
Sequence Diversity of Tp1 and Tp2 Antigens and Population Genetic Analysis of Theileria parva in Unvaccinated Cattle in Zambia’s Chongwe and Chisamba Districts

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results
2.1. Theileria parva PCR Screening
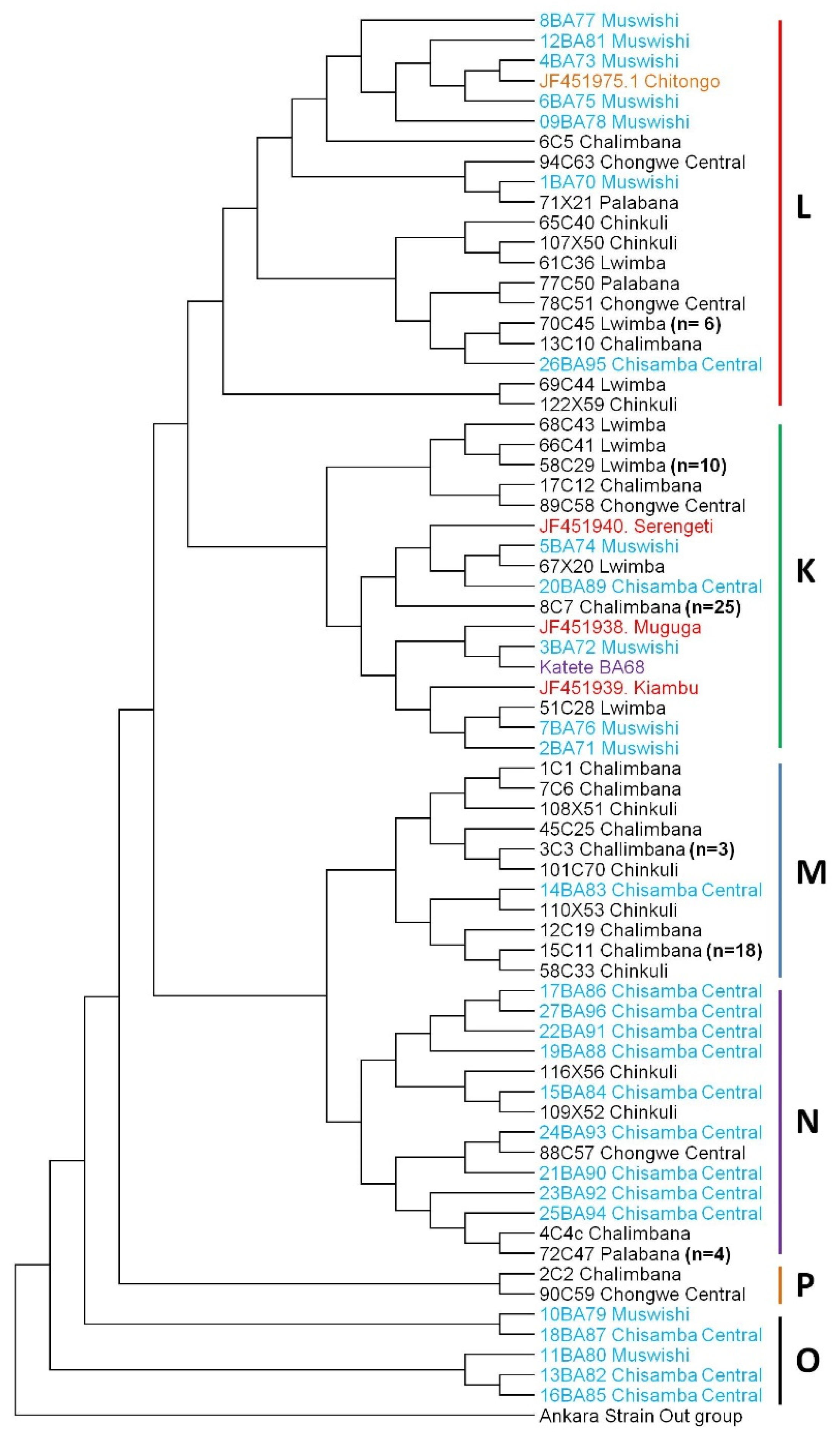
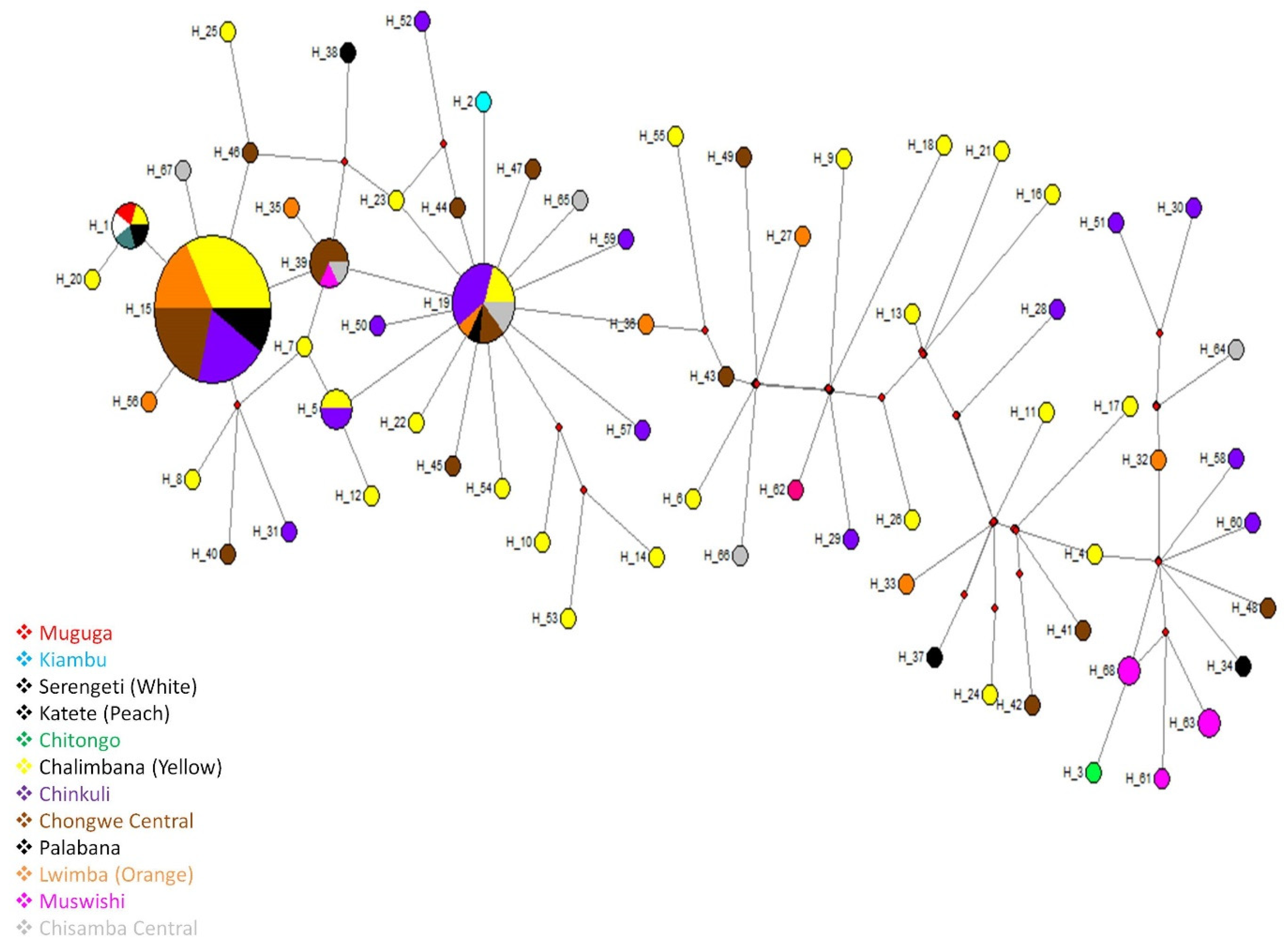
2.2. Sequence Diversity, Phylogenetic and Similarity Analyses of the Tp1 Gene Locus
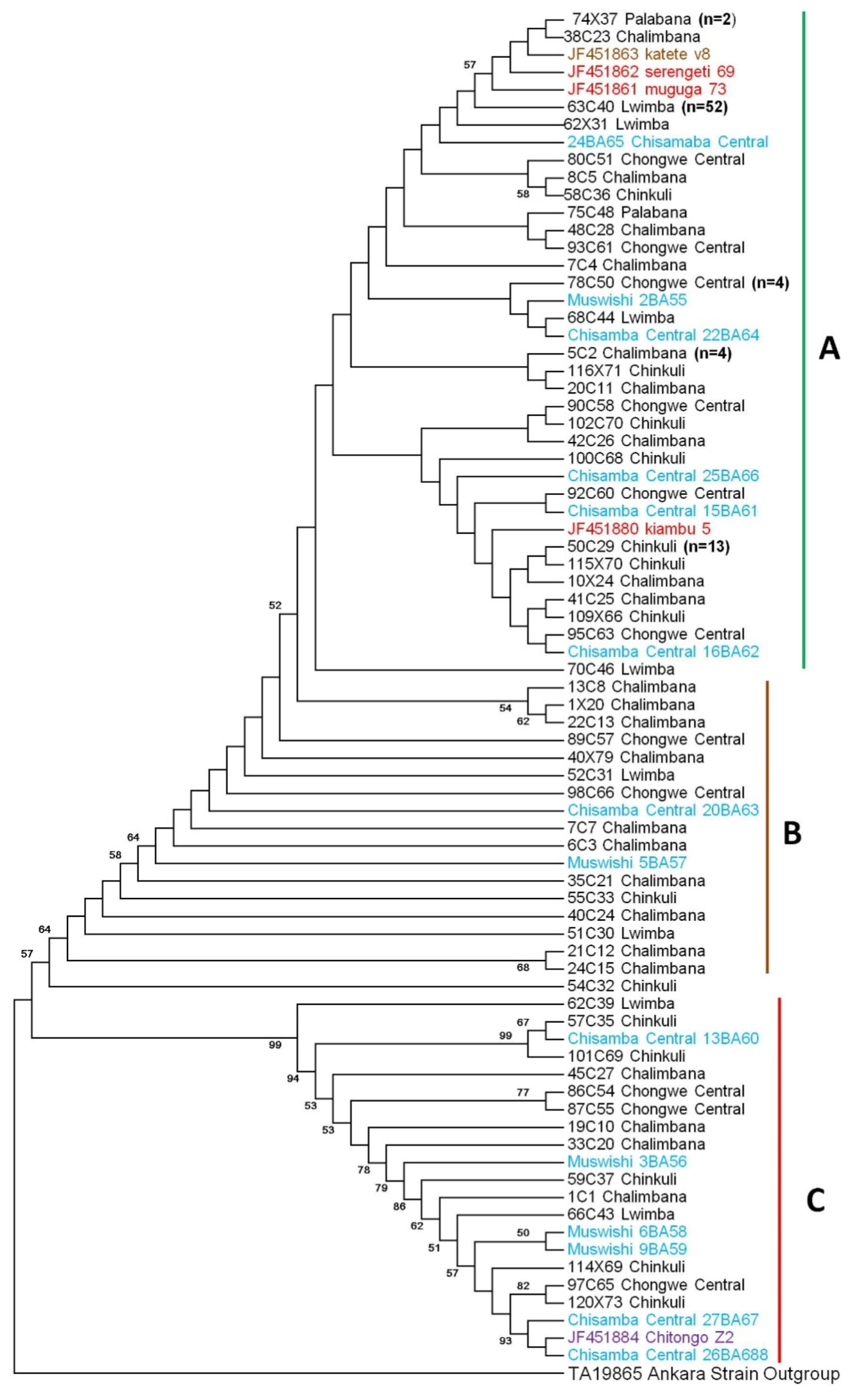
2.3. Sequence Diversity, Phylogenetic and Similarity Analyses of Tp2 Gene Locus
2.4. Microsatellite Marker and Similarity Analysis
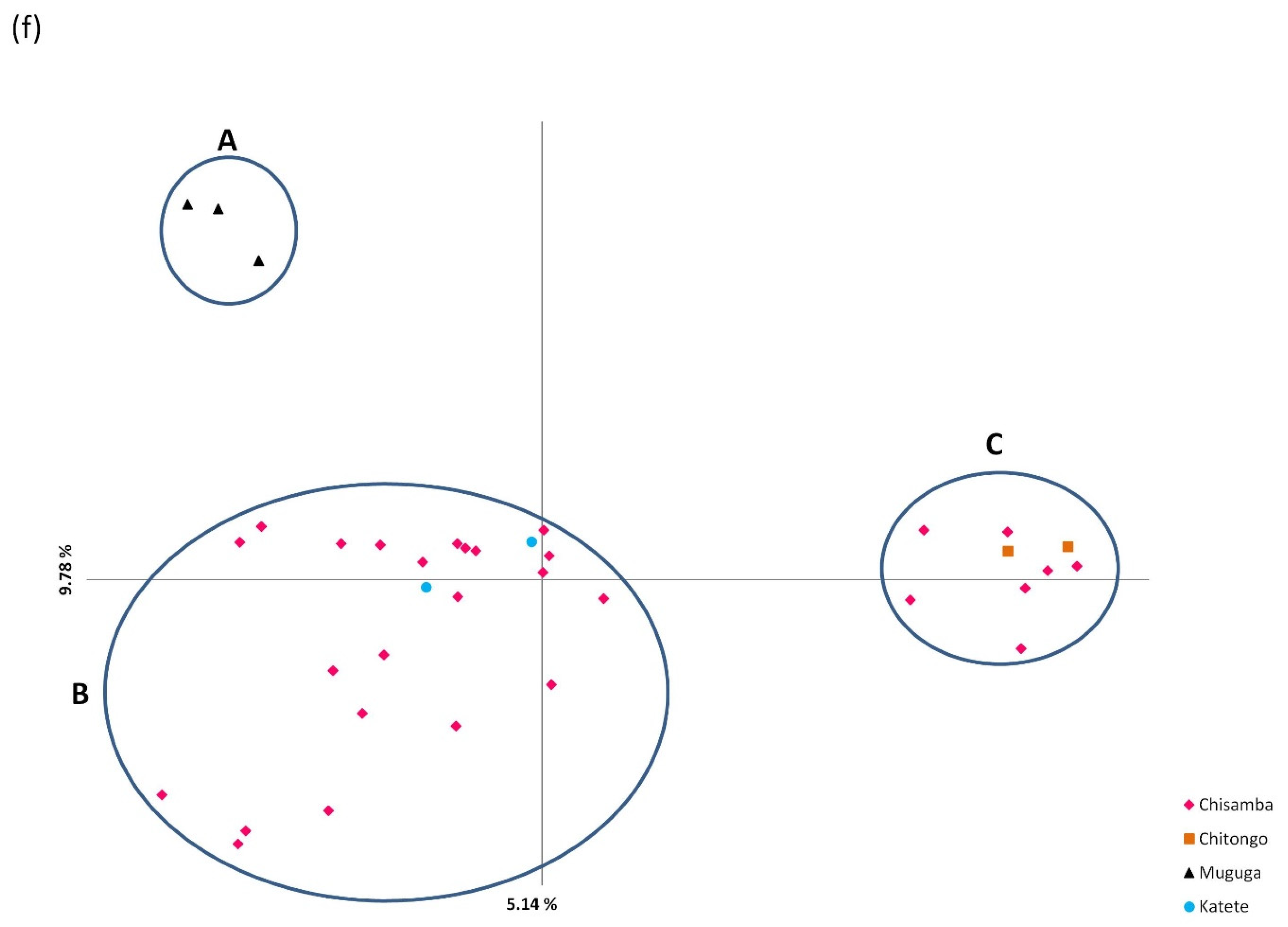
2.5. Population Differentiation and Genetic Analysis
3. Discussion
4. Materials and Methods
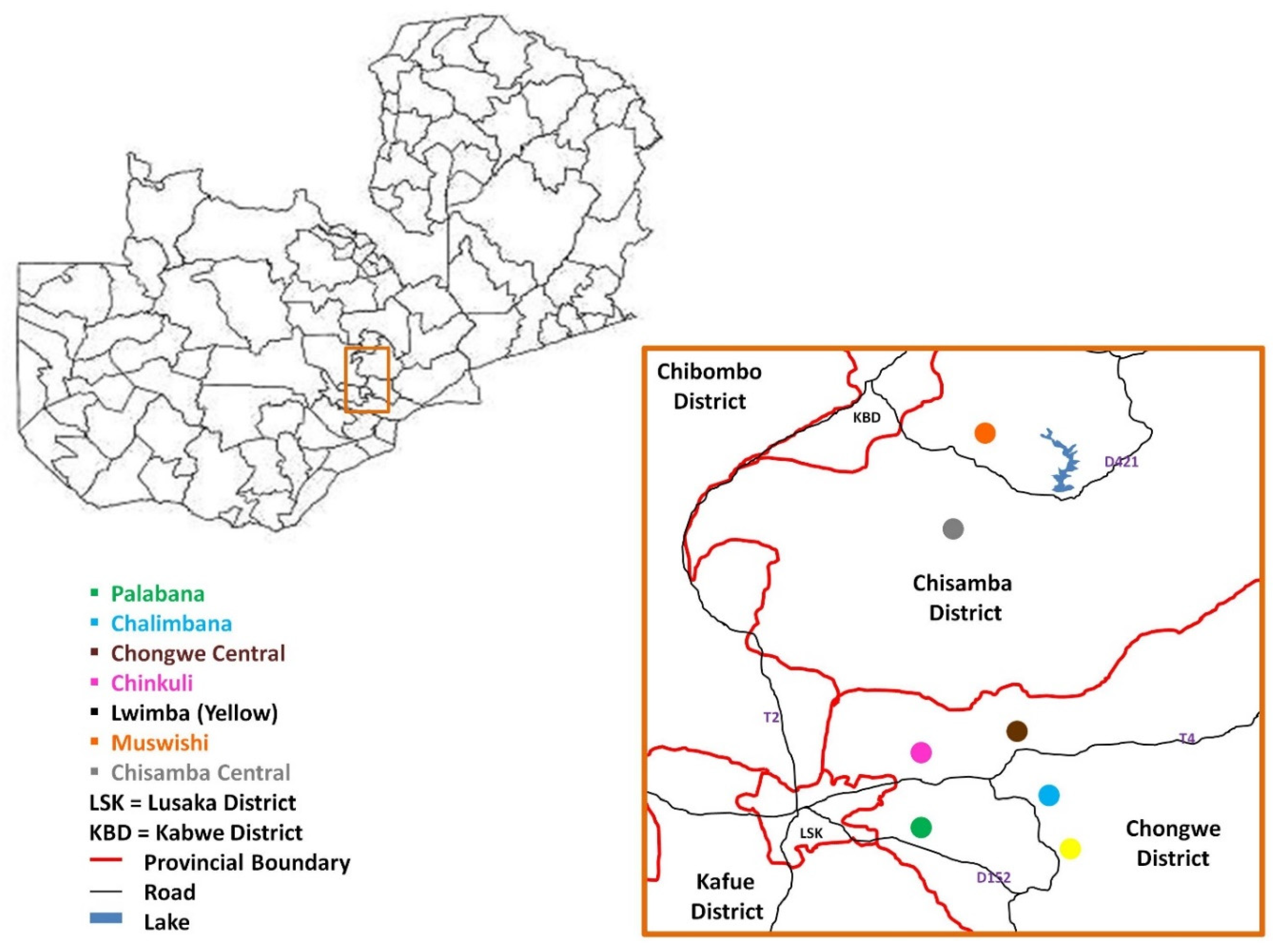
4.1. Study Sites
4.2. Sample Collection and DNA Extraction
4.3. PCR Screening of Theileria parva
4.4. PCR Amplification of Tp1 and Tp2 Genes
4.5. Cycle Sequencing
4.6. Microsatellite PCR
5. Data Analysis
