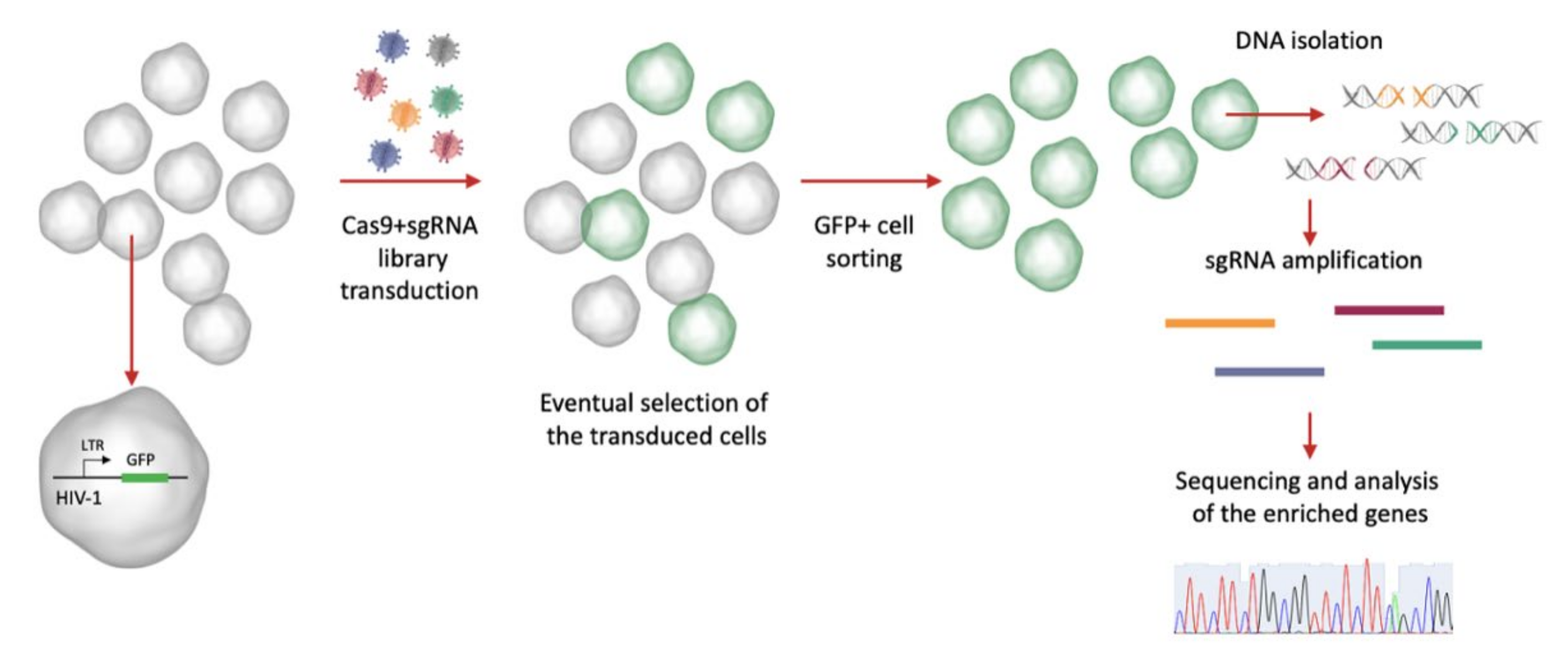
5. CRISPR Screening to Find Host Dependency Factors Involved in HIV-1 Latency
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Dieleman, J.L.; Haakenstad, A.; Micah, A.; Moses, M.; Abbafati, C.; Acharya, P.; Adhikari, T.B.; Adou, A.K.; Ahmad Kiadaliri, A.; Alam, K.; et al. Spending on Health and HIV/AIDS: Domestic Health Spending and Development Assistance in 188 Countries, 1995–2015. Lancet 2018, 391, 1799–1829. [Google Scholar] [CrossRef] [Green Version]
- HIV-CAUSAL Collaboration; Ray, M.; Logan, R.; Sterne, J.A.; Hernández-Díaz, S.; Robins, J.M.; Sabin, C.; Bansi, L.; van Sighem, A.; de Wolf, F.; et al. The Effect of Combined Antiretroviral Therapy on the Overall Mortality of HIV-Infected Individuals. AIDS 2010, 24, 123–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arts, E.J.; Hazuda, D.J. HIV-1 Antiretroviral Drug Therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef] [PubMed]
- Delelis, O.; Carayon, K.; Saïb, A.; Deprez, E.; Mouscadet, J.-F. Integrase and Integration: Biochemical Activities of HIV-1 Integrase. Retrovirology 2008, 5, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeks, S.G.; Overbaugh, J.; Phillips, A.; Buchbinder, S. HIV Infection. Nat. Rev. Dis Primers 2015, 1, 15035. [Google Scholar] [CrossRef] [PubMed]
- Gasper, D.J.; Tejera, M.M.; Suresh, M. CD4 T-Cell Memory Generation and Maintenance. Crit. Rev. Immunol. 2014, 34, 121–146. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, R.F.; Greene, W.C. HIV Latency. Cold Spring Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coiras, M.; López-Huertas, M.R.; Pérez-Olmeda, M.; Alcamí, J. Understanding HIV-1 Latency Provides Clues for the Eradication of Long-Term Reservoirs. Nat. Rev. Microbiol. 2009, 7, 798–812. [Google Scholar] [CrossRef]
- Khanal, S.; Schank, M.; El Gazzar, M.; Moorman, J.P.; Yao, Z.Q. HIV-1 Latency and Viral Reservoirs: Existing Reversal Approaches and Potential Technologies, Targets, and Pathways Involved in HIV Latency Studies. Cells 2021, 10, 475. [Google Scholar] [CrossRef]
- Dahabieh, M.S.; Battivelli, E.; Verdin, E. Understanding HIV Latency: The Road to an HIV Cure. Annu. Rev. Med. 2015, 66, 407–421. [Google Scholar] [CrossRef] [Green Version]
- Olson, A.; Basukala, B.; Lee, S.; Gagne, M.; Wong, W.W.; Henderson, A.J. Targeted Chromatinization and Repression of HIV-1 Provirus Transcription with Repurposed CRISPR/Cas9. Viruses 2020, 12, 1154. [Google Scholar] [CrossRef]
- Darcis, G.; Das, A.T.; Berkhout, B. Tackling HIV Persistence: Pharmacological versus CRISPR-Based Shock Strategies. Viruses 2018, 10, 157. [Google Scholar] [CrossRef] [Green Version]
- Allen, A.G.; Chung, C.-H.; Atkins, A.; Dampier, W.; Khalili, K.; Nonnemacher, M.R.; Wigdahl, B. Gene Editing of HIV-1 Co-Receptors to Prevent and/or Cure Virus Infection. Front. Microbiol. 2018, 9, 2940. [Google Scholar] [CrossRef]
- Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Carette, J.E. A CRISPR Toolbox to Study Virus–Host Interactions. Nat. Rev. Microbiol. 2017, 15, 351–364. [Google Scholar] [CrossRef]
- Ernst, M.P.T.; Broeders, M.; Herrero-Hernandez, P.; Oussoren, E.; van der Ploeg, A.T.; Pijnappel, W.W.M.P. Ready for Repair? Gene Editing Enters the Clinic for the Treatment of Human Disease. Mol. Ther.-Methods Clin. Dev. 2020, 18, 532–557. [Google Scholar] [CrossRef]
- Cyranoski, D.; Ledford, H. Genome-Edited Baby Claim Provokes International Outcry. Nature 2018, 563, 607–608. [Google Scholar] [CrossRef]
- Morrison, M.; de Saille, S. CRISPR in Context: Towards a Socially Responsible Debate on Embryo Editing. Palgrave Commun. 2019, 5, 110. [Google Scholar] [CrossRef] [Green Version]
- Davenport, M.P.; Khoury, D.S.; Cromer, D.; Lewin, S.R.; Kelleher, A.D.; Kent, S.J. Functional Cure of HIV: The Scale of the Challenge. Nat. Rev. Immunol. 2019, 19, 45–54. [Google Scholar] [CrossRef]
- Allers, K.; Hütter, G.; Hofmann, J.; Loddenkemper, C.; Rieger, K.; Thiel, E.; Schneider, T. Evidence for the Cure of HIV Infection by CCR5Δ32/Δ32 Stem Cell Transplantation. Blood 2011, 117, 2791–2799. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Peppa, D.; Hill, A.L.; Gálvez, C.; Salgado, M.; Pace, M.; McCoy, L.E.; Griffith, S.A.; Thornhill, J.; Alrubayyi, A.; et al. Evidence for HIV-1 Cure after CCR5Δ32/Δ32 Allogeneic Haemopoietic Stem-Cell Transplantation 30 Months Post Analytical Treatment Interruption: A Case Report. Lancet HIV 2020, 7, e340–e347. [Google Scholar] [CrossRef] [Green Version]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene Editing of CCR5 in Autologous CD4 T Cells of Persons Infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Schwarze, L.I.; Głów, D.; Sonntag, T.; Uhde, A.; Fehse, B. Optimisation of a TALE Nuclease Targeting the HIV Co-Receptor CCR5 for Clinical Application. Gene Ther. 2021, 28, 588–601. [Google Scholar] [CrossRef]
- Xu, L.; Wang, J.; Liu, Y.; Xie, L.; Su, B.; Mou, D.; Wang, L.; Liu, T.; Wang, X.; Zhang, B.; et al. CRISPR-Edited Stem Cells in a Patient with HIV and Acute Lymphocytic Leukemia. N. Engl. J. Med. 2019, 381, 1240–1247. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. The New Frontier of Genome Engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A Review of the Challenges and Approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Qi, L.S. A CRISPR–DCas Toolbox for Genetic Engineering and Synthetic Biology. J. Mol. Biol. 2019, 431, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.K.; Strelchenko, N.; Park, M.A.; Kim, Y.H.; Mean, K.D.; Schotzko, M.L.; Kang, H.J.; Golos, T.G.; Slukvin, I.I. Genome Editing of CCR5 by CRISPR-Cas9 in Mauritian Cynomolgus Macaque Embryos. Sci. Rep. 2020, 10, 18457. [Google Scholar] [CrossRef]