5.1. Sequence Analysis
5.2. Haplotype Similarity Analysis
5.3. Microsatellite Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waladde, S.M.; Young, A.S.; Ochieng’, S.A.; Mwaura, S.N.; Mwakima, F.N. Transmission of Theileria parva to cattle by Rhipicephalus appendiculatus adults fed as nymphae in vitro on infected blood through an artificial membrane. Parasitology 1993, 107, 249–256. [Google Scholar] [CrossRef]
- Guilbride, P.D.; Opwata, B. Observations on the resistance of Jersey-Nganda calves to east coast fever (Theileria parva). Bull. Epizoot. Dis. Afr. 1963, 11, 289–298. [Google Scholar] [PubMed]
- Uilenberg, G. International collaborative research: Significance of tick-borne hemoparasitic diseases to world animal health. Vet. Parasitol. 1995, 57, 19–41. [Google Scholar] [CrossRef]
- Nambota, A.; Samui, K.; Sugimoto, C.; Kakuta, T.; Onuma, M. Theileriosis in Zambia: Etiology, epidemiology and control measures. Jpn. J. Vet. Res. 1994, 42, 1–18. [Google Scholar] [PubMed]
- Thomas, S.E.; Wilson, D.E.; Mason, T.E. Babesia, Theileria and Anaplasma spp. infecting sable antelope, Hippotragus niger (Harris, 1838), in southern Africa. Onderstepoort J. Vet. Res. 1982, 49, 163–166. [Google Scholar]
- Carmichael, I.H.; Hobday, E. Blood parasites of some wild bovidae in Botswana. Onderstepoort J. Vet. Res. 1975, 42, 55–62. [Google Scholar]
- Marcellino, W.L.; El Hussein, A.M.; Salih, D.A.; Berkvens, D.; Geysen, D. Molecular characterization of Theileria parva parasites from South Sudan using the PCR-RFLP approach on antigen genes. Arch. Razi Inst. 2015, 70, 13–20. [Google Scholar]
- Atuhaire, D.K.; Muleya, W.; Mbao, V.; Bazarusanga, T.; Gafarasi, I.; Salt, J.; Namangala, B.; Musoke, A.J. Sequence diversity of cytotoxic T cell antigens and satellite marker analysis of Theileria parva informs the immunization against East Coast fever in Rwanda. Parasit. Vectors 2020, 13, 1–20. [Google Scholar] [CrossRef]
- Chatanga, E.; Hayashida, K.; Muleya, W.; Kusakisako, K.; Moustafa, M.A.M.; Salim, B.; Katakura, K.; Sugimoto, C.; Nonaka, N.; Nakao, R. Genetic diversity and sequence polymorphism of two genes encoding Theileria parva antigens recognized by CD8+ T cells among vaccinated and unvaccinated cattle in malawi. Pathogens 2020, 9, 334. [Google Scholar] [CrossRef]
- Atuhaire, D.K.; Muleya, W.; Mbao, V.; Niyongabo, J.; Nyabongo, L.; Nsanganiyumwami, D.; Salt, J.; Namangala, B.; Musoke, A.J. Molecular characterization and population genetics of Theileria parva in Burundi’s unvaccinated cattle: Towards the introduction of East Coast fever vaccine. PLoS ONE 2021, 16, e0251500. [Google Scholar] [CrossRef]
- Nene, V.; Musoke, A.; Gobright, E.; Morzaria, S. Conservation of the sporozoite p67 vaccine antigen in cattle-derived Theileria parva stocks with different cross-immunity profiles. Infect. Immun. 1996, 64, 2056–2061. [Google Scholar] [CrossRef] [Green Version]
- Bishop, R.P.; Spooner, P.; Kanhai, G.K.; Kiarie, J.; Latif, A.A.; Hove, T.; Masaka, S.; Dolan, T.T. Molecular characterization of Theileria parasites: Application to the epidemiology of theileriosis in Zimbabwe. Parasitology 1994, 109, 573–581. [Google Scholar] [CrossRef] [Green Version]
- Geysen, D.; Bazarusanga, T.; Brandt, J.; Dolan, T.T. An unusual mosaic structure of the PIM gene of Theileria parva and its relationship to allelic diversity. Mol. Biochem. 2003, 133, 163–173. [Google Scholar] [CrossRef]
- Oura, C.A.L.; Bishop, R.; Wampande, E.M.; Lubega, G.W.; Tait, A. The persistence of component Theileria parvastocks in cattle immunized with the ‘Muguga cocktail’ live vaccine against East Coast fever in Uganda. Parasitology 2004, 129, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Hemmink, J.D. Antigenic Diversity in Theileria parva in Vaccine Stabilate and African Buffalo. Available online: http://www.era.lib.ed.ac.uk/handle/1842/9622 (accessed on 14 January 2021).