- Hultquist, J.F.; Schumann, K.; Woo, J.M.; Manganaro, L.; McGregor, M.J.; Doudna, J.; Simon, V.; Krogan, N.J.; Marson, A. A Cas9 Ribonucleoprotein Platform for Functional Genetic Studies of HIV-Host Interactions in Primary Human T Cells. Cell Rep. 2016, 17, 1438–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petris, G.; Casini, A.; Montagna, C.; Lorenzin, F.; Prandi, D.; Romanel, A.; Zasso, J.; Conti, L.; Demichelis, F.; Cereseto, A. Hit and Go CAS9 Delivered through a Lentiviral Based Self-Limiting Circuit. Nat. Commun. 2017, 8, 15334. [Google Scholar] [CrossRef]
- Dash, P.K.; Kaminski, R.; Bella, R.; Su, H.; Mathews, S.; Ahooyi, T.M.; Chen, C.; Mancuso, P.; Sariyer, R.; Ferrante, P.; et al. Sequential LASER ART and CRISPR Treatments Eliminate HIV-1 in a Subset of Infected Humanized Mice. Nat. Commun. 2019, 10, 2753. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, P.; Chen, C.; Kaminski, R.; Gordon, J.; Liao, S.; Robinson, J.A.; Smith, M.D.; Liu, H.; Sariyer, I.K.; Sariyer, R.; et al. CRISPR Based Editing of SIV Proviral DNA in ART Treated Non-Human Primates. Nat. Commun. 2020, 11, 6065. [Google Scholar] [CrossRef]
- Gao, Z.; Fan, M.; Das, A.T.; Herrera-Carrillo, E.; Berkhout, B. Extinction of All Infectious HIV in Cell Culture by the CRISPR-Cas12a System with Only a Single CrRNA. Nucleic Acids Research 2020, 48, 5527–5539. [Google Scholar] [CrossRef]
- Yin, L.; Zhao, F.; Sun, H.; Wang, Z.; Huang, Y.; Zhu, W.; Xu, F.; Mei, S.; Liu, X.; Zhang, D.; et al. CRISPR-Cas13a Inhibits HIV-1 Infection. Mol. Ther.-Nucleic Acids 2020, 21, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Li, H.; Wang, Q.; Hua, C.; Zhang, H.; Li, W.; Jiang, S.; Lu, L. Advancements in Developing Strategies for Sterilizing and Functional HIV Cures. BioMed Res. Int. 2017, 2017, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ophinni, Y.; Inoue, M.; Kotaki, T.; Kameoka, M. CRISPR/Cas9 System Targeting Regulatory Genes of HIV-1 Inhibits Viral Replication in Infected T-Cell Cultures. Sci. Rep. 2018, 8, 7784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, C.; Zhang, T.; Qu, X.; Zhang, Y.; Putatunda, R.; Xiao, X.; Li, F.; Xiao, W.; Zhao, H.; Dai, S.; et al. In Vivo Excision of HIV-1 Provirus by SaCas9 and Multiplex Single-Guide RNAs in Animal Models. Mol. Ther. 2017, 25, 1168–1186. [Google Scholar] [CrossRef] [Green Version]