- Taracha, E.L.; Goddeeris, B.; Morzaria, S.P.; Morrison, W.I. Parasite strain specificity of precursor cytotoxic T cells in individual animals correlates with cross-protection in cattle challenged with Theileria parva. Infect. Immun. 1995, 63, 1258–1262. [Google Scholar] [CrossRef] [Green Version]
- Graham, S.; Pelle, R.; Honda, Y.; Mwangi, D.M.; Tonukari, N.J.; Yamage, M.; Glew, E.J.; de Villiers, E.P.; Shah, T.; Bishop, R. Theileria parva candidate vaccine antigens recognized by immune bovine cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA 2006, 103, 3286–3291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, S.P.; Honda, Y.; Pellé, R.; Mwangi, D.M.; Glew, E.J.; De Villiers, E.P.; Shah, T.; Bishop, R.; Van Der Bruggen, P.; Nene, V. A novel strategy for the identification of antigens that are recognised by bovine MHC class I restricted cytotoxic T cells in a protozoan infection using reverse vaccinology. Immunome Res. 2007, 3, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, S.P.; Pellé, R.; Yamage, M.; Mwangi, D.M.; Honda, Y.; Mwakubambanya, R.S.; de Villiers, E.P.; Abuya, E.; Awino, E.; Gachanja, J. Characterization of the Fine Specificity of Bovine CD8 T-Cell Responses to Defined Antigens from the Protozoan Parasite Theileria parva. Infect. Immun. 2008, 76, 685–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacHugh, N.D.; Connelley, T.; Graham, S.; Pelle, R.; Formisano, P.; Taracha, E.L.; Ellis, S.A.; McKeever, D.J.; Burrells, A.; Morrison, W.I. CD8+ T-cell responses to Theileria parva are preferentially directed to a single dominant antigen: Implications for parasite strain-specific immunity. Eur. J. Immunol. 2009, 39, 2459–2469. [Google Scholar] [CrossRef] [Green Version]
- Pelle, R.; Graham, S.P.; Njahira, M.N.; Osaso, J.; Saya, R.M.; Odongo, D.O.; Toye, P.G.; Spooner, P.R.; Musoke, A.J.; Mwangi, D.M. Two Theileria parva CD8 T Cell Antigen Genes Are More Variable in Buffalo than Cattle Parasites, but Differ in Pattern of Sequence Diversity. PLoS ONE 2011, 6, e19015. [Google Scholar] [CrossRef] [Green Version]
- Salih, D.A.; Pelle, R.; Mwacharo, J.M.; Njahira, M.N.; Marcellino, W.L.; Kiara, H.; Malak, A.K.; El Hussein, A.R.M.; Bishop, R.; Skilton, R.A. Genes encoding two Theileria parva antigens recognized by CD8+ T-cells exhibit sequence diversity in South Sudanese cattle populations but the majority of alleles are similar to the Muguga component of the live vaccine cocktail. PLoS ONE 2017, 12, e0171426. [Google Scholar] [CrossRef] [Green Version]
- Berkvens, D.L.; Geysen, D.M.; Lynen, G.M. East Coast fever Immunisation in the Eastern Province of Zambia, in Theileriosis in Eastern, Central and Southern Africa. In Proceedings of the Workshop on East Coast Fever Immunisation, Lilongwe, Malawi, 20–22 September 1988; pp. 83–86. [Google Scholar]
- Lynen, G.; Yrjö-Koskinen, A.E.; Bakuname, C.; Di Giulio, G.; Mlinga, N.; Khama, I.; Hanks, J.; Taylor, N.M.; James, A.D.; McKeever, D. East Coast fever immunisation field trial in crossbred dairy cattle in Hanang and Handeni districts in northern Tanzania. Trop. Anim. Health Prod. 2011, 44, 567–572. [Google Scholar] [CrossRef]
- Geysen, D.; Bishop, R.; Skilton, R.; Dolan, T.T.; Morzaria, S. Molecular epidemiology of Theileria parva in the field. Trop. Med. Int. Health 1999, 4, A21–A27. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Goddeeris, B.M.; Morrison, W.I.; Teale, A.J.; Bensaid, A.; Baldwin, C.L. Bovine cytotoxic T-cell clones specific for cells infected with the protozoan parasite Theileria parva: Parasite strain specificity and class I major histocompatibility complex restriction. Proc. Natl. Acad. Sci. USA 1986, 83, 5238–5242. [Google Scholar] [CrossRef] [Green Version]
- Morrison, W.I.; Goddeeris, B.M.; Teale, A.J.; Groocock, C.M.; Kemp, S.J.; Stagg, D.A. Cytotoxic T-cells elicited in cattle challenged with Theileria parva (Muguga): Evidence for restriction by class I MHC determinants and parasite strain specificity. Parasite Immunol. 1987, 9, 563–578. [Google Scholar] [CrossRef]
- Goddeeris, B.; Morrison, W.I.; Toye, P.G.; Bishop, R. Strain specificity of bovine Theileria parva-specific cytotoxic T cells is determined by the phenotype of the restricting class I MHC. Immunology 1990, 69, 38–44. [Google Scholar] [PubMed]
- Taracha, E.L.N.; Goddeeris, B.M.; Scott, J.R.; Morrison, W.I. Standardization of a technique for analysing the frequency of parasite-specific cytotoxic T lymphocyte precursors in cattle immunized with Theileria parva. Parasite Immunol. 1992, 14, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Amzati, G.S.; Djikeng, A.; Odongo, D.O.; Nimpaye, H.; Sibeko, K.P.; Muhigwa, J.-B.B.; Madder, M.; Kirschvink, N.; Marcotty, T. Genetic and antigenic variation of the bovine tick-borne pathogen Theileria parva in the great lakes region of Central Africa. Parasit. Vectors 2019, 12, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Muleya, W.; Namangala, B.; Simuunza, M.; Nakao, R.; Inoue, N.; Kimura, T.; Ito, K.; Sugimoto, C.; Sawa, H. Population genetic analysis and sub-structuring of Theileria parva in the northern and eastern parts of Zambia. Parasit. Vectors 2012, 5, 255-11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skilton, R.A.; Bishop, R.P.; Katende, J.M.; Mwaura, S.; Morzaria, S.P. The persistence of Theileria parva infection in cattle immunized using two stocks which differ in their ability to induce a carrier state: Analysis using a novel blood spot PCR assay. Parasitology 2002, 124, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Poon, A.F.; Frost, S.D.; Pond, S.L.K. Detecting Signatures of Selection from DNA Sequences Using Datamonkey. In Bioinformatics for DNA Sequence Analysis; Springer: Berlin/Heidelberg, Germany, 2009; Volume 537, pp. 163–183. [Google Scholar]
- Delport, W.; Poon, A.F.Y.; Frost, S.D.W.; Pond, S.L.K. Datamonkey 2010: A suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peakall, R.O.D.; Smouse, P.E. genalex 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. 2005, 1, 47–50. [Google Scholar] [CrossRef] [Green Version]
- Haubold, B.; Hudson, R.R. LIAN 3.0: Detecting linkage disequilibrium in multilocus data. Bioinformatics 2000, 16, 847–849. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CTL 1 | CTL 2 | CTL 3 | CTL 4 | CTL 5 | CTL 6 | |
|---|---|---|---|---|---|---|
| SHEELNKLGML (9) | PDLDKNTLF (9) | LTSHGMGKIGR (3) | LAPSIKCVS (2) | ASIKCVAQK (1) | KPSVPNPCIW (9) | |
| SDEELDNLGMF (1) | HGLDKNTLF (1) | LSSHGMGRIGR (8) | LAASIKCVS (1) | PSIKCVSHH (2) | KTSIPNPCEW (1) | |
| SDEELDNLGML (1) | PDLDRNTLF (3) | LSSHGMGRIGK (5) | FAASIKCVS (2) | ASIKCVSHH (1) | KNSIPNPCKW (1) | |
| SDDELDNLGML (4) | DDLDRNALF (1) | LSSHGMGKVGK (3) | FAPSIKCVA (1) | ASIKCVSQY (2) | KPSVPNPCKW (2) | |
| SHEELKKMGMV (1) | HDFDKNTLF (1) | LPSHGMGRIGR (2) | FAQSLMCVS (1) | PSIKCVAQY (1) | KTDIPNPCKW (2) | |
| SDEGLNKLGML (1) | QDLDKNTLF (1) | RSSHGMGKVGK (1) | FAQSLMCVL (10) | QSLMCVSQK (1) | KPSIPNPCKW (1) | |
| SDDELNNLGML (4) | PDSDRNTLF (1) | LSSHGMGKIGK (2) | FAPSLMCVL (1) | QSLMCVLQK (1) | KPSGPNPCIW (1) | |
| SDNELDTLGLL (1) | HDFDRNTLF (1) | LSSHGMGKIGK (1) | FAASLMCVS (1) | QSLMCVLLK (1) | VTDIPNPCKW (1) | |
| SDEELDTLGML (1) | EGFDKDALF (1) | ISSHGMGKVGK (1) | FAQSLKCVL (1) | PSLMCVLLN (1) | ||
| SDNELDTLGLL (1) | HGLARNTLF (1) | KSSQSMGIVGR (1) | FAASIKCVL (2) | ASLMCVSLK (1) | ||
| SEDELDTLGML (1) | DGLDRNTLF (1) | FVQSIMCVI (1) | QSLMCVLMK (9) | |||
| SDDELNKLGML (2) | DGLDRDALF (1) | QSLKCVLLK (1) | ||||
| SDDELKNLGLL (1) | DDFDRDALF (1) | ASIKCVLQY (2) | ||||
| SDNELDTLGLL (1) | DGFDRNALF (1) | QSLVCVLLK (1) | ||||