- Karpel, M.E.; Boutwell, C.L.; Allen, T.M. BLT Humanized Mice as a Small Animal Model of HIV Infection. Curr. Opin. Virol. 2015, 13, 75–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenstiel, P.; Gharavi, A.; D’Agati, V.; Klotman, P. Transgenic and Infectious Animal Models of HIV-Associated Nephropathy. J. Am. Soc. Nephrol. 2009, 20, 2296–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. CRISPR-Cas9 Can Inhibit HIV-1 Replication but NHEJ Repair Facilitates Virus Escape. Mol. Ther. 2016, 24, 522–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. A Combinatorial CRISPR-Cas9 Attack on HIV-1 DNA Extinguishes All Infectious Provirus in Infected T Cell Cultures. Cell Rep. 2016, 17, 2819–2826. [Google Scholar] [CrossRef] [Green Version]
- Dampier, W.; Sullivan, N.T.; Mell, J.C.; Pirrone, V.; Ehrlich, G.D.; Chung, C.-H.; Allen, A.G.; DeSimone, M.; Zhong, W.; Kercher, K.; et al. Broad-Spectrum and Personalized Guide RNAs for CRISPR/Cas9 HIV-1 Therapeutics. AIDS Res. Hum. Retrovir. 2018, 34, 950–960. [Google Scholar] [CrossRef]
- Chung, C.-H.; Allen, A.G.; Atkins, A.; Link, R.W.; Nonnemacher, M.R.; Dampier, W.; Wigdahl, B. Computational Design of GRNAs Targeting Genetic Variants Across HIV-1 Subtypes for CRISPR-Mediated Antiviral Therapy. Front. Cell. Infect. Microbiol. 2021, 11, 593077. [Google Scholar] [CrossRef]
- Akcakaya, P.; Bobbin, M.L.; Guo, J.A.; Malagon-Lopez, J.; Clement, K.; Garcia, S.P.; Fellows, M.D.; Porritt, M.J.; Firth, M.A.; Carreras, A.; et al. In Vivo CRISPR Editing with No Detectable Genome-Wide off-Target Mutations. Nature 2018, 561, 416–419. [Google Scholar] [CrossRef]
- Sanches-da-Silva, G.D.N.; Medeiros, L.F.S.; Lima, F.M. The Potential Use of the CRISPR-Cas System for HIV-1 Gene Therapy. Int. J. Genom. 2019, 2019, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ebina, H.; Misawa, N.; Kanemura, Y.; Koyanagi, Y. Harnessing the CRISPR/Cas9 System to Disrupt Latent HIV-1 Provirus. Sci. Rep. 2013, 3, 2510. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Lei, R.; Le Duff, Y.; Li, J.; Guo, F.; Wainberg, M.A.; Liang, C. The CRISPR/Cas9 System Inactivates Latent HIV-1 Proviral DNA. Retrovirology 2015, 12, 22. [Google Scholar] [CrossRef] [Green Version]
- Liao, H.-K.; Gu, Y.; Diaz, A.; Marlett, J.; Takahashi, Y.; Li, M.; Suzuki, K.; Xu, R.; Hishida, T.; Chang, C.-J.; et al. Use of the CRISPR/Cas9 System as an Intracellular Defense against HIV-1 Infection in Human Cells. Nat. Commun. 2015, 6, 6413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminski, R.; Chen, Y.; Fischer, T.; Tedaldi, E.; Napoli, A.; Zhang, Y.; Karn, J.; Hu, W.; Khalili, K. Elimination of HIV-1 Genomes from Human T-Lymphoid Cells by CRISPR/Cas9 Gene Editing. Sci. Rep. 2016, 6, 22555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitney, J.B.; Brad Jones, R. In Vitro and In Vivo Models of HIV Latency. In HIV Vaccines and Cure; Zhang, L., Lewin, S.R., Eds.; Advances in Experimental Medicine and Biology; Springer Singapore: Singapore, 2018; Volume 1075, pp. 241–263. ISBN 9789811304835. [Google Scholar]
- Hu, W.; Kaminski, R.; Yang, F.; Zhang, Y.; Cosentino, L.; Li, F.; Luo, B.; Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Karn, J.; et al. RNA-Directed Gene Editing Specifically Eradicates Latent and Prevents New HIV-1 Infection. Proc. Natl. Acad. Sci. USA 2014, 111, 11461–11466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front. Cell. Infect. Microbiol. 2019, 9, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunze, C.; Börner, K.; Kienle, E.; Orschmann, T.; Rusha, E.; Schneider, M.; Radivojkov-Blagojevic, M.; Drukker, M.; Desbordes, S.; Grimm, D.; et al. Synthetic AAV/CRISPR Vectors for Blocking HIV-1 Expression in Persistently Infected Astrocytes. Glia 2018, 66, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Berkhout, B.; Pasternak, A.O. The Quest for Cellular Markers of HIV Reservoirs: Any Color You Like. Front. Immunol. 2019, 10, 2251. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, R.; Bella, R.; Yin, C.; Otte, J.; Ferrante, P.; Gendelman, H.E.; Li, H.; Booze, R.; Gordon, J.; Hu, W.; et al. Excision of HIV-1 DNA by Gene Editing: A Proof-of-Concept in Vivo Study. Gene Ther. 2016, 23, 690–695. [Google Scholar] [CrossRef]
- Kim, Y.; Anderson, J.L.; Lewin, S.R. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe 2018, 23, 14–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vansant, G.; Bruggemans, A.; Janssens, J.; Debyser, Z. Block-And-Lock Strategies to Cure HIV Infection. Viruses 2020, 12, 84. [Google Scholar] [CrossRef] [Green Version]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W.; et al. RNA-Guided Gene Activation by CRISPR-Cas9–Based Transcription Factors. Nat. Methods 2013, 10, 973–976. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yin, C.; Zhang, T.; Li, F.; Yang, W.; Kaminski, R.; Fagan, P.R.; Putatunda, R.; Young, W.-B.; Khalili, K.; et al. CRISPR/GRNA-Directed Synergistic Activation Mediator (SAM) Induces Specific, Persistent and Robust Reactivation of the HIV-1 Latent Reservoirs. Sci. Rep. 2015, 5, 16277. [Google Scholar] [CrossRef]
- Bialek, J.K.; Dunay, G.A.; Voges, M.; Schäfer, C.; Spohn, M.; Stucka, R.; Hauber, J.; Lange, U.C. Targeted HIV-1 Latency Reversal Using CRISPR/Cas9-Derived Transcriptional Activator Systems. PLoS ONE 2016, 11, e0158294. [Google Scholar] [CrossRef] [Green Version]
- Limsirichai, P.; Gaj, T.; Schaffer, D.V. CRISPR-Mediated Activation of Latent HIV-1 Expression. Mol. Ther. 2016, 24, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Saayman, S.M.; Lazar, D.C.; Scott, T.A.; Hart, J.R.; Takahashi, M.; Burnett, J.C.; Planelles, V.; Morris, K.V.; Weinberg, M.S. Potent and Targeted Activation of Latent HIV-1 Using the CRISPR/DCas9 Activator Complex. Mol. Ther. 2016, 24, 488–498. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Arango, G.; Li, F.; Xiao, X.; Putatunda, R.; Yu, J.; Yang, X.-F.; Wang, H.; Watson, L.T.; Zhang, L.; et al. Comprehensive Off-Target Analysis of DCas9-SAM-Mediated HIV Reactivation via Long Noncoding RNA and MRNA Profiling. BMC Med. Genom. 2018, 11, 78. [Google Scholar] [CrossRef]