| SDEELNKSGML (1) | DDFDRDTLF (1) | QSIMCVINK (1) | ||||
| TEEELNNMGMV (1) | EGFDKDALF (1) | |||||
| Total | 31 | 26 | 27 | 24 | 26 | 18 |
| Population | N | MS2 | MS7 | ms9 | MS14 | MS15 | MS19 | MS25 | MS33 | MS39 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Alleles within the Populations | Chongwe | 96 | 13 | 23 | 17 | 55 | 14 | 27 | 17 | 40 | 21 |
| Chisamba | 29 | 8 | 14 | 4 | 6 | 5 | 6 | 5 | 14 | 4 | |
| Muguga cocktail | 3 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | |
| Katete | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | |
| Chitongo | 2 | 1 | 2 | 1 | 2 | 1 | 1 | 2 | 2 | 1 | |
| Gene Diversity | Chongwe | 96 | 0.856 | 0.907 | 0.784 | 0.970 | 0.708 | 0.942 | 0.878 | 0.953 | 0.894 |
| Chisamba | 29 | 0.833 | 0.897 | 0.554 | 0.374 | 0.613 | 0.719 | 0.495 | 0.921 | 0.707 | |
| Muguga cocktail | 3 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.667 | 0.00 | 0.00 | 0.00 | |
| Katete | 2 | 1.00 | 0.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | |
| Chitongo | 2 | 0.00 | 1.00 | 0.00 | 1.00 | 0.00 | 0.00 | 1.00 | 1.00 | 0.00 | |
| Overall | 132 | 0.538 | 0.561 | 0.468 | 0.669 | 0.464 | 0.666 | 0.675 | 0.775 | 0.520 |
| Population | N | Effective Alleles | Ht | VD | L | p-Value | ISA | Linkage | FST |
|---|---|---|---|---|---|---|---|---|---|
| Chongwe | 96 | 6.086 | 0.8767 | 1.1059 | 0.9673 | <0.01 | 0.0260 | LD | 0.036 |
| Chisamba | 29 | 3.574 | 0.6719 | 2.0003 | 1.8916 | <0.01 | 0.0233 | LD | −0.012 |
| Chongwe + Chisamba | 125 | 7.426 | 0.8629 | 1.3044 | 1.0620 | <0.01 | 0.0373 | LD | 0.0960 |
| Chongwe + Chisamba + Vaccines | 132 | 3.855 | 0.8641 | 1.3357 | 1.0518 | <0.01 | 0.0421 | LD | 0.1205 |
| Marker | Chromosome | Colour | Annealing Temp | Bp |
|---|---|---|---|---|
| MS2 | 1 | Red | 60 | 342–562 |
| MS3 | 1 | Red | 60 | 193–380 |
| MS8 | 1 | Red | 60 | 160–330 |
| MS14 | 2 | Yellow | 60 | 360–600 |
| MS15 | 2 | Red | 60 | 120–220 |
| MS21 | 3 | Red | 60 | 170–400 |
| MS27 | 3 | Yellow | 60 | 130–220 |
| MS30 | 3 | Red | 60 | 170–230 |
| MS33 | 4 | Yellow | 60 | 150–220 |
| MS40 | 4 | Red | 60 | 150–250 |
| MS25 | 3 | Red | 50 | 324 |
| MS39 | 4 | Yellow | 50 | 263 |
| MS7 | 1 | Red | 50 | 372 |
| MS19 | 2 | Yellow | 50 | 304 |
| ms9 | 3 | Yellow | 50 | 230 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muleya, W.; Atuhaire, D.K.; Mupila, Z.; Mbao, V.; Mayembe, P.; Kalenga, S.; Fandamu, P.; Namangala, B.; Salt, J.; Musoke, A.J. Sequence Diversity of Tp1 and Tp2 Antigens and Population Genetic Analysis of Theileria parva in Unvaccinated Cattle in Zambia’s Chongwe and Chisamba Districts. Pathogens 2022, 11, 114. https://doi.org/10.3390/pathogens11020114
Muleya W, Atuhaire DK, Mupila Z, Mbao V, Mayembe P, Kalenga S, Fandamu P, Namangala B, Salt J, Musoke AJ. Sequence Diversity of Tp1 and Tp2 Antigens and Population Genetic Analysis of Theileria parva in Unvaccinated Cattle in Zambia’s Chongwe and Chisamba Districts. Pathogens. 2022; 11(2):114. https://doi.org/10.3390/pathogens11020114
Chicago/Turabian StyleMuleya, Walter, David Kalenzi Atuhaire, Zachariah Mupila, Victor Mbao, Purity Mayembe, Sydney Kalenga, Paul Fandamu, Boniface Namangala, Jeremy Salt, and Antony Jim Musoke. 2022. "Sequence Diversity of Tp1 and Tp2 Antigens and Population Genetic Analysis of Theileria parva in Unvaccinated Cattle in Zambia’s Chongwe and Chisamba Districts" Pathogens 11, no. 2: 114. https://doi.org/10.3390/pathogens11020114
APA StyleMuleya, W., Atuhaire, D. K., Mupila, Z., Mbao, V., Mayembe, P., Kalenga, S., Fandamu, P., Namangala, B., Salt, J., & Musoke, A. J. (2022). Sequence Diversity of Tp1 and Tp2 Antigens and Population Genetic Analysis of Theileria parva in Unvaccinated Cattle in Zambia’s Chongwe and Chisamba Districts. Pathogens, 11(2), 114. https://doi.org/10.3390/pathogens11020114