- Jin, D.-Y. Special Issue: Applications of CRISPR Technology in Virology 2018. Viruses 2019, 11, 839. [Google Scholar] [CrossRef] [Green Version]
- Tough, R.H.; McLaren, P.J. Interaction of the Host and Viral Genome and Their Influence on HIV Disease. Front. Genet. 2019, 9, 720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, H.-Y.; Ji, L.-J.; Gao, A.-M.; Liu, P.; He, J.-D.; Lu, X.-J. CRISPR-Cas9 for Medical Genetic Screens: Applications and Future Perspectives. J. Med. Genet. 2016, 53, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evers, B.; Jastrzebski, K.; Heijmans, J.P.M.; Grernrum, W.; Beijersbergen, R.L.; Bernards, R. CRISPR Knockout Screening Outperforms ShRNA and CRISPRi in Identifying Essential Genes. Nat. Biotechnol. 2016, 34, 631–633. [Google Scholar] [CrossRef]
- Park, R.J.; Wang, T.; Koundakjian, D.; Hultquist, J.F.; Lamothe-Molina, P.; Monel, B.; Schumann, K.; Yu, H.; Krupzcak, K.M.; Garcia-Beltran, W.; et al. A Genome-Wide CRISPR Screen Identifies a Restricted Set of HIV Host Dependency Factors. Nat. Genet. 2017, 49, 193–203. [Google Scholar] [CrossRef]
- Jin, S.; Liao, Q.; Chen, J.; Zhang, L.; He, Q.; Zhu, H.; Zhang, X.; Xu, J. TSC1 and DEPDC5 Regulate HIV-1 Latency through the MTOR Signaling Pathway. Emerg. Microbes Infect. 2018, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved Vectors and Genome-Wide Libraries for CRISPR Screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wu, J.; Chavez, L.; Hoh, R.; Deeks, S.G.; Pillai, S.K.; Zhou, Q. Reiterative Enrichment and Authentication of CRISPRi Targets (REACT) Identifies the Proteasome as a Key Contributor to HIV-1 Latency. PLoS Pathog. 2019, 15, e1007498. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Kong, W.; Jean, M.; Fiches, G.; Zhou, D.; Hayashi, T.; Que, J.; Santoso, N.; Zhu, J. A CRISPR/Cas9 Screen Identifies the Histone Demethylase MINA53 as a Novel HIV-1 Latency-Promoting Gene (LPG). Nucleic Acids Res. 2019, 47, 7333–7347. [Google Scholar] [CrossRef]
- Rathore, A.; Iketani, S.; Wang, P.; Jia, M.; Sahi, V.; Ho, D.D. CRISPR-Based Gene Knockout Screens Reveal Deubiquitinases Involved in HIV-1 Latency in Two Jurkat Cell Models. Sci. Rep. 2020, 10, 5350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasnopolsky, S.; Kuzmina, A.; Taube, R. Genome-Wide CRISPR Knockout Screen Identifies ZNF304 as a Silencer of HIV Transcription That Promotes Viral Latency. PLoS Pathog. 2020, 16, e1008834. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, Y.; Lu, P.; Shen, Y.; Zhao, X.; Zhu, Y.; Jiang, Z.; Yang, H.; Pan, H.; Zhao, L.; et al. PEBP 1 Suppresses HIV Transcription and Induces Latency by Inactivating MAPK/NF-κB Signaling. EMBO Rep. 2020, 21, e49305. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Wang, P.; Ding, D.; Li, L.; Wang, H.; Ma, L.; Zhou, X.; Liu, S.; Lin, S.; Wang, X.; et al. Zinc-Finger-Nucleases Mediate Specific and Efficient Excision of HIV-1 Proviral DNA from Infected and Latently Infected Human T Cells. Nucleic Acids Res. 2013, 41, 7771–7782. [Google Scholar] [CrossRef] [PubMed]
- Jordan, A.; Bisgrove, D.; Verdin, E. HIV Reproducibly Establishes a Latent Infection after Acute Infection of T Cells in Vitro. EMBO J. 2003, 22, 1868–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbay, B.; Shmakova, A.; Vassetzky, Y.; Dokudovskaya, S. Modulation of MTORC1 Signaling Pathway by HIV-1. Cells 2020, 9, 1090. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.; Kim, Y.K.; Hokello, J.; Lassen, K.; Friedman, J.; Tyagi, M.; Karn, J. Epigenetic Silencing of Human Immunodeficiency Virus (HIV) Transcription by Formation of Restrictive Chromatin Structures at the Viral Long Terminal Repeat Drives the Progressive Entry of HIV into Latency. J. Virol. 2008, 82, 12291–12303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, N.; Liu, M.; Hsu, J.; Xue, Y.; Chou, S.; Burlingame, A.; Krogan, N.J.; Alber, T.; Zhou, Q. HIV-1 Tat and Host AFF4 Recruit Two Transcription Elongation Factors into a Bifunctional Complex for Coordinated Activation of HIV-1 Transcription. Mol. Cell 2010, 38, 428–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Zhao, X.; Zhu, Y.; Shen, Y.; Wang, Y.; Lu, P.; Jiang, Z.; Pan, H.; Yang, J.; Xun, J.; et al. FKBP3 Induces Human Immunodeficiency Virus Type 1 Latency by Recruiting Histone Deacetylase 1/2 to the Viral Long Terminal Repeat. mBio 2021, 12, e00795-21. [Google Scholar] [CrossRef]
- Telwatte, S.; Morón-López, S.; Aran, D.; Kim, P.; Hsieh, C.; Joshi, S.; Montano, M.; Greene, W.C.; Butte, A.J.; Wong, J.K.; et al. Heterogeneity in HIV and Cellular Transcription Profiles in Cell Line Models of Latent and Productive Infection: Implications for HIV Latency. Retrovirology 2019, 16, 32. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-C.; Martinez, J.P.; Zorita, E.; Meyerhans, A.; Filion, G.J. Position Effects Influence HIV Latency Reversal. Nat. Struct. Mol. Biol. 2017, 24, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Abner, E.; Stoszko, M.; Zeng, L.; Chen, H.-C.; Izquierdo-Bouldstridge, A.; Konuma, T.; Zorita, E.; Fanunza, E.; Zhang, Q.; Mahmoudi, T.; et al. A New Quinoline BRD4 Inhibitor Targets a Distinct Latent HIV-1 Reservoir for Reactivation from Other “Shock” Drugs. J. Virol. 2018, 92, e02056-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golumbeanu, M.; Cristinelli, S.; Rato, S.; Munoz, M.; Cavassini, M.; Beerenwinkel, N.; Ciuffi, A. Single-Cell RNA-Seq Reveals Transcriptional Heterogeneity in Latent and Reactivated HIV-Infected Cells. Cell Rep. 2018, 23, 942–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| References | Strategy Employed | Cell Line | Gene(s) Identified | Gene Function |
|---|---|---|---|---|
| Jin S, et al. [69] | Genome-scale CRISPR Knock-Out (GeCKO) lentiCRISPRv2.0 genome-wide pooled sgRNA library [70] | Jurkat-derived C11 cell line | SUV39H1, TSC1 and DEPDC5 | Heterochromatin modulation, mTOR signaling pathway modulator |
| Li Z, et al. [71] | Tet-On dCas9-KRAB-mCherry stable cells transduced with a genome-wide pooled sgRNA library, application of Reiterative Enrichment and Authentication of CRISPRi Targets (REACT) | Jurkat-derived 2D10 cell line | PSMD1, NFKBIA, CYLD, GON4L, PSMD3, and PSMD8 | Transcriptional suppression/co-repression, proteasome subunit |
| Huang H, et al. [72] | Lentiviral transduction of a sgRNA sub-pool library targeting nuclear proteins | J-Lat A2 (Tat-GFP) cell line | MINA53 | Histone demethylase |
| Rathore A, et al. [73] | Genome-scale CRISPR Knock-Out (GeCKO) lentiCRISPRv2.0 genome-wide pooled sgRNA library [70] | J-Lat 10.6 cell line | IWS1, POLE3, POLR1B, PSMD1, and TGM2 | Transcriptional repressor, proteasome subunit, heterochromatin remodeling, component of RNA Pol I, enzyme which cross-links proteins |
| Krasnopolsky S., et al. [74] | Genome-scale CRISPR Knock-Out (GeCKO) lentiCRISPRv2.0 genome-wide pooled sgRNA library [70] | Jurkat T cell line (transduced with a HIV-BFP vector) | ZNF304 | KRAB-containing zinc finger protein |
| Yang X, et al. [75] | Genome-scale CRISPR Knock-Out (GeCKO) lentiCRISPRv2.0 genome-wide pooled sgRNA library [70] | Jurkat-derived C11 cell line | PEBP1 | Kinase inhibitor protein involved in MAPK and NF-κB signaling pathways |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magro, G.; Calistri, A.; Parolin, C. Targeting and Understanding HIV Latency: The CRISPR System against the Provirus. Pathogens 2021, 10, 1257. https://doi.org/10.3390/pathogens10101257
Magro G, Calistri A, Parolin C. Targeting and Understanding HIV Latency: The CRISPR System against the Provirus. Pathogens. 2021; 10(10):1257. https://doi.org/10.3390/pathogens10101257
Chicago/Turabian StyleMagro, Gloria, Arianna Calistri, and Cristina Parolin. 2021. "Targeting and Understanding HIV Latency: The CRISPR System against the Provirus" Pathogens 10, no. 10: 1257. https://doi.org/10.3390/pathogens10101257
APA StyleMagro, G., Calistri, A., & Parolin, C. (2021). Targeting and Understanding HIV Latency: The CRISPR System against the Provirus. Pathogens, 10(10), 1257. https://doi.org/10.3390/pathogens10101257